
Abstract
In the biosynthesis sterols an enzyme-catalyzed demethylation is achieved via a stepwise oxidative transformation of alcohols to olefins. The overall demethylation proceeds through two sequential monooxygenation reactions and a subsequent dehydroformylative saturation. To mimic the desaturation processes observed in nature, we have successfully integrated photoredox proton-coupled electron transfer (PCET) and cobaloxime chemistry for the acceptorless dehydrogenation of alcohols. The state-of-the-art remote and precise desaturation of ketones proceeds efficiently through the activation of cyclic alcohols using bond-dissociation free energy (BDFE) as thermodynamic driving force. The resulting transient alkoxyl radical allows C-C bond scission to generate the carbon-centered radical remote to the carbonyl moiety. This key intermediate is subsequently combined with cobaloxime photochemistry to furnish the alkene. Moreover, the mild protocol can be extended to desaturation of linear alcohols as well as aromatic hydrocarbons. Application to bioactive molecules and natural product derivatives is also presented.
Similar content being viewed by others

Introduction
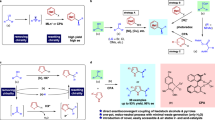
In the biosynthesis of sterols, a central reaction is the enzyme-catalyzed demethylation that involves a stepwise oxidative transformation of alcohols to olefins1,2,3,4. To trigger the C–C bond cleavage, the generation of an alkoxyl radical is believed to be crucial (Fig. 1a). Inspired by this intriguing enzymatic process, we sought an analogous methodology for desaturation of alcohols in organic synthesis. The access of alkoxyl radicals via hydrogen atom transfer from the hydroxyl groups of aliphatic alcohols is straightforward but remarkably challenging. This is mainly attributed to the intrinsic difficulty of activating the kinetically inert O–H bonds5. In recent years, visible light photoredox catalysis has emerged as a powerful technique in organic synthesis that relies upon energetic electron transfer processes to facilitate previously thermally inaccessible or kinetically inert transformations6,7,8,9,10. In this context, the activation of O–H bonds has found broad utility in a number of reactions for the construction of C–C 11,12,13,14,15,16, C–N 17,18,19, C–S 20, and C–X 21,22 bonds (Fig. 1b). Despite these efforts, it is surprising to consider that the bioinspired olefin synthesis through acceptorless desaturation of alcohols remains an unmet challenge, to the best of our knowledge23.

a 14α-Demethylase (CYP51)-catalyzed demethylation of lanosterol and possible reaction mechanism. The CYP51 belongs to the family of cytochrome P450 enzymes. The demethylation proceeds through two sequential monooxygenation reactions and subsequently a dehydroformylation. b Ring-opening and functionalization of cycloalkanols enabled by photoredox catalysis. c Possible mechanism for stearoyl Δ9 desaturation. d Recent notable work on dehydrogenation via photoredox cobaloxime dual catalysis. e Desaturation of alcohols and aliphatic C–H bond via PCET/cobaloxime dual catalysis.
Unsaturated carbonyl compounds are not only versatile synthetic building blocks, but also ubiquitous in natural products and biologically relevant molecules24,25,26,27,28,29. Although protocols for carbonyl desaturation at the adjacent sites (α-/β-) have been widely established, a mild and general strategy for remote site desaturation of ketone would be very appealing, but has not been reported. In nature, desaturase enzyme fascinates us with the regio- and stereoselective olefinic bond formation during the biosynthesis of fatty acid (Fig. 1c)30. Notably, Baran31 and Gevorgyan32,33 recently reported auxiliaries assisted remote desaturation of alcohols and amines, through TEMPO mediated process or palladium photoredox catalysis, respectively. Given state-of-the-art remote functionalization, we considered that it is highly desirable to exploit readily accessible reaction partners with efficient catalyst systems to produce unsaturated ketones that are difficult to synthesize via conventional pathways.
