
Abstract
Microbial lipid metabolism is an attractive route for producing oleochemicals. The predominant strategy centers on heterologous thioesterases to synthesize desired chain-length fatty acids. To convert acids to oleochemicals (e.g., fatty alcohols, ketones), the narrowed fatty acid pool needs to be reactivated as coenzyme A thioesters at cost of one ATP per reactivation - an expense that could be saved if the acyl-chain was directly transferred from ACP- to CoA-thioester. Here, we demonstrate such an alternative acyl-transferase strategy by heterologous expression of PhaG, an enzyme first identified in Pseudomonads, that transfers 3-hydroxy acyl-chains between acyl-carrier protein and coenzyme A thioester forms for creating polyhydroxyalkanoate monomers. We use it to create a pool of acyl-CoA’s that can be redirected to oleochemical products. Through bioprospecting, mutagenesis, and metabolic engineering, we develop three strains of Escherichia coli capable of producing over 1 g/L of medium-chain free fatty acids, fatty alcohols, and methyl ketones.
Similar content being viewed by others
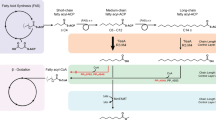
Introduction
Oleochemicals are a class of aliphatic hydrocarbons that are industrially derived from petroleum, animal fats, or oilseed crops. Depending on the terminal reductive state of the acyl chain, oleochemicals can be classified as fatty acids, alcohols, aldehydes, ketones, esters, olefins, alkanes, etc. Oleochemicals are also categorized by size (i.e., acyl-chain length). Medium-chain oleochemicals are defined as compounds with an acyl moiety of 8–12 carbons and are of particular interest due to their applications as a commodity and specialty chemicals1,2. For example, medium-chain fatty acids are used as herbicides, antimicrobials, and precursors for lubricant synthesis3. Medium-chain methyl ketones are used as flavors, floral fragrances, insecticides, and renewable liquid transportation fuels4,5. Medium-chain alcohols are used as surfactants, as additives in biodiesel, cosmetics, and consumer products. The unique molecular and chemical properties of medium-chain oleochemicals and their limited natural abundance make them attractive targets for biosynthesis.
The final enzymatic reactions in oleochemical biosynthesis use acyl-thioesters as substrates, making high-flux to desired chain length acyl-thioesters a key element to any metabolic engineering strategy. Thus far, two approaches have been applied to produce medium-chain acyl-thioesters in microbes: thiolase-driven β-reduction cycles (i.e., reversal of β-oxidation)6 and thioesterase-driven diversion of fatty acid biosynthesis intermediates2,7. The reverse β-oxidation pathway incorporates a reversible, thiolase-catalyzed Claisen condensation (e.g., FadA) with acetyl-CoA as the donor to bypass the energy consumption required for producing malonyl-CoA in fatty acid biosynthesis. The 1-ATP savings per elongation provides reversal of β-oxidation with the highest theoretical yield of all oleochemical biosynthesis routes at the cost of losing the strong driving force provided by the decarboxylative Claisen condensation in fatty acid biosynthesis. β-reduction was used to demonstrate the production of both medium-chain fatty acids and fatty alcohols6,8,9,10,11 in high yields, albeit with poor selectivity to specific chain length products. This is in part due to the competition between termination reactions (cleavage or reduction, respectively) and thiolase-catalyzed extension of acyl-CoA thioesters. To date, strategies to bias termination at a particular chain length have yet to be demonstrated and better results have come from finding ways to accumulate pools of desired chain-length acyl-CoAs made via fatty acid biosynthesis to avoid the competition entirely.
Plants synthesize novel oils containing medium-chain acyl groups by expressing selective acyl-ACP thioesterases in the chloroplast to generate free fatty acids of the desired size7. Once made, the free fatty acids are transported to the cytosol for reactivation as acyl-CoA and subsequent incorporation into storage lipids12. In microbes, plant acyl-ACP thioesterases can be leveraged to produce free fatty acids of desired chain length. Additional genetic modifications are needed to enable the conversion of the free fatty acids to the desired oleochemical form (e.g., alcohol, ketone, ester). Unlike plants where biochemistries can be compartmentalized, bacteria express enzymes for both fatty acid biosynthesis and catabolism in the same locations. In order to accumulate a pool of desired acyl-CoA’s β-oxidation must be blocked by eliminating all enzymes that catalyze one of the four reactions in the cycle. Depending on the desired product form, different β-oxidation steps are targeted (e.g., acyl-CoA dehydrogenase, FadE, for fatty alcohol production; thiolase, FadA/FadI, for methyl ketone production) to produce the substrate for termination enzymes (e.g., saturated acyl-CoA for acyl-CoA reductase conversion to fatty alcohol; β-ketoacyl-CoA for β-ketothioesterase conversion to methyl ketone). The last steps in the metabolic engineering strategy are an expression of a specialized acyl-ACP thioesterase (to produce desired free fatty acids), expression of acyl-CoA synthetase (to activate the free fatty acid), and expression of the desired termination enzyme. Highly active acyl-ACP thioesterases have been identified from natural sources and others have been engineered or evolved in the laboratory to produce octanoic acid, decanoic acid, dodecanoic acid, and tetradecanoic acid13,14,15,16,17. For instance, a highly active variant of a Cuphea palustris thioesterase FatB (referred to CpFatB*) was identified using a CpFatB random mutagenesis library and a growth selection based on the lipoic acid requirement of Escherichia coli. E. coli strain NHL17 (MG1655 ΔaraBAD ΔfadD::Ptrc-CpfatB*) produced 1.7 g/L octanoic acid with >90% specificity from 20 g/L glycerol14. The highly active C8-specific CpFatB* enzyme was utilized to produce 1-octanol by expressing an acyl-CoA synthetase (MtFadD6) from Mycobacterium tuberculosis to reactivate octanoic acid at the cost of 1 mole ATP and expressing an acyl-CoA reductase from Marinobacter aquaeolei (MaACR) to convert octanoyl-CoA to 1-octanol. The resulting E. coli strain NHL24 produced 1.3 g/L 1-octanol18. In a separate study, 2-heptanone was produced by converting octanoyl-CoA to β-ketooctanoyl-CoA via an acyl-CoA oxidase from Micrococcus luteus (Mlut_11700) and an endogenous bi-functional dehydrogenase EcFadB. Subsequently, β-ketooctanoyl-CoA was hydrolyzed by an β-ketoacyl-CoA thioesterase PsFadM from Providencia sneebia (PsFadM) and the resulting β-ketooctanoic acid was decarboxylated non-enzymatically to yield 2-heptanone. Strain E. coli TRS12 (MG1655 ΔaraBAD ΔfadD::Ptrc-CpfatB1* ΔfadA ΔfadE ΔfadI ΔfadR) harboring pTRC99a-MtfadD6-PsfadM and pACYC-Mlmlut_11700 plasmids produced up to 4 g/L 2-heptanone in fed-batch bioreactor experiments4. Although successful, this thioesterase strategy could be further improved by replacing the futile cycle of thioester cleavage and formation with a direct acyl transfer.
