
Abstract
When molecular transitions strongly couple to photon modes, they form hybrid light-matter modes called polaritons. Collective vibrational strong coupling is a promising avenue for control of chemistry, but this can be deterred by the large number of quasi-degenerate dark modes. The macroscopic occupation of a single polariton mode by excitations, as observed in Bose-Einstein condensation, offers promise for overcoming this issue. Here we theoretically investigate the effect of vibrational polariton condensation on the kinetics of electron transfer processes. Compared with excitation with infrared laser sources, the vibrational polariton condensate changes the reaction yield significantly at room temperature due to additional channels with reduced activation barriers resulting from the large accumulation of energy in the lower polariton, and the many modes available for energy redistribution during the reaction. Our results offer tantalizing opportunities to use condensates for driving chemical reactions, kinetically bypassing usual constraints of fast intramolecular vibrational redistribution in condensed phase.
Similar content being viewed by others

Introduction
Light and matter couple strongly when a large number of molecules are placed within optical cavities that confine light1,2,3. As a result, hybrid light-matter excitations called polaritons form when a collective molecular transition and a photon mode coherently exchange energy faster than the individual decay from each component. Light-matter strong coupling (SC) opens up a new path to modify material properties by controlling their electromagnetic environment4,5. For instance, vibrational strong coupling (VSC), where an infrared cavity mode couples to an ensemble of localized molecular vibrations in a film or solution, influences chemical reactivity even without external pumping6,7. However, the microscopic mechanism for modification of molecular processes through hybridization with light is still poorly understood8,9,10, since it could be limited by the presence of a large number of quasi-degenerate dark modes that do not possess any photonic character and are likely to behave similarly to uncoupled molecules10.
A Bose–Einstein condensate of polaritons11 offers a solution to this problem since the macroscopic occupation of polaritonic states enhances the effects from SC. In the last decade, Bose–Einstein condensation has been demonstrated in several organic exciton-polariton systems at room temperature12,13,14,15. Recently, organic polariton condensates were used to build polariton transistors16, and theoretical predictions suggest they may also modify incoherent charge transport17. Interestingly, the consequences of polariton condensation on chemical reactivity have not been addressed in the literature prior to the present study.
Ideas of using Bose–Einstein condensates of molecules in chemistry have been previously proposed, but they require ultracold temperatures due to the large mass of the condensing entities18,19. The low effective mass that polaritons inherit from their photonic component, along with the large binding energy of Frenkel excitons, enables room-temperature condensation20. The partly photonic character of polaritons also offers additional benefits such as delocalization and remote action for manipulating chemistry21.
Here, we investigate the effect of polariton condensation on electron transfer (ET) (Fig. 1). While the reaction yield under infrared laser excitation, without SC, already differs from that under thermal equilibrium conditions22,23, polariton condensation amplifies this difference by changing the activation barrier for the forward and backward reactions unevenly, tilting the equilibrium towards either reactants or products.

A large number of molecules are placed inside an optical cavity where their vibrations strongly couple to the cavity mode. The system is constantly pumped to create a polariton condensate and the right side of the figure depicts the occupation of different modes under condensation (frequencies of the upper polariton, dark modes, and lower polariton are ω+, ωvib, and ω−, respectively). The rate of intramolecular electron transfer under vibrational polariton condensation is investigated.
Results
Bose–Einstein condensation of vibrational polaritons
Bose–Einstein condensation of vibrational polaritons has not been experimentally achieved yet; however, as we shall argue, there are compelling reasons to believe that they are presently within reach. Most theoretical investigations on polariton condensation with organic microcavities involve systems under electronic strong coupling (ESC)24,25; polariton condensation under VSC requires a separate treatment due to the difference in energy scales and the involved relaxation pathways26. While typical bare exciton energies range from 2–3 eV with Rabi splitting ~200 meV under ESC, the bare frequency of vibrations is 100–300 meV with Rabi splitting ~20−40 meV under VSC. Since the Rabi splitting is of the order of kBT at room temperature, thermal effects are crucial for vibrational polaritons. Under ESC, polariton relaxation is assisted by high-frequency intramolecular vibrations27, whereas, under VSC, low-frequency solvent modes play a key role in this process28,29, similar to what happens in THz phonon Fröhlich condensation in biomolecules30,31.
We model the polariton system as a set of N vibrational modes (\({\hat{a}}_{{{{{{{{\rm{vib}}}}}}}},j}\)), with frequency ωvib, strongly coupled to a single photon mode (\({\hat{a}}_{{{{{{{{\rm{ph}}}}}}}}}\)) with frequency ωph. In the Hamiltonian of the system,
we have applied the rotating wave approximation. Upon diagonalizing this Hamiltonian, we get normal modes: lower and upper polaritons, and N − 1 dark mode with frequencies ω−, ω+, and \({\omega }_{{{{{{{{\rm{D}}}}}}}}}^{k}\), respectively:
where \({{\Omega }}=\sqrt{{{{\Delta }}}^{2}+4{g}^{2}N}\) is the Rabi splitting and Δ = ωph − ωvib the detuning between cavity and molecular vibrations. To model polariton population dynamics, we use Boltzmann rate equations where the polariton system is weakly coupled to a low-frequency solvent bath, which enables scattering between modes32. These rate equations also account for final-state stimulation,
where ni is the population, γi is the decay rate and Pi is the external pumping rate of the ith mode. The scattering rate from mode j to i, Wij, satisfies detailed balance: \({W}_{ij}/{W}_{ji}={e}^{-\beta ({\epsilon }_{i}-{\epsilon }_{j})}\). Here, β = 1/(kBT), kB is the Boltzmann constant, T is the temperature, and ϵi = ℏωi where ωi is the frequency of the ith mode. The decay from different modes is \({\gamma }_{i}=| {c}_{{{{{{{{\rm{vib}}}}}}}}}^{i}{| }^{2}{{{\Gamma }}}_{\downarrow }+| {c}_{{{{{{{{\rm{ph}}}}}}}}}^{i}{| }^{2}\kappa\), where \(| {c}_{{{{{{{{\rm{vib}}}}}}}}}^{i}{| }^{2}\) and \(| {c}_{{{{{{{{\rm{ph}}}}}}}}}^{i}{| }^{2}\) are the molecular and photon fraction, respectively, Γ↓ is the decay rate of the molecular vibrations, and κ is the cavity leakage rate.