Recently, our group established a dual Nickel photoredox catalysis reaction to enable the remote cross coupling of tertiary alcohols16. This catalytic manifold provides a general and facile access to carbon-centered radicals remote to the carbonyl moiety via multiple site proton-coupled electron transfer (PCET)34,35. In line with the insight gained from this dual catalytic system, as well as recent developments on the dehydrogenative functionalization36,37,38,39,40,41, we envisioned the alkyl radical generated in this manner could in principle be merged with metallaphotoredox to realize the bioinspired acceptorless desaturation of alcohols. Enlighted by the most recent development on cobaloxime-based photoredox catalysis by Ritter, Sorensen and Leonori groups (Fig. 1d)42,43,44,45,46,47,48,49,50,51,52,53,54,55, we became interested in the combination of photoredox PCET with the proton reduction reactivity of cobaloximes (Fig. 1e). Here, we describe the development of a bioinspired acceptorless remote desaturation of tertiary, as well as secondary alcohols, via the photoredox PCET and cobalt synergistic catalysis and the extension to the desaturation to aromatic hydrocarbons, as well as silyl enol ethers.
Results
Rational design
Our mechanistic proposal is shown in Fig. 2. Upon visible light irradiation, a single electron transfer from the cyclohexanol derivative (Ep/2 = 1.57 V vs. SCE)16 to the highly oxidizing singlet excited state *Mes–Acr–Me+ (E1/2red = +2.06 V vs. SCE)6 would generate the corresponding arene radical cation along with the reduced form of the photocatalyst Mes–Acr–Me•. Subsequent multiple site PCET reaction between the hydroxyl group and the radical cation in the presence of base would give the key alkoxyl radical species, which readily cleaves into a carbonyl moiety and a distal carbon-centered radical through β scission of the neighboring C–C bond. The C-centered radical would subsequently be intercepted by the CoII species (I) to yield an alkyl–CoIII intermediate (II), which can undergo C-cobalt bond homolysis upon light irradiation. Next, a, β-hydrogen abstraction by CoII at this stage would deliver the desired olefin and a cobaltIII hydride species (III). Hydrogen gas evolves through the interaction between (III) and a proton generated in the PCET step. The cobalt and photoredox catalytic cycles then simultaneously complete via a single electron transfer event between CoIII intermediate (IV) (E1/2red = –0.68 V vs. SCE) and reduced form of photocatalyst C56,57.

Photoredox PCET/cobaloxime dual catalysis enabled desaturation of alcohols. PMP p-methoxy phenyl group.
Optimization of the reaction conditions
The reaction conditions optimization of the synergistic combination of photoredox and cobalt catalysis is briefly summarized in Table 1. The initial evaluation focused on readily available substrate 1a to mimic the enzymatic process, namely Δ9 desaturation of stearoyl-CoA (Fig. 1c). Optimized reaction conditions were readily established, using 7.5 mol% PC-I Mes–Acr–Me+, 10 mol% Co(dmgH)2(py)2PF6 and 2 equiv. 2,4,6-collidine in a 0.1 M solution of 1,2-dichloroethane (DCE) at room temperature with blue light-emitting diodes (LEDs) irradiation. Under these conditions, the desired product was formed in 93% NMR yield and very good selectivity (22:1) (Table 1, entry 1). Use of less oxidizing photosensitizers such as [Ru(bpy)3](PF6)2 (PC-II, E1/2[Ru*III/RuII = + 0.77 V vs. SCE in CH3CN])58 and [Ir(dF(CF3)ppy)2(bpy)]PF6 (PC-III, E1/2[Ir*III/IrII = + 1.21 V vs. SCE in CH3CN])59 resulted in no product or trace amounts of product, while the strong oxidizing [Ir(dF(CF3)ppy)2(5,5’(CF3)bpy)]PF6 (PC-IV, E1/2[Ir*III/IrII = + 1.68 V vs. SCE in CH3CN]) gave 2a in 71% yield (entries 2 to 4). A screening of different organic and inorganic bases revealed that collidine was the best choice (entries 5 and 6). The use of Co(dmgH)(dmgH2)Cl2 and Co(dmgH)2PyCl as cobaloxime sources leads to a decrease of yields and selectivities (entries 7 and 8). When the reaction was conducted in HFIP or MeCN, the amount of product could be negligible. The reaction efficiency diminished dramatically when toluene was employed, while a comparable result was observed using DCM (entries 9–12). The inverse relationship between solvent polarity and yield can be taken as support for the intermediacy of hydrogen bonding, because the polar interaction is generally disfavored in polar solvent. It is not surprising to observe that control experiments performed without photocatalyst, Co catalyst, base or light each failed to give any desired product (Table 1, entry 13).