In nature, some bacteria such as Pseudomonads, accumulate polyhydroxyalkanoate (PHA) as a means of storing carbon and energy. PHA polymerization requires a supply of (R)-3-hydroxyacyl-CoAs (PHA monomers), which can be derived from either fatty acid biosynthesis or β-oxidation. In 1998, PhaG, an enzyme found in Pseudomonas putida and Pseudomonas aeruginosa, was identified as the enzymatic link between fatty-acid biosynthesis and PHA biosynthesis19,20. PhaG was hypothesized to catalyze the transfer of the (R)-3-hydroxyacyl moiety from the ACP thioester to CoA. In vitro experimental results showed a time course of CoA release by incubating purified PhaG, (R)-3-hydroxydecanoyl-CoA and holo-ACP, indicating PhaG catalyzes a reversible transacylase reaction21. In subsequent studies, researchers overexpressed PhaG and observed increased PHA content in cells and an increased fraction of medium-chain length (mcl) 3-hydroxyalkanoate units in the polymer22,23,24,25,26. Given these results, we hypothesized that PhaG could provide a similar role in linking fatty acid biosynthesis with the creation of tailored pools of acyl-CoAs and oleochemical products.
In the present study, we demonstrate a PhaG-dependent pathway as an alternative strategy to link fatty acid biosynthesis and oleochemical production. We validate the ability of P. putida PhaG to direct flux towards oleochemical synthesis at rates comparable to thioestereases. Using computational bioprospecting tools, we identify seven homologs of PpuPhaG and evaluate their in vivo activities. The PhaG variant from Pseudomonas koreensis produces 1.6-fold more methyl ketones than the PpuPhaG variant. We construct a random mutagenesis library of PkPhaG and isolate seventeen beneficial mutations that increase octanoic acid production 3.3–16.3-fold above strains expressing the original PkPhaG. We use these improved enzymes to construct strains capable of producing three demonstration oleochemicals—free fatty acids, fatty alcohols, and methyl ketones. Strains expressing the PhaG-dependent pathway are capable of producing 1.1 g/L C8–C14 free fatty acids, 1.5 g/L C7–C15 methyl ketones, and 1.1 g/L C8–C16 fatty alcohols depending on the tailoring enzymes co-expressed. These titers demonstrate that PhaG is a useful alternative for medium chain length oleochemical synthesis and a promising target for future protein engineering to guide substrate selectivity. Yields remain ~50% of the theoretical limit, on par with demonstrations of many thioesterase-utilizing strategies. Continued improvement of PhaG-driven pathways will allow strains to access higher theoretical yields than current thioesterase strategies (Fig. 1, Supplementary Table 1 and Supplementary Method 1).

Oleochemical products such as methyl ketones (MK), fatty acid ethyl esters (FAEE), fatty alcohols (FAOH), and alkanes are made by enzymes that act on acyl-thioesters. Many oleochemical-producing enzymes can act on long-chain acyl-ACPs (Option 1) but fail to produce medium or short-chain products in high amounts without additional genetic modifications. The reduced activity is likely caused by lower concentrations of shorter chain length acyl-ACPs and/or reduced specific activity on these substrates, particularly when compared to enzymes in fatty acid biosynthesis that elongate the same substrates. Alternatively, if β-oxidation is blocked (often by deleting enzymes catalyzing the appropriate step in the cycle), higher concentrations of mcl-acyl-CoA’s can be provided to saturate oleochemical-producing enzymes. This acyl-CoA pool can be created either by the combination of thioesterase (TE) and acyl-CoA synthetase (e.g., FadD) activity (Option 2) or by acyl-transferase activity (Option 3). Transferase activity (e.g., PhaG) saves one ATP per product relative to Option 2. Detailed pathways for Options 2 and 3 are illustrated in Supplementary Fig. 1.
Results and discussion
Genetic studies support PhaG has acyl-ACP:CoA transacylase activity
Homologs of PpuPhaG have been used extensively as a means of enhancing the production of mcl PHA in bacteria19,27,28,29,30,31. That said, the specific activities catalyzed by PpuPhaG are debated. In vitro studies have confirmed the ability of PhaG to generate 3-hydroxyacyl-ACP from the corresponding CoA species and holo-ACP20,21. This is the reverse reaction of the one desired for oleochemical production studies and no in vitro data on acyl-ACPs, the substrate in the forward direction is available. From 1998 to 2012, PhaG was generically called a 3-hydroxyacyl ACP:CoA transacylase, based on in vitro data. In 2012, Nomura and co-workers challenged the name and the ability of the enzyme to perform the transferase reaction30. In this study, E. coli BL21 cells harboring a plasmid for expressing an mcl-PHA polymerase (PhaC1) were transformed with plasmids expressing PhaG and/or a CoA ligase from P. putida. Cells expressing both enzymes produced more than ten times the amount of PHA than those lacking the CoA ligase. The conclusion drawn from this study is that the ligase is needed for high-flux PHA generation and PhaG acts primarily as a thioesterase. Subsequent papers have used the name thioesterase, but have not provided further evidence to support the presence of thioesterase activity. In contrast, prior studies demonstrated that co-expression of TesB, a promiscuous CoA thioesterase enhanced the production of 3-hydroxy-fatty acids in both E. coli and P. putida32,33. TesB was similarly used to produce 3-hydroxy fatty acids as precursors to methyl esters34.
The complicated history motivated us to confirm that PhaG had substantial transferase activity. Therefore, we compared the metabolic product profiles (looking for production of methyl ketones or 3-hydroxy fatty acids) of specifically engineered strains of E. coli (MG1655 ΔfadA, ΔfadI, ΔfadD, ΔfadR, pTRC99a-PpuphaG-EcfadM) to determine if heterologously expressed PhaG demonstrated more thioesterase or transacylase activity. Strains were designed to create a 3-hydroxy fatty acid product sink to indicate potential PhaG thioesterase activity and a methyl ketone product sink for PhaG transacylase activity (Fig. 2A). Deletion of fadD removes the dominant acyl-CoA synthetase activity and prevents the reactivation of free fatty acids generated by either FadM or PhaG. Deletion of fadA and fadI removes known thiolase activities from E. coli and blocks β-oxidation from catabolizing any acyl-CoAs produced in vivo. Deletion of fadR removes repression of fadB expression and thereby upregulates a bi-functional enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase responsible for converting 3-hydroxyacyl-CoA thioesters to 3-ketoacyl-CoA thioesters. FadM4,35 is overexpressed to provide 3-ketoacyl-CoA thioesterase activity, resulting in the conversion of any 3-hydroxyacyl-CoAs generated by PhaG to the corresponding methyl ketones. Cultures of E. coli RADI strain harboring pTRC99a-PpuphaG-EcfadM were grown in Clomburg media at 30 °C for 48 h. Culture samples were extracted and derivatized for GC/FID and GC/MS analysis. The samples contained a total of 170 mg/L C7–C13 methyl ketones but no detectable 3-hydroxy methyl esters (Fig. 2B) indicating that PpuPhaG functions primarily as a 3-hydroxyacyl ACP:CoA transacylase. Strains expressing FadD (E. coli RAI strain pTRC99a-PpuphaG-EcfadM) produced equivalent amounts of methyl ketones, indicating that the carbon flux for methyl ketone synthesis is not enhanced by FadD-catalyzed free fatty acid reactivation. Strains lacking PhaG overexpression (E. coli RADI strain pTRC99a-EcfadM) produced small amounts (<1 mg/L) of methyl ketones (Fig. 2B) that have been previously observed in strains expressing FadM4,35. Strains lacking EcFadM overexpression (E. coli RADI strain pTRC99a-PpuphaG) contained a total of 55 mg/L C8–C12 3-hydroxy methyl esters (Fig. 2C) consistent with prior studies32,33. Together, these data suggest that 3-hydroxy fatty acid production observed in past studies likely comes from thioesterase activities encoded by native enzymes (e.g., YciA, FadM, TesB can potentially catalyze the cleavage of (R)-3-hydroxyacyl-CoA to 3-hydroxy fatty acids) that are outcompeted by the methyl ketone synthesis pathway we introduced.