Two factors play a determining role in the condensation threshold: (i) the rate of scattering between polariton and dark modes relative to losses from the system, i.e., the rate of thermalization and (ii) the abundance of modes close in energy to the condensing mode33. For all calculations, we assume fast thermalization. As mentioned in (ii), the presence of many modes close to the lower polariton would deter condensation by distributing the energy pumped into the system among all these modes. Thus, the energetic proximity between the dark state manifold, which has a large density of states (DOS), and the lower polariton pose one of the biggest challenges for polariton condensation under VSC.
The distribution of excitations between the polariton and dark modes is shown in Fig. 2 for different detunings and we observe a condensation transition at ℏΔ ≈ −1.5kBT (see Supplementary Note 2 for details). Above the condensation threshold, a large fraction of excitations resides in the lower polariton \(\frac{{n}_{-}}{(\mathop{\sum }\nolimits_{k = 2}^{\infty }{n}_{{{{{{{{\rm{D}}}}}}}}}^{k})}\gg \frac{1}{(N-1){e}^{-\beta \hslash ({{\Omega }}-{{\Delta }})/2}}\).

Fraction of excitations in different modes as a function of cavity detuning ℏΔ = ℏωph − ℏωvib while keeping the pumping rate P− fixed. The black curve includes the excitations in all dark modes taken together while the green and orange curves show the fraction excitations in the lower and upper polariton, respectively. The condensation transition takes place close to ℏΔ ≈ −1.5kBT. Here, the lower polariton is pumped with rate P− = 0.16NΓ↓, the light-matter coupling \(2\hslash g\sqrt{N}=0.72{k}_{{{{{{{{\rm{B}}}}}}}}}T\), the temperature kBT = 0.1389ℏωvib (T = 298 K when ℏωvib = 185 meV), number of molecules N = 107 and cavity leakage rate κ = Γ↓.
The average population per molecule at the condensation threshold \(\bar{n}={P}_{{{{{{{{\rm{th}}}}}}}}}/N{{{\Gamma }}}_{\downarrow }\) is a good measure of the feasibility of vibrational polariton condensation. For instance, demanding population inversion, \(\bar{n} \, > \, 0.5\), would be experimentally difficult to achieve in general. In Fig. 3, we plot \(\bar{n}\) for different light-matter coupling strengths, \(2\hslash g\sqrt{N}\), and detunings, ℏΔ. Here, we numerically obtain Pth as the pumping rate when 10% of the excitations are in the lower polariton. The threshold obtained this way closely corresponds with the theoretical condition for condensation
where, \({\bar{n}}_{{{{{{{{\rm{D}}}}}}}}}^{k}=\frac{1}{N-1}\mathop{\sum }\nolimits_{k = 2}^{N}{n}_{{{{{{{{\rm{D}}}}}}}}}^{k}\) is the average occupation of a dark mode, and nsolvent(E) is the Bose–Einstein population of a solvent mode with energy E at room temperature Troom33. The energy difference between the lower polariton and the dark state reservoir ℏ(Ω − Δ)/2 determines the condensation threshold. From Fig. 3 we see that vibrational polariton condensation is feasible for water even at room temperature for up to zero detuning.

Numerically obtained average population per molecule at the condensation threshold \(\bar{n}={P}_{{{{{{{{\rm{th}}}}}}}}}/N{{{\Gamma }}}_{\downarrow }\) (10% of the excitations are in the lower polariton), for a range of light-matter coupling strengths \(2\hslash g\sqrt{N}\) and cavity detunings ℏΔ = ℏωph − ℏωvib. In the black and purple regions of the plot (Δ > 0 and \(2\hslash g\sqrt{N}/{k}_{{{{{{{{\rm{B}}}}}}}}}T \, < \, 2\)), the threshold for condensation is high, \(\bar{n}\gg 1\), and polariton condensation is difficult to achieve experimentally. The threshold for condensation is much lower, \(\bar{n} \, < \, 0.1\), for the lighter colored (yellow, orange) regions. In our plot above only the upper polariton is pumped and we use cavity leakage rate κ = Γ↓. The vertical lines correspond to \(2\hslash g\sqrt{N}/{k}_{{{{{{{{\rm{B}}}}}}}}}T\) at room temperature for H2O (\(2\hslash g\sqrt{N}\approx 700{{{{{{{{\rm{cm}}}}}}}}}^{-1}\)) and W(CO)6 (\(2\hslash g\sqrt{N}\approx 50{{{{{{{{\rm{cm}}}}}}}}}^{-1}\)). Calculations in Figs. 4–6 are presented for the conditions in point A.
Our model does not include disorder; as a result, all dark modes are degenerate at frequency ωvib, but in experimental systems, inhomogeneous broadening of transitions can lead to a nonzero density of dark states even at the bottom of the lower polariton branch34. This fact will affect the condensation threshold and should be considered in the future while looking for experimental systems that can demonstrate vibrational polariton condensation. Stimulating the lower polariton directly by shining a resonant laser on it16 or using a Raman scattering scheme35 can help overcome this issue by dynamically lowering the condensation threshold.