Substrate scope
As illustrated in Fig. 3, we found the desaturation protocol was tolerant to a wide range of alcohols and gave the corresponding olefinic products in moderate to very good yields. To begin with, it was found that cyclic tertiary alcohols with different ring sizes (1a–1h) all reacted smoothly to selectively generate remote desaturated ketones regardless of their ring strains (2a–h, 51–91% yields). Notably, due to the base mediated isomerization, the α-/β-sites desaturation product was obtained exclusively with prolonged reaction time in the reaction of cyclobutanol 1b. Symmetrical substituted cyclohexanols (1i to 1p) behaved well to give products 2i to 2p in moderate yields (42–68%). Functional groups including trifluoromethyl, silyl ether, geminal difluoride, nitrile and amide could be well tolerated. The ring-opening of unsymmetrical cyclohexanols 1r and 1s took place regioselectively and, the remarkable selectivity can be explained by C–C bond cleavage favors the formation of more stabilized carbon-centered radical (2r and 2s, 60% and 82% yields).

General reaction conditions: 1 (1 equiv.), 2,4,6-collidine (2 equiv.), PC-I (7.5 mol%), Co(dmgH)2(py)2PF6 (10 mol%), DCE (0.1 M), rt, blue LEDs, 48–72 h. a 1 or 3 mol% [Ir(dF(CF3)ppy)2(5,5′d(CF3)bpy)](PF6) PC-IV was used. b NBu4OP(O)(OPh)2 as base. c Toluene or PhCF3 as solvent. d 5 mol% Co catalyst was used in 2u to 2z. e 1H NMR yield with internal standard.
For the norcamphor-derived bridged bicyclic substrate 1t, mixture of isomers was observed as a result of poor selectivity in the ring-opening. In contrast, a menthone derivative displays excellent regioselectivity in the ring-opening step (1v, 84%). Moreover, we successfully extended the scope to linear tertiary alcohols derived from pregnenolone and oleate ester, affording the corresponding olefins 2u and 2w in moderate to excellent yields. In addition to tertiary alcohols, naturally occurring secondary alcohol 1x proved to be competent substrate for the PCET enabled regioselective ring-opening/desaturation sequence. Remarkably, the dehydroxymethylative desaturation of 2,2-diphenylpropan-1-ol could take place, affording the diphenylethylene in 22% yield. Next, we found that the cascade ring cyclization/desaturation was feasible, a moderate yield of cyclized product was obtained (2z, 56%).
With the above success, we next examined the generality of the photoredox-cobalt desaturation with respect to the arene substituent on the cyclic ring. As shown in Fig. 4, a variety of arene-substituted cyclohexanols performed well. Monosubstituted aromatics such as tert-butyl–, phenyl–, and tert-butyldimethylsilyl (TBS)-protected phenols as well as biphenyl are suitable candidates (4a–4d, 58–74% yields). Disubstituted anisole derivative was also suitable substrate for this transformation (4e, 72% yield). Both naphthalene- and phenanthrene-substituted cyclohexanols smoothly underwent ring-opening/desaturation under these conditions (4f and 4g, 52% and 47% yields). Interestingly, minor amount of α-/β-sites desaturation product was obtained in the case of 4f, this can be accounted for by CoIII-hydride mediated chain walking process51. Following the success, privileged heteroaromatics including benzofuran and thiobenzofuran could also be used (4h and 4i, 43% and 63% yields).