A Metabolic pathways used to test for the presence of PhaG-dependent thioesterase and/or transferase activity. B In cells lacking FadR, FadA, FadI, and FadD (E. coli RAID harboring pTRC99a-PpuphaG), PhaG expression leads to the production of 50 mg/L medium-chain 3-hydroxy fatty acids (n = 3 biologically independent samples). These products could be generated by either direct PhaG thioesterase activity on medium-chain acyl-ACPs or by CoA thioesterase activity on stranded pools of 3-hydoxyacyl-CoAs made via PhaG transferase activity. ***P = 0.0005 was analyzed based on student two-tailed t test assuming unequal variances. C Co-expression of PhaG, and FadM in E. coli RAI (MG1655 ΔfadR ΔfadA ΔfadI harboring pTRC99a-PpuphaG-EcfadM) results in the production of 150 mg/L of medium-chain methyl ketones (n = 3 biologically independent samples). FadD expression did not impact methyl ketone production indicating that free fatty acid activation is not required for PhaG-dependent methyl ketone production. D A two-dimensional cluster map created with the Enzyme Similarity Tool37 displays the sequence similarity of PhaG variants tested in bioprospecting studies. Quantitative pairwise percent amino-acid identity of each homolog can be found in Supplementary Figure 2. Colored boxes and dots are used to indicate the sequences tested. Mean methyl ketone titers for constructs of PhaG homologs using E. coli RAI harbor pTRC99a-‘PpuphaG’-EcfadM (n = 3 biologically independent samples). All cultures were grown in Clomburg medium containing 20 g/L glycerol at 30 °C and shaking at 250 r.p.m. **P = 0.009 was analyzed based on student two-tailed t test assuming unequal variances. All data represent the mean ± s.d. of biological triplicates. Source data underlying B–D are provided as a Source Data file.
Bioprospecting identifies active PhaG variants
Next, we sought to identify higher activity variants through bioprospecting. We conducted a homology search based on the Pseudomonas putida KT2440 PhaG (PpuPhaG) sequence using the Basic Local Alignment Search Tool (BLAST)36 to identify candidate protein sequences. BLAST hits were sorted using the Enzyme Similarity Tool37. Among the homologous sequences that have similarity >45%, we found >95% of sequences belong to Pseudomonas species, indicating that PhaG provides activity unique from other PHA-producing bacteria38. We selected 13 PhaG homologs, which had a protein sequence similarity range of 24–88% based on a pairwise comparison, shown in Fig. 2D and Supplementary Figure 2. We were particularly interested in homologs from Mycobacteria and Corynebacteria species because of their potential to interface with substrates linked to the ACP domain of type I fatty acid synthase (FAS) found in these species39. The activity of PhaG homologs was assayed in vivo by monitoring methyl ketone production using E. coli RADI harboring a pTRC99a-’PkphaG’-EcfadM plasmid. Most PhaG variants generated similar methyl ketone titers to PpuPhaG albeit with reduced levels of 2-heptanone. The PhaG variants from Mycobacteria and Corynebacteria failed to produce methyl ketones. We found that the P. koreensis PkPhaG showed the highest production at 350 mg/L total methyl ketone, 1.6 times higher than the PpuPhaG, shown in Fig. 2D. The methyl ketone profile included 34 mg/L 2-heptanone, 85 mg/L 2-nonanone, 85 mg/L 2-undecanone, and 145 mg/L 2-tridecanone indicating a broad activity against medium-chain 3-hydroxyacyl-ACPs. The product distribution did not vary significantly across the tested variants. Therefore, substrate preference will need to be addressed with protein engineering efforts analogous to those targeted to acyl-ACP thioesterases15.
Metabolic engineering to enhance methyl ketone production
A central tenet of metabolic engineering states that enzyme activity must be balanced across a metabolic pathway to minimize unwanted accumulation of intermediates and maximize pathway flux. In order to assess the relative activity of PhaG to pathway flux, we varied co-overexpression of PpuPhaG, the more-active PkPhaG, and enzymes that convert 3-hydroxyacyl-CoAs to 3-ketoacyl-CoAs. In particular, we were concerned about the relative activity of ketoreductases on the two 3-hydroxyacyl-CoA stereoisomers. PhaG generates (R)-3-hydroxyacyl-CoA for direct polymerization into PHA. In contrast, β-oxidation passes through (S)-3-hydroxyacyl-CoAs. FadB, the dual-function enoyl-CoA hydratase, and dehydrogenase, can isomerize (R)-3-hydroxyacyl-CoA via the corresponding enoyl-acyl-CoA, likely at a reduced rate relative to its regular substrate isomer40,41,42,43. PHA-producing bacteria solve this problem by expressing an alternative enoyl-CoA hydratase (PhaJ) that can generate the preferred R-isomer (Fig. 3A) from β-oxidation intermediates. We selected EcFadB and EcFadJ, an anaerobically expressed FadB homolog, and four previously studied PhaJ variants from P. aeruginosa44. These enzymes were important optimization points because deletion of fadB and fadJ eliminated PhaG-dependent methyl ketone production (Fig. 3B).

A Detailed metabolic pathway is used to enable PhaG-dependent production of methyl ketones. B Evaluation of alternative routes of isomerizing 3-hydroxyacyl-CoAs with respect to PhaG activity. A series of mutations were made to the base strain, E. coli RAI (MG1655, ΔaraBAD ΔfadR ΔfadA ΔfadI) (n = 3 biologically independent samples). Cultures of each strain harboring pTRC99a-PpuphaG-EcfadM or pTRC99a-PkphaG-EcfadM were grown for 48 hrs in Clomburg medium containing 20 g/L glycerol at 30 °C and shaking at 250 r.p.m. All data represent the mean ± s.d. of biological triplicates. ***P = 0.0001, **P = 0.003, *P = 0.02 were analyzed based on student two-tailed t test assuming unequal variances. Source data underlying B are provided as a Source Data file.