Chemical reactions and vibrational polariton condensation
Electron transfer has been theoretically studied under both ESC36,37 and VSC38,39. Here, we look at how vibrational polariton condensation affects the rate of intramolecular nonadiabatic electron transfer using the VSC version38 of the Marcus–Levich–Jortner (MLJ) model40,41,42.
Our system consists of N molecules placed inside an optical cavity supporting a single photon mode with a bosonic operator \({\hat{a}}_{{{{{{{{\rm{ph}}}}}}}}}\) and frequency ωph. The molecules can be in the reactant R or product P electronic state; for the ith molecule, these states are denoted by \(\left|{{{{{{{{\rm{R}}}}}}}}}_{i}\right\rangle\) and \(\left|{{{{{{{{\rm{P}}}}}}}}}_{i}\right\rangle\), respectively. Each electronic state is dressed with a high-frequency intramolecular vibrational mode with a bosonic operator \({\hat{a}}_{x,i}\) and frequency ωvib where x = R, P; this mode couples to the photon mode. The equilibrium geometry of this vibrational mode depends on the electronic state according to,
where \({\hat{D}}_{i}=\exp (({\hat{a}}_{{{{{{{{\rm{P}}}}}}}},i}^{{{{\dagger}}} }-{\hat{a}}_{{{{{{{{\rm{P}}}}}}}},i})\sqrt{S})\) and S is the Huang–Rhys factor.
Apart from the intramolecular vibrations, an effective low-frequency solvent mode surrounding each molecule facilitates ET. It is treated classically, with qS,i and pS,i being its position and momentum.
The Hamiltonian \(\hat{H}\) for the full system is a generalization of Eq. (1) to account for the chemical reaction,
and
where \({\hat{H}}_{0}\) describes the photon (\({\hat{H}}_{{{{{{{{\rm{ph}}}}}}}}}\)), intramolecular vibrations and solvent modes of the ith molecule (\({\hat{H}}_{x,i}\)), and light-matter couplings (\({\hat{V}}_{x,i}\)). The diabatic coupling \({\hat{V}}_{{{{{{{{\rm{react}}}}}}}}}\) is a perturbation that couples R and P electronic states with coupling strength JRP. We have taken the dipole moment to be zero when the vibrational coordinate is in its equilibrium position in both R and P electronic states. Relaxing this assumption will add terms of the form \({c}_{x}({\hat{a}}_{{{{{{{{\rm{ph}}}}}}}}}+{\hat{a}}_{{{{{{{{\rm{ph}}}}}}}}})\left|{x}_{i}\right\rangle \left\langle {x}_{i}\right|\) to the Hamiltonian and will not affect the reaction rates calculated.
where ΔG is the free-energy difference of each individual molecule reaction.
We construct potential energy surfaces (PES) by parametrically diagonalizing \({\hat{H}}_{0}\) as a function of the solvent coordinate qS,i. The operators \({\hat{N}}_{{{{{{{{\rm{R}}}}}}}}}=\mathop{\sum }\nolimits_{i = 1}^{N}\left|{{{{{{{{\rm{R}}}}}}}}}_{i}\right\rangle \left\langle {{{{{{{{\rm{R}}}}}}}}}_{i}\right|\) and \({\hat{N}}_{{{{{{{{\rm{P}}}}}}}}}=\mathop{\sum }\nolimits_{i = 1}^{N}\left|{{{{{{{{\rm{P}}}}}}}}}_{i}\right\rangle \left\langle {{{{{{{{\rm{P}}}}}}}}}_{i}\right|\) commute with H0 and correspond to the number of R and P molecules, respectively. While dynamics under \({\hat{H}}_{0}\) conserves NR, NP, the effect of \({\hat{V}}_{{{{{{{{\rm{react}}}}}}}}}\) is to induce reactive transitions that modify those quantities while keeping N = NR + NP constant. We assign the label 1 ≤ i ≤ NR to R molecules, and NR + 1 ≤ i ≤ N to P molecules. We also reorganize the intramolecular vibrations into a single bright mode,
that possesses the correct symmetry to couple with light and N − 1 dark modes (Dk),
labeled by an additional index 2 ≤ k ≤ N, which do not couple with light. The dark modes are orthogonal to the bright mode \({g}_{{{{{{{{\rm{R}}}}}}}}}\mathop{\sum }\nolimits_{i = 1}^{{N}_{{{{{{{{\rm{R}}}}}}}}}}{c}_{k,i}+{g}_{{{{{{{{\rm{P}}}}}}}}}\mathop{\sum }\nolimits_{i = {N}_{{{{{{{{\rm{R}}}}}}}}}+1}^{N}{c}_{k,i}=0\), and to each other \(\mathop{\sum }\nolimits_{i = 1}^{N}{c}_{k,i}{c}_{k^{\prime} ,i}^{* }={\delta }_{k,k^{\prime} }\). Unless mentioned otherwise, the number of R and P molecules is NR and NP, respectively, and for brevity, we will drop (NR, NP) dependence in the subscript. The bright and photon modes combine to form the upper polariton (UP) \({\hat{a}}_{+}\), and lower polariton (LP) \({\hat{a}}_{-}\), modes:
with mixing angle,
where \({{\Omega }}=\sqrt{{{{\Delta }}}^{2}+4({g}_{{{{{{{{\rm{R}}}}}}}}}^{2}{N}_{{{{{{{{\rm{R}}}}}}}}}+{g}_{{{{{{{{\rm{P}}}}}}}}}^{2}{N}_{{{{{{{{\rm{P}}}}}}}}})}\) is the Rabi splitting, and Δ = ωph − ωvib the detuning between the cavity and molecular vibrations. The eigenstates of \({\hat{H}}_{0}\) are the dark, upper, and lower polariton modes with frequencies given in Eq. (2).