Reaction conditions: 3 (0.2 mmol, 1 equiv.), 2,4,6-collidine (0.4 mmol, 2 equiv.), PC-I (7.5 mol%), Co(dmgH)2(py)2PF6 (10 mol%), DCE (2.0 mL, 0.1 M), rt, blue LEDs, 48–72 h.
To further highlight the robustness of this protocol, tetrahydronaphthalene 5a was subjected to the reaction conditions (Fig. 5). The desaturation takes place selectively to release 2 molar equiv. hydrogen gas and no dihydronaphthalene product was observed, indicating the second desaturation should be easier than the initial one. Therefore, two styrene derivatives were examined next, to give products such as 6b and 6c in good to excellent yields. Heterocycle such as 6d can also be prepared in good yield (74%). Moreover, when benzylic alcohols were employed in the PCET enabled acceptorless desaturation, the corresponding aldehyde and ketone products were obtained in excellent yields (6e and 6f). Importantly, this protocol was able to transform silyl enol ethers into silyloxyarenes in moderate to good yields60. For example, desaturation of 5g leads to the formation of tert-butyl(dimethyl)silyl (TBS) ether of α-naphthol with excellent efficiency (6g, 84% yield). Silyl enol ethers derived from cyclohexanones were also amenable to the current protocol via the removal of two molecules of hydrogen gas, with no detection of α, β-desaturated ketone product (6h and 6i, 51% and 72% yields, respectively). This intriguing selectivity stands in stark contrast to previous dehydrogenation reactions that exclusively affording cyclohexenones61,62.

Reaction conditions: 5 (0.2 mmol, 1 equiv.), 2,4,6-collidine (0.2 mmol, 1 equiv.), [Ir(dF(CF3)ppy)2(5,5′d(CF3)bpy)](PF6) PC-IV(1 mol%), Co(dmgH)2(py)2PF6 (10 mol%), DCE (1.0 mL, 0.2 M), rt, blue LEDs, 36 h.
Next, we carried out the synthesis of remote unsaturated ketone 2a on a preparative scale (2 mmol) under our optimal conditions, providing the expected olefinic product 1a in 81% yield (Fig. 6a). To showcase the synthetic utility of the product provided by this methodology, 2a was converted efficiently to a fatty acid ester epoxide during a Baeyer-Villiger oxidation (Fig. 6b). Following a Grignard reaction of 2a, the linear product 8 was subjected further to the reaction conditions, an interesting diene 9 was isolated in 51% yield (Fig. 6c).

a Reaction on large scale, under standard conditions: tertiary alcohol (1 equiv.), 2,4,6-collidine (2 equiv.), PCI (7.5 mol%), Co(dmgH)2(py) 2PF6 (10 mol%), DCE (0.1 M), rt, blue LEDs, 48 h. b Baeyer-villiger oxidation. c sequential desaturation to synthesize diene.
Apart from the control experiments shown in Table 1, we conducted some preliminary mechanistic experiments to gain some insight of the metallaphotoredox desaturation protocol. When the reaction mixture was subjected to a radical scavenger 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO, 1 equiv.) under the standard conditions, no product was detected. A remote TEMPO-trapped ketone 10 was instead formed, implying that a radical process is involved in the catalytic cycle (Fig. 7a). The generation of carbon-centered radical remote to the carbonyl group was further supported by the Heck-Type coupling with styrene, affording 11 (Fig. 7b). To gain more insight into the reaction, the formation of molecular hydrogen was quantitatively analyzed by gas chromatography. Importantly, we observed that more than 1 equiv. hydrogen gas was produced with substrate 5a, in contrast the generation of H2 is less than 1 equiv. in the case of 1d. The kinetic profile of H2 evolution of substrate 1d shows a fast gas production rate in the first hours, however, it becomes very sluggish after 6 h (Fig. 7c, d). This result is consistent with our observation that the desaturation of alcohols generally required long reaction time, we assume that the increasing amount of free base has a marked effect on the hydrogen production63.