A combination of EcFadB, EcFadB/EcFadJ, and four PpuPhaJ homologs was cloned into an operon linked to a PTRC promoter on a pACYC vector. Each of these vectors was co-expressed with pTRC99a-PpuphaG-EcfadM or pTRC99a-PkphaG-EcfadM in E. coli RAI. Cultures of each strain were grown at 30 °C for 48 h. Methyl ketones were extracted from culture samples and quantified by GC/FID (Fig. 3). Strains expressing PpuPhaG all produced ~160 mg/L of methyl ketones with similar distributions to prior experiments. In contrast, when PkPhaG was expressed, methyl ketone titers increased two- threefold relative to the corresponding PpuPhaG strains. In this series, co-expression of PhaJ1 and PhaJ3 had the biggest impact on methyl ketone titer, surpassing 0.5 g/L in cultures of the best strains. These experiments indicate that 3-hydroxyacyl-CoA isomerization can be a limiting step when PhaG transacylase activity is increased. Further, these experiments suggested that titers could be improved with more-active PhaG enzymes.
Random mutagenesis improves PhaG activity
We initiated a protein engineering study to seek variants with enhanced activity altered and if possible altered product specificity. We constructed an error-prone PCR library of PkphaG ORFs and screened for the ability to complement a lipoic acid auxotrophy; this approach was previously used to isolate C8-specific thioesterases with enhanced Vmax14. E. coli strains lacking LipB require supplementation of lipoic acid or octanoic acid in the media to enable the formation of active pyruvate dehydrogenase and aerobic growth on glucose. We constructed a lipoic acid auxotrophic strain, E. coli CM23-ΔlipB, that also lacks β-oxidation genes and many fermentation pathways. To link the octanoic acid selection to PhaG activity, we added heterologous enzymes to convert the 3-hydroxyoctanoyl-CoA to octanoic acid (Fig. 4A). These enzymes include PkPhaG, PaPhaJ3, a Treponema denticola trans-enoyl-CoA reductase (TdTER)8 and a Mycobacterium sp. acyl-CoA thioesterase MaTesB A197D (referred as to MaTesB*)45 (Fig. 4A). Purified Mycobacterium avium MaTesB* has been shown to hydrolyze octanoyl-CoA and generate octanoic acid in vitro45. We neglected the octanoic acid synthesis activities by endogenous E. coli acyl-CoA thioesterase because EcTesB generally has activities toward longer chain acyl-CoA (>C10)41 and EcYciA has activities toward shorter chain acyl-CoA (<C8)46,47. The base strain, expressing the wild-type PkphaG, produced ~20 mg/L octanoic acids after 48 h, whereas the corresponding strain without PkPhaG produced <1 mg/L octanoic acid. In order to reduce the baseline octanoic acid titer, we subcloned PkphaG onto a low-copy vector (pBTRCK)8 and MatesB* onto a high copy number vector pTRC99a. The latter was performed to ensure that octanoic acid production would be limited solely by PhaG activity. After tuning the copy number of PkphaG and MatesB*, E. coli CM23-ΔlipB harboring pTRC99a-MatesB*-TdTER + pACYC-PaphaJ3 + pBTRCK-’PkphaG’ plasmids produced ~7 mg/L octanoic acids after 48 h. This strain was used to perform selections of the error-prone PhaG library.

A Metabolic pathways involved in PhaG complementation of a ΔlipB driven lipoic acid auxotrophy in E. coli CM23 ΔlipB. EcFadD deletion blocks the reactivation of fatty acids. EcFadBJ deletion blocks hydration and dehydrogenation of octenoyl-CoA to β-ketooctanoyl-CoA. EcFadAI deletion blocks thiolase-driven elongation or reduction of acyl-CoA chains. LipB deletion blocks activation of apo pyruvate dehydrogenase complex (PDC) E2 domain to octanoylated PDC E2 domain. (R)-3-hydroxyoctanoyl-CoA is dehydrated and hydrogenated by PaPhaJ3 and TdTER to generate octanoyl-CoA. Octanoyl-CoA is hydrolyzed by MaTesB* to release octanoic acid and CoA. Octanoic acid is ligated to Apo PDC E2 domain by LplA. LipA creates the functional lipoylated E2 domain restoring PDH activity. A functional selection was performed by introducing PhaG variants into the selection strain and plating cells on minimal MOPS-glucose agar. In the selection strain, MaTesB* and TdTER were expressed from pTRC99a, PaPhaJ3 was expressed from pACYC, PkPhaG, and other variants, were expressed from a low-copy vector, pBTRCK. B FAME analysis of cultures harboring PhaG variants containing combinations of point mutations identified in selection experiments (n = 3 biologically independent samples). E. coli CM23 harboring pBTRCK-’PkphaG’ + pACYC-PaphaJ3 + pTRC99a-TdTER-MatesB* were cultured in test tubes containing 5 mL Clomburg 20 g/L glycerol and 1 mM IPTG at 30 °C for 48 hrs. All data represent the mean ± s.d. of biological triplicates. ***P = 0.0001 (Q45R Y138F G142V vs. wild-type), ***P = 0.0006 (PkPhaG Q45R G142V vs. PkPhaG), **P = 0.004 (PkPhaG Y138F vs. PkPhaG), *P = 0.02 (PkPhaG Q45R vs. PkPhaG) and *P = 0.03 (PkPhaG G142V vs. PkPhaG) were analyzed based on two-tailed student t test assuming unequal variances. Source data underlying B are provided as a Source Data file.
In the first-round of mutagenesis, hundreds of colonies appeared three days after plating on MOPS-glucose minimal agar containing 20 μM isopropyl β-d-1-thiogalactopyranoside (IPTG). On day 4, we picked ~180 of the largest colonies and quantified the octanoic acid titer from individual liquid cultures grown in Clomburg liquid media containing 20 g/L glycerol. We found 17 PkPhaG variants increased octanoic acid titer (3.3–16.3-fold) and total fatty acid titer (1.8–8.3-fold) relative to the parent PkPhaG (Supplementary Figures 3–5 and Supplementary Method 2). The 17 improved PkPhaG variants contained a total of 28 point mutations that we recreated individually. We co-expressed each PkPhaG variant from the high copy number pTRC99a-’PkphaG’-TdTER vector with pACYC-PaphaJ3 + pBTRCK-MatesB* in E. coli CM23. Each strain was cultured for 48 hrs at 30 °C and samples were harvested for fatty-acid quantification. We found that variants containing 6 of the 28 single point mutations (e.g., Q45R, R66H, H76Y, Y138F, G142V, Q277X) increased octanoic acid titer more than twofold over strains expressing the parent PkPhaG (Supplementary Figure 6). Among these, three individual PkPhaG mutants Q45R, G142V and Y138F strains had 1.9-, 1.8-, and 1.5-fold higher total fatty acid titers than strains expressing wild-type PkPhaG.