According to the MLJ theory, the rate constant for ET outside of an optical cavity depends on properties of the intramolecular and solvent modes40,41,42. Under laser driving, this rate constant is,
where
Here, \({P}_{\bar{n}}(n)\) is the Poisson distribution with average mode population \(\bar{n}\), λS is the solvent reorganization energy, \({E}_{f}^{{{{\ddagger}}} }\) is the activation energy, and \(| \left\langle n| n+f\right\rangle ^{\prime} {| }^{2}\) is the Franck–Condon (FC) factor, where \(\left|n\right\rangle\) and \(\left|n+f\right\rangle ^{\prime}\) are the intramolecular initial and final states, respectively. \({P}_{\bar{n}}(n)\) has been taken to correspond to the ideal laser-driven-damped harmonic oscillator, leading to a coherent state in the vibrational mode. The presence of anharmonic couplings would lead to intramolecular vibrational energy redistribution (IVR)43, reducing the value of \({P}_{\bar{n}}(n)\) for high-lying Fock states. However, as we shall see below, even under these ideal circumstances, the condensate can outcompete the laser-driven situation in terms of reactivity. We thus expect the benefits of the condensate to be enhanced when IVR processes are taken into account.
Apart from vibrations within the reacting molecule, under VSC, the ET process also depends on vibrations in all other molecules and the photon mode, and can be represented by,
Here and hereafter, the primed and unprimed quantities refer to electronic states with (NR, NP) and (NR − 1, NP + 1) reactant-product distributions, respectively. The symmetry of the light-matter coupling allows us to use the dark state basis introduced in44 and38 to reduce the number of modes involved in the reaction from N + 1 to three,
Here, the cth molecule is reacting, while Dx,c and \({{{{{{{{\rm{D}}}}}}}}_{x,c}^{\prime}}\) are dark modes highly localized in it, with corresponding operators \({\hat{a}}_{{{{{{{{\rm{D}}}}}}}}}^{(R,c)}\) and \({\hat{a}}_{{{{{{{{\rm{D}}}}}}}}}^{(P,c)^{\prime} }\) (see Supplementary Note 1).
We perform all our calculations in this subsection using parameters from point A in Fig. 3 but while pumping the lower polariton. Here, ℏΔ = −0.5kBT, \(2\hslash g\sqrt{N}=0.72{k}_{{{{{{{{\rm{B}}}}}}}}}T\), kBT = 0.1389ℏωvib (T = 298 K when ℏωvib = 185 meV) and N = 107; we choose pumping rate P− = 0.08NΓ↓, which leads to average mode populations N+ = 0.064, N− = 1.94 × 104 and ND = 0.078 under symmetric coupling gR = gP = g. Here, 2.4% of all excitations reside in the lower polariton. To compare the reaction rates under polariton condensation and outside the cavity under pumping, we take \(\bar{n}=0.08\) in Eq. (13). Under condensation, the initial vibrational state of the system can be described by \(\rho ={\sum }_{{n}_{+},{n}_{-},{n}_{{{{{{{{\rm{D}}}}}}}}}}P({n}_{+},{n}_{-},{n}_{{{{{{{{\rm{D}}}}}}}}})\left|{n}_{+},{n}_{-},{n}_{{{{{{{{\rm{D}}}}}}}}}\right\rangle \left\langle {n}_{+},{n}_{-},{n}_{{{{{{{{\rm{D}}}}}}}}}\right|\), where the entries in \(\left|{n}_{+},{n}_{-},{n}_{{{{{{{{\rm{D}}}}}}}}}\right\rangle\) label number of quanta in the UP, LP, and DR,c modes, respectively. The results from the rate equations Eq. (3) provide us only with the average steady-state mode populations, N+, N−, and ND, and not the distribution P(n+, n−, nD). For simplicity, we assume the semiclassical approximation \(P({n}_{+},{n}_{-},{n}_{{{{{{{{\rm{D}}}}}}}}})\approx {\delta }_{{n}_{+},0}{P}_{{N}_{{{{{{{{\rm{D}}}}}}}}}}^{{{{{{{{\rm{th}}}}}}}}}({n}_{{{{{{{{\rm{D}}}}}}}}}){\delta }_{{n}_{-},{N}_{-}}\), where \({P}_{{N}_{{{{{{{{\rm{D}}}}}}}}}}^{{{{{{{{\rm{th}}}}}}}}}(n)\) is the thermal distribution with average population ND. This approximation is reasonable for populations N+ < ND ≪ 1 ≪ N−,
The product vibrational states are \(\left|{\nu }_{+},{\nu }_{-},{\nu }_{{{{{{{{\rm{D}}}}}}}}}\right\rangle ^{\prime}\).
We assume that cavity leakage and the rate of scattering between modes is much faster than the rate of the chemical reaction. For a cavity with ~100 ps lifetime and ET reactions with 1/kR→P ~ 106 − 102 ps45, this assumption is valid. Therefore, if the populations of polariton modes change during the course of the reaction, they quickly reach a steady state before the next molecule reacts. Similarly, we also assume that the polariton and dark mode populations reach a steady state before the backward reaction takes place while computing the rate constant \({k}_{{{{{{{{\rm{P}}}}}}}}\to {{{{{{{\rm{R}}}}}}}}}^{{{{{{{{\rm{cond}}}}}}}}}\). Generalizing the cavity MLJ theory presented in ref. 38, we calculate the rate constant
for the forward reaction under polariton condensation, where
The FC factor \(| {F}_{{\nu }_{+},{\nu }_{-},{\nu }_{{{{{{{{\rm{D}}}}}}}}}}^{f,n}{| }^{2}=\big| \left\langle 0,{N}_{-},n| {\nu }_{+},{\nu }_{-},{\nu }_{{{{{{{{\rm{D}}}}}}}}}\right\rangle ^{\prime}\big| ^{2}\), and activation energy \({E}_{{\nu }_{+},{\nu }_{-},{\nu }_{{{{{{{{\rm{D}}}}}}}}}}^{f,n{{{\ddagger}}} }\) play an important role in determining the rate constant.