a Radical-trapping experiment with free-radical scavenger. b Heck-Type coupling with styrene. c Reaction conditions and results for H2 production of substrates 1d and 5a. d H2 production rate monitoring and quantification for substrates 1d and 5a.
Discussion
In conclusion, we have developed a bioinspired acceptorless desaturation of tertiary as well as secondary alcohols via the photoredox PCET and cobalt synergistic catalysis. The manifold provides a concise access to remotely dehydrogenated ketones that are difficult to synthesize with current methods, through ring-opening/desaturation of cyclic alcohols. We also demonstrated the strategy could be applied to linear alcohol, aromatic hydrocarbons as well as silyl enol ethers. Importantly, a variety of bioactive molecules and natural product derivatives were all well tolerated under such mild conditions. In consideration with numerous findings about the essential role of PCET in biological redox processes, this contribution expands the less-developed applications of PCET in organic synthesis.
Methods
General procedure for bioinspired dehydrogenation of alcohols
To a 15 mL vial equipped with a stir bar was added Co(dmgH)2(py)2PF6 (12 mg, 0.02 mmol, 10 mol%), and photocatalyst (9-mesityl-10-methylacridinium perchlorate 7.5 mol% or [Ir(dF(CF3)ppy)2(5,5′d(CF3)bpy)](PF6) 1 mol%), collidine (1 or 2 equiv.) and tertiary alcohol (0.2 mmol, 1 equiv.). The vial was sealed, evacuated and backfilled with Argon three times, then 2 mL of DCE was added. After degassing with Freeze–Pump–Thaw methods for three cycles, it was stirred and irradiated with the corresponding blue LEDs photoreactor. Upon completion, the reaction mixture was concentrated in vacuo and purified with column chromatography to afford the desired product.
Data availability
The authors declare that all data generated in this study are available within the article and the Supplementary Information.
References
Shyadehi, A. Z. et al. The mechanism of the acyl-carbon bond cleavage reaction catalyzed by recombinant sterol 14α-demethylase of candida albicans (other names are: lanosterol 14α-demethylase, P-45014DM, and CYP51). J. Biol. Chem. 271, 12445–12450 (1996).
Lepesheva, G. I. & Waterman, M. R. Sterol 14α-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim. Biophys. Acta 1770, 467–477 (2007).
Lepesheva, G. I. & Waterman, M. R. Structural basis for conservation in the CYP51 family. Biochim. Biophys. Acta 1814, 88–93 (2011).
Yoshida, Y. in Cytochrome P450 (eds Schenkman, J. B. & Greim, H.) 627–639 (Springer Berlin Heidelberg, 1993).
Blanksby, S. J. & Ellison, G. B. Bond dissociation energies of organic molecules. Acc. Chem. Res. 36, 255–263 (2003).
Romero, N. A. & Nicewicz, D. A. Organic photoredox catalysis. Chem. Rev. 116, 10075–10166 (2016).
Shaw, M. H., Twilton, J. & MacMillan, D. W. C. Photoredox catalysis in organic chemistry. J. Org. Chem. 81, 6898–6926 (2016).
Xie, J., Jin, H. & Hashmi, A. S. K. The recent achievements of redox-neutral radical C–C cross-coupling enabled by visible-light. Chem. Soc. Rev. 46, 5193–5203 (2017).
Chen, Y., Lu, L.-Q., Yu, D.-G., Zhu, C.-J. & Xiao, W.-J. Visible light-driven organic photochemical synthesis in China. Sci. China Chem. 62, 24–57 (2019).
Kancherla, R., Muralirajan, K., Sagadevan, A. & Rueping, M. Visible Light-induced excited-state transition-metal catalysis. Trends Chem. 1, 510–523 (2019).