We constructed all four possible combinations of Q45R, G142V, and Y138F mutations in a vector pTRC99a-’PkphaG’-TdTER and repeated analogous fatty-acid production experiments. The best variant, PkPhaG Q45R G142V, produced 1.1 g/L of C8–C14 free fatty acids, a 4.0-fold increase compared with the original PkPhaG (Fig. 4B). The fatty-acid pool contained 41% tetradecanoic acid, 22% dodecanoic acid, 11% decanoic acid, and 26% octanoic acid, similar to the original PkPhaG-expressing strain. This indicates that the increased production of octanoic acid was due to a general increase in activity, not selectivity.
We next repeated the experiments described in Fig. 3 to confirm that the more-active PkPhaG* (referred as to PkPhaG Q45R G142V) variant was not exceeding the downstream FadB and PhaJ activities. Strains expressing PaPhaJ3, which was present in all protein engineering experiments, and PaPhaJ1 produced the highest fatty acid titers (Supplementary Figure 7) and more than any other alternative dehydratase. Similar to the observation in Fig. 2C, deletion of FadD did not substantially affect methyl ketone titer (Fig. 5A) generated by the enhanced PkPhaG*, suggesting that the modified enzyme does not have enhanced thioesterase activity. Note, these experiments were performed with a FadM thioesterase from P. sneebia that we found had higher activity than E. coli FadM4.

A Methyl ketone production by E. coli RAI or RADI harboring pTRC99a-PkphaG*-PsfadM + pACYC-PaphaJ3 plasmids. B Fatty alcohol production by E. coli RADI harboring pTRC99a-PkphaG*-TdTER + pACYC-PaphaJ3 + pBTRCK-MaACR. The corresponding negative control harbored a pTRC99a empty vector in place of the PhaG* vector. All cells were grown in 50 mL of Clomburg media containing 20 g/L glycerol, 20% (v/v) dodecane, and 1 mM IPTG at 30 °C for 48 hrs (n = 3 biologically independent samples). The shadings and non-shadings in graphs represented products in the dodecane organic layer and aqueous phase, respectively. All data represent the mean ± s.d. of biological triplicates. Source data underlying A, B are provided as a Source Data file.
Benchmarking PhaG-driven oleochemical production
Next, we benchmarked PhaG-driven production of methyl ketones and fatty alcohols as model oleochemical products. To produce methyl ketones, we cultured E. coli RADI harboring pTRC99a-PkphaG*-PsfadM and pACYC-PaphaJ3. To produce fatty alcohols, we cultured strain E. coli RADI harboring pTRC99a-PkphaG*-TdTER, pACYC-PaphaJ3, and pBTRCK-MaACR. A bi-functional alcohol forming acyl-CoA reductase MaACR from M. aquaeolei VT8 was chosen because of its successful use in prior studies8. Each strain was cultured for 72 hr at 30 °C in shake flasks containing rich glycerol media. The cultures produced 1.5 g/L total methyl ketone and ~1.1 g/L total fatty alcohol from 20 g/L glycerol (Fig. 5). The chain length distribution of each product was different. Methyl ketones were evenly distributed across 8–16-carbon chain lengths, whereas, fatty alcohols were dominated by 14- and 16-carbon alcohols. The titer and yield of fatty acids, methyl ketones, and fatty alcohols from the PhaG-dependent strategies are comparable to values reported in the literature using thioesterase strategies or reverse β-oxidation strategies to produce products in shake flasks (Table 1).
To further improve methyl ketone titers, we performed discontinuous fed-batch cultivations in a stirred bioreactor by adding media pulses after cells reached high cell densities. Enhanced aeration in bioreactors can lead to loss of volatile oleochemical products in the off-gas. Therefore, we added a 20% dodecane overlay to the culture4 to trap products in the reactor and designed a gas trap to recover products from the off-gas. Details of off-gas methyl ketone capture and ASPEN analysis can be found in the Supplementary information file (Supplementary Figure 8 and Supplementary Method 3). After 96 hrs of cultivation in reactors lacking an off-gas scrubber, cells consumed 90.0 g/L of glycerol, reached an OD600 ~60, and produced 6.8 g/L total methyl ketone (Fig. 6A). In a separate experiment, we bubbled the bioreactor off-gas through a jacketed gas dryer filled with dodecane maintained at 5 °C. After 96 hrs of induced cultivation, cells reached a density of OD600 ~60, consumed 95.2 g/L glycerol, and produced 6.7 g/L total methyl ketone (Fig. 6B). At the endpoint of the cultivation, we observed 3.4 g/L methyl ketone contained in the condensed dodecane phase of the off-gas trap, corresponding to an additional 0.51 g/L (per L of culture volume) of total methyl ketone captured from the bioreactor (Fig. 6C).

Time course of glycerol consumption, OD600, and methyl ketone titer from fed-batch bioreactor cultures (n = 3 biologically independent samples). A and bioreactor coupling a condenser (n = 2 biologically independent samples) B using E. coli RADI harboring pTRC99a-PkphaG*-PsfadM and pACYC-PaphaJ3 plasmids. C Evaluation of methyl ketone concentration from samples in the aqueous phase (dark gray), dodecane layer (light gray), and condenser (shading) after 96 h fermentation. All data represent the mean ± s.d. of biological triplicates. Source data underlying A–C are provided as a Source Data file.
This study demonstrated that PhaG is capable of supporting high-flux to three medium-chain oleochemical products, fatty acids, fatty alcohols, and methyl ketones. Through bioprospecting, we identified a PhaG from P. koreensis that demonstrated higher activity when producing methyl ketones. Random mutagenesis of PkPhaG produced 17 enhanced PhaG variants that were isolated by complementing lipoic acid auxotrophy. The improved activity was demonstrated by 3–16-fold more octanoic acid being produced when the PhaG variants were co-expressed in a cell designed to direct flux to octanoic acid (co-expressing TesB, PhaJ3, and Ter). Reconstitution of the individual point mutations led to the creation of a double mutant, PkPhaG Q45R G142V, that showed 4.0-fold higher activity relative to the parent enzyme. Finally, we demonstrated the production of 1.1 g/L C8–C14 free fatty acids, 1.5 g/L C7–C15 methyl ketones, and 1.1 g/L C8–C16 fatty alcohols in shake flasks and 7.2 g/L of methyl ketones in a fed-batch. These results demonstrate that PhaG is a viable alternative strategy that should be considered for oleochemical production. The PhaG-dependent strategy has the potential to achieve higher theoretical yields compared with the well-established thioesterase route. However, this impact, saving one ATP per product, is not likely to be observed until cells approach theoretical limits. Given the current state of the field, additional work is needed on both fronts. Our work motivates continued strain development as well as additional protein engineering15,48,49 to narrow the PhaG product profile and further increase activity.