While many methods have been developed for computing multimode FC factors46,47,48, the focus has been on increasing the number of modes while keeping their occupation small. The current problem, however, offers a new technical challenge: the large occupation of LP makes the aforementioned methods computationally expensive. Instead, we draw inspiration from previous work that employs generating functions47 and combine those techniques with the powerful Lagrange–Bürmann formula49 to obtain analytical expressions for the required three-dimensional FC factors (see details in Supplementary Section S3).
The activation energies for the various channels of reactivity are,
where
When condensation takes place, the number of quanta in the lower polariton N− ~105 is so large that the summation in \({k}_{{{{{{{{\rm{R}}}}}}}}\to {{{{{{{\rm{P}}}}}}}}}^{{{{{{{{\rm{cond}}}}}}}}}(n)\) becomes difficult to estimate. To simplify the computation and gain intuition, we group channels into sets with the same change in the total number of intramolecular vibrational quanta f = ν+ + ν− + νD − N− − n upon ET. The closeness in energy between PES with the same f, and hence similar activation barriers, is the rationale for this grouping. \({k}_{{{{{{{{\rm{R}}}}}}}}\to {{{{{{{\rm{P}}}}}}}}}^{{{{{{{{\rm{cond}}}}}}}}}(n)\) then goes from a free summation over three indices ν+, ν− and νD into a summation over four indices f, ν+, ν− and νD with the constraint ν+ + ν− + νD = N− + n + f,
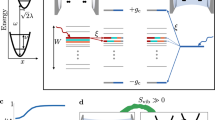
To understand the qualitative difference between reactions under polariton condensation and external pumping without SC, in Fig. 4a, b we plot the PESs (not to scale) showing the forward reaction under symmetric light-matter coupling and zero detuning. The yellow (black) parabolas in Fig. 4a, b represent PESs for a molecule in the electronic state \(\left|{{{{{{{\rm{R}}}}}}}}\right\rangle\) (\(\left|{{{{{{{\rm{P}}}}}}}}\right\rangle\)) and vibrational state \(\left|2\right\rangle\) (\(\left|2+f\right\rangle ^{\prime}\)) in Fig. 4a and \(\left|0,{N}_{-},2\right\rangle\) (\(\left|0,{N}_{-},2+f\right\rangle ^{\prime}\)) in Fig. 4b. The red parabolas in Fig. 4b are additional final PESs provided by the vibrational polariton condensate (hereafter referred to solely as “condensate”) that account for all other final vibrational states \(\left|{\nu }_{+},{\nu }_{-},{\nu }_{{{{{{{{\rm{D}}}}}}}}}\right\rangle ^{\prime}\).

a, c, e are results for a laser-driven system without light-matter strong coupling (SC) and b, d, f are for the same system under SC and 2.4% of the population in the lower polariton (condensation). All these plots are for symmetric light-matter coupling gR = gP. a, b For a clearer qualitative picture, we plot the PESs under zero detuning Δ = 0. Initial (yellow) and final (black) PESs for a molecule undergoing the forward reaction with solvent coordinate qS,c. While the energy separation between black PESs is ℏωvib, the condensate provides many additional final PESs (red, separated by ℏΩ/2 at resonance). c Reaction yield \({N}_{{{{{{{{\rm{P}}}}}}}}}^{{{{{{{{\rm{ss}}}}}}}}}/N\) at temperature kBT = 0.1389ℏωvib (T = 298 K when ℏωvib = 185 meV), Huang–Rhys factor S = 3.5, and average occupation of the intramolecular vibrational mode \(\bar{n}=0.08\). d Reaction yield \({N}_{{{{{{{{\rm{P}}}}}}}}}^{{{{{{{{\rm{ss}}}}}}}}}/N\) with Δ = − 0.0695ωvib, \(2{g}_{{{{{{{{\rm{R}}}}}}}}}\sqrt{N}=2{g}_{{{{{{{{\rm{P}}}}}}}}}\sqrt{N}=0.1{\omega }_{{{{{{{{\rm{vib}}}}}}}}}\), P− = 0.08NΓ↓, N = 107, temperature and Huang–Rhys factor are the same as c. The contributions of the red PESs through the condensate provide a broader tunability of reaction yields with respect to ΔG than under laser driving without SC. Notice that originally endergonic (exergonic) reactions in the absence of optical pumping can become exergonic (endergonic) under the featured nonequilibrium conditions. e, f A cross-section of plot (c, d) when λS = 10−2ℏωvib. The pink shaded regions correspond to cases where the dominant forward (backward) channel is in the inverted (normal) regime; the opposite is true for the green shaded regions. The condensate amplifies the forward (backward) reaction in the pink (green) shaded regions.
The net rate of ET is,
where \({k}_{{{{{{{{\rm{R}}}}}}}}\to {{{{{{{\rm{P}}}}}}}}}^{z}\) and \({k}_{{{{{{{{\rm{P}}}}}}}}\to R}^{z}\) (z = IR, cond) are the rate constants for the forward and backward reactions, respectively, which are themselves functions of NR and NP when gR ≠ gP. We find the steady-state solution \({N}_{{{{{{{{\rm{R}}}}}}}}}^{{{{{{{{\rm{ss}}}}}}}}}\) from this equation and compute the reaction yield \({N}_{{{{{{{{\rm{P}}}}}}}}}^{{{{{{{{\rm{ss}}}}}}}}}/N\).
The difference in yield between the condensate and bare case is particularly large when λS ≪ ℏωvib < ∣ΔG∣ (see Fig. 4c, d for symmetric coupling gR = gP). To understand the underlying reason, we define the dominant channel \({f}_{\min }\) as the one with minimum activation barrier outside of the cavity.