Jia, K., Zhang, F., Huang, H. & Chen, Y. Visible-light-induced alkoxyl radical generation enables selective C(sp3)–C(sp3) bond cleavage and functionalizations. J. Am. Chem. Soc. 138, 1514–1517 (2016).
Hu, A. et al. Cerium-catalyzed formal cycloaddition of cycloalkanols with alkenes through dual photoexcitation. J. Am. Chem. Soc. 140, 13580–13585 (2018).
Hu, A., Guo, J.-J., Pan, H. & Zuo, Z. Selective functionalization of methane, ethane, and higher alkanes by cerium photocatalysis. Science 361, 668–672 (2018).
Schwarz, J. & König, B. Visible-light mediated C–C bond cleavage of 1,2-diols to carbonyls by cerium-photocatalysis. Chem. Commun. 55, 486–488 (2019).
Zhang, K., Chang, L., An, Q., Wang, X. & Zuo, Z. Dehydroxymethylation of alcohols enabled by cerium photocatalysis. J. Am. Chem. Soc. 141, 10556–10564 (2019).
Huang, L., Ji, T. & Rueping, M. Remote nickel-catalyzed cross-coupling arylation via proton-coupled electron transfer-enabled C–C bond cleavage. J. Am. Chem. Soc. 142, 3532–3539 (2020).
Guo, J.-J. et al. Photocatalytic C−C bond cleavage and amination of cycloalkanols by cerium(III) chloride complex. Angew. Chem. Int. Ed. 55, 15319–15322 (2016).
Hu, A. et al. δ-selective functionalization of alkanols enabled by visible-light-induced ligand-to-metal charge transfer. J. Am. Chem. Soc. 140, 1612–1616 (2018).
Chen, Y., Du, J. & Zuo, Z. Selective C-C bond scission of ketones via visible-light-mediated cerium. Catal. Chem. 6, 266–279 (2020).
Ji, T., Chen, X.-Y., Huang, L. & Rueping, M. Remote trifluoromethylthiolation enabled by organophotocatalytic C–C bond cleavage. Org. Lett. 22, 2579–2583 (2020).
Wang, D., Mao, J. & Zhu, C. Visible light-promoted ring-opening functionalization of unstrained cycloalkanols via inert C–C bond scission. Chem. Sci. 9, 5805–5809 (2018).
Zhao, R. et al. Visible-light-enhanced ring opening of cycloalkanols enabled by brønsted base-tethered acyloxy radical induced hydrogen atom transfer-electron transfer. Org. Lett. 20, 1228–1231 (2018).
Wu, X., Cruz, F. A., Lu, A. & Dong, V. M. Tandem catalysis: transforming alcohols to alkenes by oxidative dehydroxymethylation. J. Am. Chem. Soc. 140, 10126–10130 (2018).
Conia, J. M. & Le Perchec, P. The thermal cyclisation of unsaturated carbonyl compounds. Synthesis 1975, 1–19 (1975).
Houk, K. N. The photochemistry and spectroscopy of β,γ-unsaturated carbonyl compounds. Chem. Rev. 76, 1–74 (1976).
Shiraki, T. et al. α,β-unsaturated ketone is a core moiety of natural ligands for covalent binding to peroxisome proliferator-activated receptor γ. J. Biol. Chem. 280, 14145–14153 (2005).
LoPachin, R. M., Barber, D. S. & Gavin, T. Molecular mechanisms of the conjugated α,β-unsaturated carbonyl derivatives: relevance to neurotoxicity and neurodegenerative diseases. Toxicol. Sci. 104, 235–249 (2007).
Muzart, J. One-pot syntheses of α,β-unsaturated carbonyl compounds through palladium-mediated dehydrogenation of ketones, aldehydes, esters, lactones and amides. Eur. J. Org. Chem. 2010, 3779–3790 (2010).
Zhang, S., Neumann, H. & Beller, M. Synthesis of α,β-unsaturated carbonyl compounds by carbonylation reactions. Chem. Soc. Rev. 49, 3187–3210 (2020).