Methods
Bacterial strains, plasmids, oligonucleotides, and reagents
All bacterial strains used in this study are listed in Supplementary Data 1. Q5 DNA polymerase and Monarch® PCR and DNA Cleanup Kit were purchased from New England Biolabs (Ipswich, MA). Oligonucleotide primers and gBlocks were synthesized by Integrated DNA Technologies (IDT), Inc. (San Diego, CA). Chemicals including fatty acid, fatty alcohol, and methyl ketones were purchased from Sigma-Aldrich (St. Louis, MO).
E. coli DH5α strains were used for plasmid amplification and DNA assembly. E. coli RABIJ (MG1655 ΔaraBAD ΔfadR ΔfadA ΔfadB ΔfadI ΔfadJ) and E. coli CM23 (MG1655ΔaraBAD ΔFadABIJDRE ΔldhA ΔackApta ΔadhE ΔpoxB ΔfrdABCD ΔydiO ΔatoC) were created as part of prior studies11,26. E.coli RAI (MG1655 ΔaraBAD ΔfadR ΔfadA ΔfadI) and RADI (MG1655 ΔaraBAD ΔfadR ΔfadA ΔfadI ΔfadD) strains were used in those experiments presented in Figs. 2B–D, 3B, 5A, B, 6A, B. E. coli CM23 strains were used in those experiments presented in Fig. 4B.
Plasmid and strain construction
All plasmids used in this study are summarized in Supplementary Data 1. Plasmid maps are available in Source Data file folder. All plasmids were constructed by Gibson assembly (New England Biolabs). The chromosomal deletion of fadD and lipB were performed using a combination of lambda red recombination and CRISPR/Cas9-mediated selection4,8,14,18. All cloned sequences and gene deletions were confirmed by Sanger sequencing performed by Functional Biosciences (Madison, WI). Constructs expressing FadB4,8 and PhaJ homologs were obtained from prior studies44.
Oleochemical production and quantification
All oleochemical production studies were performed by growing E. coli strains at 30 °C in Clomburg medium containing 20 g/L glycerol, the appropriate antibiotics (Carbenicillin—100 μg/mL; kanamycin—50 μg/mL; chloramphenicol—34 μg/mL), and IPTG for induction as indicated. Pre-cultures for each experiment were prepared by inoculating 5 mL LB media (+antibiotics) with a single colony and incubating overnight at 30 °C with shaking at 250 r.p.m. A 2.5 vol% inoculum was transferred into production flasks with a starting OD600 0.1. Fatty alcohol and methyl ketone production cultures were supplemented with 10% (v/v) dodecane to provide a product sink. Samples from each culture were extracted after 72 h incubation at 30 °C.
Fed-batch fermentation was performed using a 1-L Infors Multifors bioreactor. Overnight pre-cultures were inoculated to an initial OD600 of 0.05 into a bioreactor containing 500 mL Clomburg medium with ~50 g/L glycerol. The bioreactor was operated at the following conditions: the temperature was controlled at 30 °C post induction, airflow was 1.5 L/min, stirrer rate was varied between 250 r.p.m. and 1000 r.p.m. to control dissolved oxygen at a value of 30%, pH was maintained at 7.0 using 2 M sulfuric acid and 2 M ammonia hydroxide. When the OD600 reached ~1.0, IPTG was added to achieve a final concentration of 1 mM and 100 mL dodecane was fed in the bioreactor. At 24 h of post induction, ~100 mL 5× concentrated Clomburg media containing 500 g/L glycerol was one-time bolus-fed into the bioreactor and fermentation terminated 96 h post induction. Measurements of methyl ketone, glycerol, optical density, and CO2 evolution were recorded for 96 h total.
The bioreactor outlet gas stream was directed through a chilled organic absorber to capture methyl ketone vapors stripped from the culture broth. The absorber was composed of a jacketed glass gas dryer with a ceramic sparge distributing the off-gas into 100 mL of chilled dodecane. The temperature of the absorber was maintained at 5 °C by an external water cooler. Methyl ketone data were taken at 96 h from the dodecane, and endpoint methyl ketone capture was taken from both the collected water and remaining dodecane 96–101 h after inducing the culture. A schematic of the absorber is described in more detail in Supplementary Figure 7.
To determine the methyl ketone and fatty alcohol concentration in the distinct organic or aqueous phases, 50 mL of cell culture was centrifuged at 4500 × g for 10 min and 0.5 mL samples from the dodecane layer and 2.5 mL samples from the aqueous phase were collected and evaluated separately. Fatty acids were extracted from culture according to an acid-based esterification method14,15. Fatty alcohols and methyl ketones were extracted from culture into n-hexane4,8. Fatty acid and methyl ketone species were separated using an Agilent RTX-5 column and fatty alcohol species were separated using Agilent DB-Fatwax column (Santa Clara, CA). Oleochemicals were quantified by comparing GC-FID peak areas against standard curves prepared with commercial standards.
Mutagenesis of PhaG
A mutagenic PhaG library was constructed by error-prone PCR using GeneMorph II from Agilent (Senta Clara, CA) with a low mutation frequency (0–4.5 mutations/kb). The plasmid backbone (pBTRCK) was PCR-amplified using a high-fidelity DNA polymerase Q5 from New England Biolabs (Ipswich, MA). The library was assembled using an isothermal assembly method50. Primers used in the creation of the library contained the start and stop codons in order to prevent mutation of them.
The sequence of improved PhaG variants was obtained by Sanger sequencing of colony PCR products made by high-fidelity Q5 DNA polymerase. To re-introduce single point mutations, we amplified PkPhaG with mutagenic primers and subcloned the fragments into pTRC99A-’PkphaG’-TdTER by an isothermal assembly method51. The resulting plasmids were transformed into E. coli CM23 harboring pBTRCK-MatesB* and pACYC-PaphaJ3 plasmids.
Lipoic acid selection
3-Hydroxyoctanoyl-CoA producing variants of PhaG were isolated using a lipoic/octanoic acid selection strategy14. In brief, Gibson assembly reaction mixtures (2 μL) containing a PhaG library (pBTRCK-PkphaG*) was transformed into 100 μL of electrocompetent E. coli CM23-ΔlipB (CM23 strain, ΔlipB) strain harboring pTRC99a-TdTER-MatesB* + pACYC-PaphaJ3. Transformants were plated on MOPS minimal media agarose plates containing 0.2% glucose, 20 μM IPTG, and kanamycin, chloramphenicol, and carbenicillin to maintain and induce plasmids14. Cells were rescued growth and appeared on the selection plates after 3 days of incubation. Transformants were patched onto LB plates for archiving and secondary screening in octanoic acid production studies.
Statistics
We used instrument software to collect and analyze most experiments. Shimadzu Labsolutions (Long Beach, CA) was used for GC and HPLC data analysis. The fluorescence intensity of YFP and RFP was detected by the Tecan 200 Plate reader software 3.9.1.0. (Männedorf, Switzerland). Error bars indicate standard deviations from three biological replicates. All P values were generated from a two-tailed Student’s t test using Microsoft Excel 2016 (Microsoft Corporation, USA).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper (and its Supplementary Information files). All strains and plasmids information are available in Supplementary Data 1. The additional reactions and metabolites used in Oleo_iML1515 than iML1515 are available in Supplementary Data 2 and 3. All plasmid maps including annotations of oligonucleotide and gene sequences are available in Supplementary Data 4. Source data are provided with this paper.