Setting the derivative in Eq. (24) equal to zero and taking into account the discrete nature of f, we find the dominant channel, \({f}_{\min }=\left\lceil\frac{-{{\Delta }}G-{\lambda }_{{{{{{{{\rm{S}}}}}}}}}}{\hslash {\omega }_{{{{{{{{\rm{vib}}}}}}}}}}\right\rceil\) or \(\left\lfloor\frac{-{{\Delta }}G-{\lambda }_{{{{{{{{\rm{S}}}}}}}}}}{\hslash {\omega }_{{{{{{{{\rm{vib}}}}}}}}}}\right\rfloor\). When λS ≪ ℏωvib, ∣ΔG∣, this channel contributes most to the rate constant because \(\frac{1}{{k}_{{{{{{{{\rm{B}}}}}}}}}T}\left|\frac{d{E}_{f}^{{{{\ddagger}}} }}{df}\right|\gg 1\). We define Marcus normal \(\frac{d{E}_{f}^{{{{\ddagger}}} }}{df}{\Big|}_{{f}_{\min }} > \, 0\) and inverted \(\frac{d{E}_{f}^{{{{\ddagger}}} }}{df}{\Big|}_{{f}_{\min }} < \, 0\) regimes with respect to the dominant channel. If the dominant forward channel is in the inverted regime, the dominant backward channel (which can be found by replacing ΔG → − ΔG in Eq. (24)) will be in the normal regime when λS ≪ ℏωvib, ∣ΔG∣.
In Fig. 4d, we see periodic yield modification in ΔG with period ~ ℏωvib, which decays for large ΔG/ℏωvib due to concomitant decline in FC factor for large changes in the number of vibrational quanta between the initial and final states. Outside of the cavity, we only see the first fringe (Fig. 4c). To observe the full periodic structure in yield, we need a large occupation of higher vibrational states which requires very large pumping rates outside of the cavity. However, under polariton condensation, the macroscopic population of the lower polariton enables these interesting periodic features to be observed at room temperature with experimentally attainable pumping rates. Additionally, polariton condensation not only modifies the reaction yield under symmetric light-matter coupling strengths, as seen in Fig. 4, it also changes the yield when the reactant and product asymmetrically couple with light (Fig. 5).

a, c The yield of the reaction when only the product (reactant) weakly couples with light. b, d Analogous plots under strong coupling \(2{g}_{{{{{{{{\rm{P}}}}}}}}}\sqrt{N}=0.1{\omega }_{{{{{{{{\rm{vib}}}}}}}}}\), gR = 0 (gP = 0, \(2{g}_{{{{{{{{\rm{R}}}}}}}}}\sqrt{N}=0.1{\omega }_{{{{{{{{\rm{vib}}}}}}}}}\)). We use parameters Δ = −0.0695ωvib, kBT = 0.1389ℏωvib (T = 298K when ℏωvib = 185 meV), S = 3.5, P− = 0.08NΓ↓ and N = 107. We assume the same scattering parameters Wij and decay rates Γ↓, κ as in Fig. 2.
Condensation provides many additional channels for the forward and backward reactions (separated by ~ℏΩ/2, see red curves in Fig. 4b, showing only the forward channels at resonance Δ = 0) due to transfer of quanta between the polariton and dark modes during the reaction. Condensation speeds up the reaction when the dominant channel is either in the inverted or in the normal regime (Fig. 6b). This is the case because there are additional channels with energy both higher (benefiting the inverted regime) and lower (benefiting the normal regime) than the dominant channel (see red curves in Fig. 4b). Apart from reduced activation energy, the additional channels provided by the condensate also have large enough FC factors to affect the rate constant. Reaction channels that involve changes in the number of quanta in the LP during the reaction have significantly larger FC factors (~1020 times) under condensation N− = 0.1N than without any pumping N− = 0 (Fig. S1 in the supplementary information compares them). Changes in the rate constant as a function of λS (Fig. 6a) and ΔG (Fig. 6b) are large for small λS/ℏωvib and when ΔG/ℏωvib = n/2 where n is an odd integer since activation energy effects are large for these set of parameters.

Ratio of the rate constants inside \({k}_{{{{{{{{\rm{R}}}}}}}}\to {{{{{{{\rm{P}}}}}}}}}^{{{{{{{{\rm{cond}}}}}}}}}\) and outside \({k}_{{{{{{{{\rm{R}}}}}}}}\to {{{{{{{\rm{P}}}}}}}}}^{{{{{{{{\rm{IR}}}}}}}}}\) of the cavity under laser excitation with Δ = −0.0695ωvib, kBT = 0.1389ℏωvib (T = 298 K when ℏωvib = 185 meV), S = 3.5, \(2g\sqrt{N}=0.1{\omega }_{{{{{{{{\rm{vib}}}}}}}}}\), P− = 0.08NΓ↓ and N = 107 for symmetric coupling gR = gP = g. a Relative rate constant as a function of reorganization energy, λS, with ΔG = −3.33ℏωvib (the pink curve; here, the dominant channel lies in the inverted regime) and ΔG = −3.73ℏωvib (the green curve; here, the dominant channel lies in the normal regime) and b as a function of ΔG with λS = 10−2ℏωvib. Here, the pink region corresponds to the dominant channel in the inverted regime and the green to the normal regime.