Buist, P. H. Fatty acid desaturases: selecting the dehydrogenation channel. Nat. Prod. Rep. 21, 249–262 (2004).
Voica, A.-F., Mendoza, A., Gutekunst, W. R., Fraga, J. O. & Baran, P. S. Guided desaturation of unactivated aliphatics. Nat. Chem. 4, 629–635 (2012).
Parasram, M., Chuentragool, P., Wang, Y., Shi, Y. & Gevorgyan, V. General, auxiliary-enabled photoinduced Pd-catalyzed remote desaturation of aliphatic alcohols. J. Am. Chem. Soc. 139, 14857–14860 (2017).
Chuentragool, P. et al. Aliphatic radical relay heck reaction at unactivated C(sp3)−H sites of alcohols. Angew. Chem. Int. Ed. 58, 1794–1798 (2019).
Yayla, H. G., Wang, H., Tarantino, K. T., Orbe, H. S. & Knowles, R. R. Catalytic ring-opening of cyclic alcohols enabled by PCET activation of strong O–H bonds. J. Am. Chem. Soc. 138, 10794–10797 (2016).
Ota, E., Wang, H., Frye, N. L. & Knowles, R. R. A redox strategy for light-driven, out-of-equilibrium isomerizations and application to catalytic C–C bond cleavage reactions. J. Am. Chem. Soc. 141, 1457–1462 (2019).
Turlik, A., Chen, Y. & Newhouse, T. R. Dehydrogenation adjacent to carbonyls using palladium–allyl intermediates. Synlett 27, 331–336 (2016).
Kato, S. et al. Hybrid catalysis enabling room-temperature hydrogen gas release from n-heterocycles and tetrahydronaphthalenes. J. Am. Chem. Soc. 139, 2204–2207 (2017).
Chen, M. & Dong, G. Copper-catalyzed desaturation of lactones, lactams, and ketones under pH-neutral conditions. J. Am. Chem. Soc. 141, 14889–14897 (2019).
Fuse, H., Mitsunuma, H. & Kanai, M. Catalytic acceptorless dehydrogenation of aliphatic alcohols. J. Am. Chem. Soc. 142, 4493–4499 (2020).
Tsukamoto, T. & Dong, G. Catalytic dehydrogenative cyclization of o-teraryls under pH-neutral and oxidant-free conditions. Angew. Chem. Int. Ed. 59, 15249–15253 (2020).
U. Dighe, S., Juliá, F., Luridiana, A., Douglas, J. J. & Leonori, D. A photochemical dehydrogenative strategy for aniline synthesis. Nature 584, 75–81 (2020).
Weiss, M. E., Kreis, L. M., Lauber, A. & Carreira, E. M. Cobalt-catalyzed coupling of alkyl iodides with alkenes: deprotonation of hydridocobalt enables turnover. Angew. Chem. Int. Ed. 50, 11125–11128 (2011).
West, J. G., Huang, D. & Sorensen, E. J. Acceptorless dehydrogenation of small molecules through cooperative base metal catalysis. Nat. Commun. 6, 10093 (2015).
Thullen, S. M. & Rovis, T. A mild hydroaminoalkylation of conjugated dienes using a unified cobalt and photoredox catalytic system. J. Am. Chem. Soc. 139, 15504–15508 (2017).
Abrams, D. J., West, J. G. & Sorensen, E. J. Toward a mild dehydroformylation using base-metal catalysis. Chem. Sci. 8, 1954–1959 (2017).
Cartwright, K. C. & Tunge, J. A. Decarboxylative elimination of n-acyl amino acids via photoredox/cobalt dual catalysis. ACS Catal. 8, 11801–11806 (2018).
Hu, X., Zhang, G., Bu, F. & Lei, A. Selective oxidative [4+2] imine/alkene annulation with H2 liberation induced by photo-oxidation. Angew. Chem. Int. Ed. 57, 1286–1290 (2018).