Code availability
All codes for calculating theoretical yields are available at the GitHub repository [https://github.com/Pfleger-Lab/Metabolic-Modeling---Yield-Analysis-of-PhaG].
References
Sarria, S., Kruyer, N. S. & Peralta-Yahya, P. Microbial synthesis of medium-chain chemicals from renewables. Nat. Biotechnol. 35, 1158–1166 (2017).
Yan, Q. & Pfleger, B. F. Revisiting metabolic engineering strategies for microbial synthesis of oleochemicals. Metab. Eng. 58, 35–46 (2020).
Vaughn, S. F. & Holser, R. A. Evaluation of biodiesels from several oilseed sources as environmental friendly contact herbicides. Ind. Crops Prod. 26, 63–68 (2007).
Yan, Q. et al. Metabolic engineering of β-oxidation to leverage thioesterases for production of 2-heptanone, 2-nonanone and 2-undecanone. Metab. Eng. 61, 335–343 (2020).
Longo, M. A. & Sanromán, M. A. Production of food aroma compounds: microbial and enzymatic methodologies. Food Technol. Biotechnol. 44, 335–353 (2006).
Dellomonaco, C., Clomburg, J. M., Miller, E. N. & Gonzalez, R. Engineered reversal of the β-oxidation cycle for the synthesis of fuels and chemicals. Nature 476, 355–359 (2011).
Lennen, R. M. & Pfleger, B. F. Engineering Escherichia coli to synthesize free fatty acids. Trends Biotechnol. 30, 659–667 (2012).
Mehrer, C. R., Incha, M. R., Politz, M. C. & Pfleger, B. F. Anaerobic production of medium-chain fatty alcohols via a β-reduction pathway. Metab. Eng. 48, 63–71 (2018).
Wu, J. et al. Construction of artificial micro-aerobic metabolism for energy- and carbon-efficient synthesis of medium chain fatty acids in Escherichia coli. Metab. Eng. 53, 1–13 (2019).
Wu, J. et al. Improving metabolic efficiency of the reverse beta-oxidation cycle by balancing redox cofactor requirement. Metab. Eng. 44, 313–324 (2017).
Wu, J., Zhang, X., Xia, X. & Dong, M. A systematic optimization of medium chain fatty acid biosynthesis via the reverse beta-oxidation cycle in Escherichia coli. Metab. Eng. 41, 115–124 (2017).
Benning, C. Mechanisms of lipid transport involved in organelle biogenesis in plant cells. Annu. Rev. Cell Dev. Biol. 25, 71–91 (2009).
Deng, X. et al. Structure-guided reshaping of the acyl binding pocket of ‘TesA thioesterase enhances octanoic acid production in E. coli. Metab. Eng. 61, 24–32 (2020).
Hernández Lozada, N. J. et al. Highly active C 8 -Acyl-ACP thioesterase variant isolated by a synthetic selection strategy. ACS Synth. Biol. 7, 2205–2215 (2018).
Grisewood, M. J. et al. Computational redesign of Acyl-ACP thioesterase with improved selectivity toward medium-chain-length fatty acids. ACS Catal. 7, 3837–3849 (2017).
Feng, Y. et al. Tuning of acyl-ACP thioesterase activity directed for tailored fatty acid synthesis. Appl. Microbiol. Biotechnol. 102, 3173–3182 (2018).
Voelker, T. A. & Davies, H. M. Alteration of the specificity and regulation of fatty acid synthesis of Escherichia coli by expression of a plant medium-chain acyl-acyl carrier protein thioesterase. J. Bacteriol. 176, 7320–7327 (1994).
Hernández Lozada, N. J., Simmons, T. R., Xu, K., Jindra, M. A. & Pfleger, B. F. Production of 1-octanol in Escherichia coli by a high flux thioesterase route. Metab. Eng. 61, 352–359 (2020).
Fiedler, S., Steinbüchel, A. & Rehm, B. H. A. PhaG-mediated synthesis of Poly(3-Hydroxyalkanoates) consisting of medium-chain-length constituents from nonrelated carbon sources in recombinant Pseudomonas fragi. Appl. Environ. Microbiol. 66, 2117–2124 (2000).
Rehm, B. H. A., Krüger, N. & Steinbüchel, A. A new metabolic link between fatty acid de novosynthesis and polyhydroxyalkanoic acid synthesis. J. Biol. Chem. 273, 24044–24051 (1998).
Hoffmann, N. et al. Biochemical characterization of the Pseudomonas putida 3-hydroxyacyl ACP:CoA transacylase, which diverts intermediates of fatty acid de novo biosynthesis. J. Biol. Chem. 277, 42926–42936 (2002).
Wang, Y. Synthesis of medium-chain-length-polyhydroxyalkanoates in tobacco via chloroplast ge-netic engineering. Chin. Sci. Bull. 50, 1113 (2005).
Romano, A., van der Plas, L. H. W., Witholt, B., Eggink, G. & Mooibroek, H. Expression of poly-3-(R)-hydroxyalkanoate (PHA) polymerase and acyl-CoA-transacylase in plastids of transgenic potato leads to the synthesis of a hydrophobic polymer, presumably medium-chain-length PHAs. Planta 220, 455–464 (2005).
Salvachúa, D. et al. Metabolic engineering of Pseudomonas putida for increased polyhydroxyalkanoate production from lignin. Microb. Biotechnol. 13, 290–298 (2020).
Matsumoto, K., Matsusaki, H., Taguchi, S., Seki, M. & Doi, Y. Cloning and characterization of the Pseudomonas sp. 61-3 phaG gene involved in polyhydroxyalkanoate biosynthesis. Biomacromolecules 2, 142–147 (2001).
Poblete-Castro, I. et al. In-silico-driven metabolic engineering of Pseudomonas putida for enhanced production of poly-hydroxyalkanoates. Metab. Eng. 15, 113–123 (2013).
Zhang, X., Lin, Y., Wu, Q., Wang, Y. & Chen, G.-Q. Synthetic biology and genome-editing tools for improving pha metabolic engineering. Trends Biotechnol. 38, 689–700 (2020).
Choi, S. Y. et al. Microbial polyhydroxyalkanoates and nonnatural polyesters. Adv. Mater. 32, 1907138 (2020).
Tobin, K. M., O’Leary, N. D., Dobson, A. D. W. & O’Connor, K. E. Effect of heterologous expression of phaG [(R)-3-hydroxyacyl-ACP-CoA transferase] on polyhydroxyalkanoate accumulation from the aromatic hydrocarbon phenylacetic acid in Pseudomonas species. FEMS Microbiol. Lett. 268, 9–15 (2007).