Discussion
Our result is a first step towards understanding the effect of Bose–Einstein condensation of polaritons on chemical reactivity. We demonstrate this effect using a simple electron transfer model (MLJ) with molecular vibrations strongly coupled to light. In particular, we show that one can counteract the massive degeneracy of dark modes and enhance polaritonic effects by having a macroscopic occupation of the lower polariton mode i.e., Bose–Einstein condensation. Our results indicate that the latter is feasible for experimentally realizable pump powers and Rabi splittings, despite the close proximity in the energy of the dark state manifold with ℏΩ ~kBT. These results can guide the choice of suitable materials for condensation under VSC. While laser driving without SC modifies the reaction yield, this change is amplified by the condensate, due to the availability of many additional reactive channels that differ in energy by ~ℏΩ/2 rather than ~ℏωvib. For a wide range of parameters, we find that this leads to a periodic dependence of reaction yield as a function of ΔG (with period ~ℏωvib), rendering a set of originally endergonic reactions exergonic, and vice versa. These effects are substantially weaker under laser driving and highlight both the energetic (availability of additional channels with lower activation energy) and entropic (redistribution of vibrational energy from the condensate into the polariton and dark modes upon reaction) advantages of exploiting polariton condensates for reactivity. To summarize, vibrational polariton condensation offers a novel strategy to accumulate energy into a well-defined normal mode, a holy-grail in the field of vibrational dynamics that has been historically hindered by IVR. Its successful demonstration could revive hopes of “mode selective chemistry”50, beyond electron transfer processes. In future work, it will be interesting to explore how the studied phenomena generalize to molecular polariton condensates in different spectral ranges.
Data availability
Datasets generated by our code are available by email upon request to the authors.
Code availability
Computational scripts used to generate the plots in the present article are available by email upon request to the authors.
References
Lidzey, D. G. et al. Strong exciton–photon coupling in an organic semiconductor microcavity. Nature 395, 53–55 (1998).
Shalabney, A. et al. Coherent coupling of molecular resonators with a microcavity mode. Nat. Commun. 6, 1–6 (2015).
Long, J. P. & Simpkins, B. Coherent coupling between a molecular vibration and fabry–perot optical cavity to give hybridized states in the strong coupling limit. ACS Photonics 2, 130–136 (2015).
Ebbesen, T. W. Hybrid light–matter states in a molecular and material science perspective. Acc. Chem. Res. 49, 2403–2412 (2016).
Li, X., Mandal, A. & Huo, P. Cavity frequency-dependent theory for vibrational polariton chemistry. Nat. Commun. 12, 1–9 (2021).
Thomas, A. et al. Tilting a ground-state reactivity landscape by vibrational strong coupling. Science 363, 615–619 (2019).
Hirai, K., Takeda, R., Hutchison, J. A. & Uji-i, H. Modulation of prins cyclization by vibrational strong coupling. Angew. Chem. 132, 5370–5373 (2020).
Galego, J., Climent, C., Garcia-Vidal, F. J. & Feist, J. Cavity casimir-polder forces and their effects in ground-state chemical reactivity. Phys. Rev. X 9, 021057 (2019).
Li, T. E., Nitzan, A. & Subotnik, J. E. On the origin of ground-state vacuum-field catalysis: equilibrium consideration. J. Chem. Phys. 152, 234107 (2020).
Campos-Gonzalez-Angulo, J. A. & Yuen-Zhou, J. Polaritonic normal modes in transition state theory. J. Chem. Phys. 152, 161101 (2020).
Proukakis, N. P, Snoke, D. W & Littlewood, P. B. Universal Themes of Bose-Einstein Condensation (Cambridge Univ. Press, 2017).
Daskalakis, K., Maier, S., Murray, R. & Kéna-Cohen, S. Nonlinear interactions in an organic polariton condensate. Nat. Mater. 13, 271–278 (2014).
Plumhof, J. D., Stöferle, T., Mai, L., Scherf, U. & Mahrt, R. F. Room-temperature bose–einstein condensation of cavity exciton–polaritons in a polymer. Nat. Mater. 13, 247–252 (2014).
Dietrich, C. P. et al. An exciton-polariton laser based on biologically produced fluorescent protein. Sci. Adv. 2, e1600666 (2016).
Väkeväinen, A. I. et al. Sub-picosecond thermalization dynamics in condensation of strongly coupled lattice plasmons. Nat. Commun. 11, 1–12 (2020).
Zasedatelev, A. V. et al. A room-temperature organic polariton transistor. Nat. Photon. 13, 378–383 (2019).
Zeb, M. A., Kirton, P. G. & Keeling, J. Incoherent charge transport in an organic polariton condensate. Preprint at https://arxiv.org/abs/2004.09790 (2020).
Moore, M. & Vardi, A. Bose-enhanced chemistry: amplification of selectivity in the dissociation of molecular bose-einstein condensates. Phys. Rev. Lett. 88, 160402 (2002).
Heinzen, D., Wynar, R., Drummond, P. & Kheruntsyan, K. Superchemistry: dynamics of coupled atomic and molecular bose-einstein condensates. Phys. Rev. Lett. 84, 5029 (2000).
Keeling, J. & Kéna-Cohen, S. Bose–einstein condensation of exciton-polaritons in organic microcavities. Annu. Rev. Phys. Chem. 71, 435–459 (2020).
Du, M., Ribeiro, R. F. & Yuen-Zhou, J. Remote control of chemistry in optical cavities. Chem 5, 1167–1181 (2019).
Delor, M. et al. Toward control of electron transfer in donor-acceptor molecules by bond-specific infrared excitation. Science 346, 1492–1495 (2014).
Hammes-Schiffer, S. & Tully, J. C. Vibrationally enhanced proton transfer. J. Phys. Chem. 99, 5793–5797 (1995).
Strashko, A., Kirton, P. & Keeling, J. Organic polariton lasing and the weak to strong coupling crossover. Phys. Rev. Lett. 121, 193601 (2018).