Kalsi, D., Dutta, S., Barsu, N., Rueping, M. & Sundararaju, B. Room-temperature C–H bond functionalization by merging cobalt and photoredox catalysis. ACS Catal. 8, 8115–8120 (2018).
Sun, X., Chen, J. & Ritter, T. Catalytic dehydrogenative decarboxyolefination of carboxylic acids. Nat. Chem. 10, 1229–1233 (2018).
Liu, W.-Q. et al. Cobaloxime catalysis: selective synthesis of alkenylphosphine oxides under visible light. J. Am. Chem. Soc. 141, 13941–13947 (2019).
Meng, Q.-Y., Schirmer, T. E., Katou, K. & König, B. Controllable isomerization of alkenes by dual visible-light-cobalt catalysis. Angew. Chem. Int. Ed. 58, 5723–5728 (2019).
Cao, H. et al. Photoinduced site-selective alkenylation of alkanes and aldehydes with aryl alkenes. Nat. Commun. 11, 1956 (2020).
Constantin, T. et al. Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides. Science 367, 1021–1026 (2020).
Takizawa, K. et al. Cobalt-catalyzed allylic alkylation enabled by organophotoredox catalysis. Angew. Chem. Int. Ed. 58, 9199–9203 (2019).
Kojima, M. & Matsunaga, S. The merger of photoredox and cobalt catalysis. Trends Chem. 2, 410–426 (2020).
Dempsey, J. L., Brunschwig, B. S., Winkler, J. R. & Gray, H. B. Hydrogen evolution catalyzed by cobaloximes. Acc. Chem. Res. 42, 1995–2004 (2009).
Artero, V., Chavarot-Kerlidou, M. & Fontecave, M. Splitting water with cobalt. Angew. Chem. Int. Ed. 50, 7238–7266 (2011).
Bock, C. R. et al. Estimation of excited-state redox potentials by electron-transfer quenching. Application of electron-transfer theory to excited-state redox processes. J. Am. Chem. Soc. 101, 4815–4824 (1979).
Lowry, M. S. et al. Single-layer electroluminescent devices and photoinduced hydrogen production from an ionic iridium(III) complex. Chem. Mater. 17, 5712–5719 (2005).
Izawa, Y., Pun, D. & Stahl, S. S. Palladium-catalyzed aerobic dehydrogenation of substituted cyclohexanones to phenols. Science 333, 209–213 (2011).
Ito, Y., Hirao, T. & Saegusa, T. Synthesis of.alpha.,.beta.-unsaturated carbonyl compounds by palladium(II)-catalyzed dehydrosilylation of silyl enol ethers. J. Org. Chem. 43, 1011–1013 (1978).
Yu, J.-Q., Wu, H.-C. & Corey, E. J. Pd(OH)2/C-mediated selective oxidation of silyl enol ethers by tert-butylhydroperoxide, a useful method for the conversion of ketones to α,β-enones or β-silyloxy-α,β-enones. Org. Lett. 7, 1415–1417 (2005).
Du, P., Knowles, K. & Eisenberg, R. A homogeneous system for the photogeneration of hydrogen from water based on a platinum(II) terpyridyl acetylide chromophore and a molecular cobalt catalyst. J. Am. Chem. Soc. 130, 12576–12577 (2008).
Acknowledgements
This work was financially supported by the King Abdullah University of Science and Technology (KAUST), Saudi Arabia, Office of Sponsored Research (URF/1/4025).
Author information
Authors and Affiliations
Contributions
L.H. and M.R. conceived and designed the project. L.H. performed and analyzed the experiments. T.J. contributed to the scope and application. C.Z., H.Y., and N.Z. conducted and analyzed the H2 quantification experiments. L.H. and M.R. co-wrote the manuscript. M.R. directed the whole research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, L., Ji, T., Zhu, C. et al. Bioinspired desaturation of alcohols enabled by photoredox proton-coupled electron transfer and cobalt dual catalysis. Nat Commun 13, 809 (2022). https://doi.org/10.1038/s41467-022-28441-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28441-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