Wang, Q., Tappel, R. C., Zhu, C. & Nomura, C. T. Development of a new strategy for production of medium-chain-length polyhydroxyalkanoates by recombinant Escherichia coli via inexpensive non-fatty acid feedstocks. Appl. Environ. Microbiol. 78, 519–527 (2012).
Le Meur, S., Zinn, M., Egli, T., Thöny-Meyer, L. & Ren, Q. Production of medium-chain-length polyhydroxyalkanoates by sequential feeding of xylose and octanoic acid in engineered Pseudomonas putida KT2440. BMC Biotechnol. 12, 53 (2012).
Zheng, Z. et al. Thioesterase II of Escherichia coli plays an important role in 3-hydroxydecanoic acid production. Appl. Environ. Microbiol. 70, 3807–3813 (2004).
Zheng, Z., Zhang, M.-J., Zhang, G. & Chen, G.-Q. Production of 3-hydroxydecanoic acid by recombinant Escherichia coli HB101 harboring phaG gene. Antonie Van. Leeuwenhoek 85, 93–101 (2004).
Nawabi, P., Bauer, S., Kyrpides, N. & Lykidis, A. Engineering Escherichia coli for biodiesel production utilizing a bacterial fatty acid methyltransferase. Appl. Environ. Microbiol. 77, 8052–8061 (2011).
Goh, E.-B., Baidoo, E. E. K., Keasling, J. D. & Beller, H. R. Engineering of bacterial methyl ketone synthesis for biofuels. Appl. Environ. Microbiol. 78, 70–80 (2012).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Gerlt, J. A. et al. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): a web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta. 1854, 1019–1037 (2015).
Muneer, F. et al. Microbial polyhydroxyalkanoates (PHAs): efficient replacement of synthetic polymers. J. Polym. Environ. 28, 2301–2323 (2020).
Schweizer, E. & Hofmann, J. Microbial type I fatty acid synthases (FAS): major players in a network of cellular FAS systems. Microbiol. Mol. Biol. Rev. 68, 501–517 (2004).
He, X.-Y. & Yang, S.-Y. Histidine-450 is the catalytic residue of L-3-hydroxyacyl coenzyme a dehydrogenase associated with the large α-subunit of the multienzyme complex of fatty acid oxidation from Escherichia coli. Biochemistry 35, 9625–9630 (1996).
Yang, S. Y. & Elzinga, M. Association of both enoyl coenzyme A hydratase and 3-hydroxyacyl coenzyme A epimerase with an active site in the amino-terminal domain of the multifunctional fatty acid oxidation protein from Escherichia coli. J. Biol. Chem. 268, 6588–6592 (1993).
Yang, S. Y., Li, J. M., He, X. Y., Cosloy, S. D. & Schulz, H. Evidence that the fadB gene of the fadAB operon of Escherichia coli encodes 3-hydroxyacyl-coenzyme A (CoA) epimerase, delta 3-cis-delta 2-trans-enoyl-CoA isomerase, and enoyl-CoA hydratase in addition to 3-hydroxyacyl-CoA dehydrogenase. J. Bacteriol. 170, 2543–2548 (1988).
Smeland, T. E., Cuebas, D. & Schulz, H. Epimerization of 3-hydroxy-4-trans-decenoyl coenzyme A by a dehydration/hydration mechanism catalyzed by the multienzyme complex of fatty acid oxidation from Escherichia coli. J. Biol. Chem. 266, 23904–23908 (1991).
Agnew, D. E., Stevermer, A. K., Youngquist, J. T. & Pfleger, B. F. Engineering Escherichia coli for production of C12–C14 polyhydroxyalkanoate from glucose. Metab. Eng. 14, 705–713 (2012).
Swarbrick, C. M. D. et al. mycobacteria encode active and inactive classes of TesB fatty-Acyl CoA thioesterases revealed through structural and functional analysis. Biochemistry 56, 1460–1472 (2017).
McMahon, M. D. & Prather, K. L. J. Functional screening and in vitro analysis reveal thioesterases with enhanced substrate specificity profiles that improve short-chain fatty acid production in Escherichia coli. Appl. Environ. Microbiol. 80, 1042–1050 (2014).
Guevara-Martínez, M. et al. The role of the acyl-CoA thioesterase “YciA” in the production of (R)-3-hydroxybutyrate by recombinant Escherichia coli. Appl. Microbiol. Biotechnol. 103, 3693–3704 (2019).
Xue, P. et al. A mass spectrometry‐based high‐throughput screening method for engineering fatty acid synthases with improved production of medium‐chain fatty acids. Biotechnol. Bioeng. 117, 2131–2138 (2020).
Ellis, B. M. et al. Accelerating strain phenotyping with desorption electrospray ionization-imaging mass spectrometry and untargeted analysis of intact microbial colonies. Proc. Natl. Acad. Sci. 118, e2109633118 (2021).
Pfleger, B. F., Gossing, M. & Nielsen, J. Metabolic engineering strategies for microbial synthesis of oleochemicals. Metab. Eng. 29, 1–11 (2015).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Youngquist, J. T. et al. Production of medium chain length fatty alcohols from glucose in Escherichia coli. Metab. Eng. 20, 177–186 (2013).
Nies, S. C. et al. High titer methyl ketone production with tailored Pseudomonas taiwanensis VLB120. Metab. Eng. 62, 84–94 (2020).
Goh, E., Chen, Y., Petzold, C. J., Keasling, J. D. & Beller, H. R. Improving methyl ketone production in Escherichia coli by heterologous expression of NADH‐dependent FabG. Biotechnol. Bioeng. 115, 1161–1172 (2018).
Acknowledgements
This work was funded by the DOE Center for Advanced Bioenergy and Bioproducts Innovation (U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Number DE-SC0018420) and the National Science Foundation (CBET-1703504).
Author information
Authors and Affiliations
Contributions
Q.Y.: conceptualization, investigation, data curation, writing original draft, writing—review & editing. M.A.J.: investigation, writing—review & editing, W.T.: investigation, writing—review & editing. D.C.: investigation, writing—review & editing. X.C.: investigation, writing—review & editing. M.K.K.: investigation, writing—review & editing. B.F.P.: conceptualization, writing original draft, writing—review & editing, supervision, funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Akihiko Kondo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yan, Q., Cordell, W.T., Jindra, M.A. et al. Metabolic engineering strategies to produce medium-chain oleochemicals via acyl-ACP:CoA transacylase activity. Nat Commun 13, 1619 (2022). https://doi.org/10.1038/s41467-022-29218-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-29218-3
This article is cited by
-
Combinatorial optimization of gene expression through recombinase-mediated promoter and terminator shuffling in yeast
Nature Communications (2024)
-
Biosynthesis pathways of expanding carbon chains for producing advanced biofuels
Biotechnology for Biofuels and Bioproducts (2023)
-
Engineering Pseudomonas putida KT2440 for chain length tailored free fatty acid and oleochemical production
Communications Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