Bittner, E. R. & Silva, C. Estimating the conditions for polariton condensation in organic thin-film microcavities. J. Chem. Phys. 136, 034510 (2012).
del Pino, J., Feist, J. & Garcia-Vidal, F. J. Quantum theory of collective strong coupling of molecular vibrations with a microcavity mode. New J. Phys. 17, 053040 (2015).
Somaschi, N. et al. Ultrafast polariton population build-up mediated by molecular phonons in organic microcavities. Appl. Phys. Lett. 99, 209 (2011).
Dunkelberger, A., Spann, B., Fears, K., Simpkins, B. & Owrutsky, J. Modified relaxation dynamics and coherent energy exchange in coupled vibration-cavity polaritons. Nat. Commun. 7, 1–10 (2016).
Xiang, B. et al. State-selective polariton to dark state relaxation dynamics. J. Phys. Chem. A 123, 5918–5927 (2019).
Fröhlich, H. Bose condensation of strongly excited longitudinal electric modes. Phys. Lett. A 26, 402–403 (1968).
Zhang, Z., Agarwal, G. S. & Scully, M. O. Quantum fluctuations in the fröhlich condensate of molecular vibrations driven far from equilibrium. Phys. Rev. Lett. 122, 158101 (2019).
Banyai, L., Gartner, P., Schmitt, O. & Haug, H. Condensation kinetics for bosonic excitons interacting with a thermal phonon bath. Phys. Rev. B 61, 8823 (2000).
Imamoglu, A. et al. Nonequilibrium condensates and lasers without inversion: exciton-polariton lasers. Phys. Rev. A 53, 4250 (1996).
Vurgaftman, I., Simpkins, B. S., Dunkelberger, A. D. & Owrutsky, J. C. Negligible effect of vibrational polaritons on chemical reaction rates via the density of states pathway. J. Phys. Chem. Lett. 11, 3557–3562 (2020).
Del Pino, J., Garcia-Vidal, F. J. & Feist, J. Exploiting vibrational strong coupling to make an optical parametric oscillator out of a raman laser. Phys. Rev. Lett. 117, 277401 (2016).
Herrera, F. & Spano, F. C. Cavity-controlled chemistry in molecular ensembles. Phys. Rev. Lett. 116, 238301 (2016).
Semenov, A. & Nitzan, A. Electron transfer in confined electromagnetic fields. J. Chem. Phys. 150, 174122 (2019).
Campos-Gonzalez-Angulo, J. A., Ribeiro, R. F. & Yuen-Zhou, J. Resonant catalysis of thermally activated chemical reactions with vibrational polaritons. Nat. Commun. 10, 1–8 (2019).
Phuc, N. T., Trung, P. Q. & Ishizaki, A. Controlling the nonadiabatic electron-transfer reaction rate through molecular-vibration polaritons in the ultrastrong coupling regime. Sci. Rep. 10, 1–11 (2020).
Marcus, R. A. Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 15, 155–196 (1964).
Levich, V. Present state of the theory of oxidation-reduction in solution (bulk and electrode reactions). Adv. Electrochem. Electrochem. Eng 4, 249–371 (1966).
Jortner, J. Temperature dependent activation energy for electron transfer between biological molecules. J. Chem. Phys. 64, 4860–4867 (1976).
Nesbitt, D. J. & Field, R. W. Vibrational energy flow in highly excited molecules: role of intramolecular vibrational redistribution. J. Phys. Chem. 100, 12735–12756 (1996).
Strashko, A. & Keeling, J. Raman scattering with strongly coupled vibron-polaritons. Phys. Rev. A 94, 023843 (2016).
Miller, J. R., Calcaterra, L. & Closs, G. Intramolecular long-distance electron transfer in radical anions. the effects of free energy and solvent on the reaction rates. J. Am. Chem. Soc 106, 3047–3049 (1984).
Roche, M. On the polyatomic franck-condon factors. Chem. Phys. Lett. 168, 556–558 (1990).
Sharp, T. & Rosenstock, H. Franck-condon factors for polyatomic molecules. J. Chem. Phys. 41, 3453–3463 (1964).
Toniolo, A. & Persico, M. Efficient calculation of franck–condon factors and vibronic couplings in polyatomics. J. Comput. Chem. 22, 968–975 (2001).
Whittaker, E. T & Watson, G. N. A Course of Modern Analysis 4th edn (Cambridge Univ. Press, 1996).
Frei, H. & Pimentel, G. C. Infrared induced photochemical processes in matrices. Annu. Rev. Phys. Chem. 36, 491–524 (1985).
Acknowledgements
S.P.-S., L.A.M.-M., and J.Y.-Z. were supported by the US Department of Energy, Office of Science, Basic Energy Sciences, CPIMS Program under Early Career Research Program Award DE-SC0019188. J.A.C.-G.-A. was supported through the AFOSR award FA9550-18-1-0289. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562, under allocation number TGASC150024. J.Y.-Z. acknowledges fruitful discussions with Wei Xiong. S.P.-S thanks Juan Pérez-Sánchez and Matthew Du for helpful discussions.
Author information
Authors and Affiliations
Contributions
S.P.-S. developed model, calculations, and interpretation of the results in the manuscript. L.A.M.-M. provided guidance in the development of the initial model. J.A.C.-G.-A. developed the optimal basis to carry out the calculations for the electron transfer reaction under condensation and provided guidance on the interpretation of phenomenology. S.S. assisted on the calculation of multidimensional Franck–Condon factors. J.Y.-Z. conceived the original version of the project and supervised the work throughout.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pannir-Sivajothi, S., Campos-Gonzalez-Angulo, J.A., Martínez-Martínez, L.A. et al. Driving chemical reactions with polariton condensates. Nat Commun 13, 1645 (2022). https://doi.org/10.1038/s41467-022-29290-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-29290-9
This article is cited by
-
Manipulating hydrogen bond dissociation rates and mechanisms in water dimer through vibrational strong coupling
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
