
Abstract
To form functional kinetochores, CENP-C and CENP-T independently recruit the KMN (Knl1C, Mis12C, and Ndc80C) network onto the kinetochores. To clarify the functions of the KMN network on CENP-T, we evaluated its roles in chicken DT40 cell lines lacking the CENP-C-KMN network interaction. By analyzing mutants lacking both CENP-T-Mis12C and CENP-C-Mis12C interactions, we demonstrated that Knl1C and Mis12C (KM) play critical roles in the cohesion of sister chromatids or the recruitment of spindle checkpoint proteins onto kinetochores. Two copies of Ndc80C (N-N) exist on CENP-T via Mis12C or direct binding. Analyses of cells specifically lacking the Mis12C-Ndc80C interaction revealed that N-N is needed for proper kinetochore-microtubule interactions. However, using artificial engineering to directly bind the two copies of Ndc80C to CENP-T, we demonstrated that N-N functions without direct Mis12C binding to Ndc80C in native kinetochores. This study demonstrated the mechanisms by which complicated networks play roles in native kinetochores.
Similar content being viewed by others


Introduction
Chromosomes replicated during the S-phase must be divided into daughter cells during mitosis to transmit the genetic information to the progeny. In eukaryotes, this process is called chromosome segregation, which is achieved by the attachment of sister chromatids to the bipolar mitotic spindles. The spindle microtubules bind to a large protein complex called kinetochore, which is formed on the centromere of each sister chromatid, to ensure the accurate segregation of the chromosomes1,2,3,4.
Kinetochores are composed of two major complexes. One complex, which consists of 16 protein components, forms a base on the centromeric chromatin for the formation of kinetochores and is called the constitutive centromere-associated network (CCAN), which constitutively localizes to the centromere throughout the cell cycle2,3,5,6. From the late G2 phase to mitosis, another large complex starts to associate with CCAN to form a fully functional kinetochore. This complex contains the Knl1 complex (Knl1C), Mis12 complex (Mis12C), and Ndc80 complex (Ndc80C), which form the KMN network2,6. In the KMN network, Ndc80C directly associates with spindle microtubules7,8,9, Knl1C appears to make an adapter for various proteins, including spindle checkpoint proteins10,11,12, and Mis12C recruits Ndc80C and Knl1C2,6. The linkage between CCAN and KMN bridges the centromeric chromatin and spindle microtubules to facilitate accurate chromosome segregation.
CCAN is divided into several subgroups13,14,15,16, including centromere protein (CENP)-C17,18,19,20,21,22, CENP-T-W-S-X5,23,24, CENP-L-N25,26,27,28,29, CENP-H-I-K-M14,30,31, and CENP-O-P-Q-R-U16,32 complexes. Among the CCAN proteins, CENP-C and the CENP-T-W-S-X complex bind to both centromeric chromatin and the KMN network1,33,34,35. Based on these studies and the results of artificial tethering of CENP-C or CENP-T into a noncentromeric region, two parallel pathways for the recruitment of the KMN network onto the kinetochores in vertebrate cells have been proposed: the CENP-C and CENP-T pathways1,3,4,34,35,36,37.
The kinetochore is a multi-protein complex with complicated protein-protein networks, which often exhibit redundant functions. Therefore, it is essential to dissect the functional roles of each complex in the native kinetochores. As some proteins contribute to multiple functions via interaction with different partners, the simple knockout approach for a target protein may cause misinterpretation of its protein function owing to the indirect effects of the knockout. Therefore, to evaluate the precise role of each complex or protein in native kinetochores, we must characterize a narrow functional domain in each protein and remove the redundancies to assess the function of an individual protein (complex).
We previously demonstrated that CENP-C-KMN network binding is dispensable for cell viability and kinetochore assembly in chicken DT40 cells35. In contrast, the CENP-T-KMN network interaction is essential for cell viability, suggesting that the CENP-T pathway is a major pathway in chicken cells35,36,37,38. However, the mechanism by which the KMN network coordinates its functions on the CENP-T pathway remains unclear. Particularly, while Mis12C binds to Knl1C and Ndc80C, it is unknown whether these interactions in the KMN network are essential for each function. Furthermore, in addition to Knl1C-Mis12C-Ndc80C (KMN) formation, an additional copy of Ndc80C directly binds to CENP-T36,37,39,40, and one KMN unit and one Ndc80C forms KMN-N in the network on CENP-T6,41. The KMN network appears to play multiple roles, including in the binding of microtubules, spindle checkpoint functions, and cohesion of sister chromatids. However, it is unclear whether each function is coordinated by the entire network or if the network can be separated into subcomplexes to perform individual functions. Moreover, even if the entire network is separable, the specific combinations of components responsible for each function remain ambiguous. Therefore, it is critical to clarify these questions to understand kinetochore functions.
In this work, to address these questions, we examined the different roles of the KMN network on the CENP-T pathway in cells lacking the CENP-C-KMN network interaction, which exhibits some redundant functions to CENP-T. We constructed cell lines lacking the CENP-T-Mis12C interaction in the absence of the CENP-C-KMN network interaction and demonstrated defects in sister chromatid cohesion, reduction in the levels of Ndc80C, or loss of Knl1C and Bub1 in these cells. These results suggest that Mis12C-Knl1C (KM) interaction on the CENP-T pathway is critical for the recruitment of checkpoint proteins and Ndc80C or the cohesion of sister chromatids. While KM on CENP-T recruits Ndc80C, CENP-T directly binds to additional Ndc80C. We then constructed cell lines that specifically lacked the Ndc80C-binding site in Mis12C and demonstrated that one copy of Ndc80C on CENP-T was insufficient, suggesting that two copies of Ndc80C (N-N) are necessary for the functional kinetochore-microtubule interaction in DT40 cells. This suggests that the Mis12C-Ndc80C interaction is necessary for the microtubule-binding function of Ndc80C. However, using artificial engineering to directly bind the two copies of Ndc80C to CENP-T, we demonstrated that Ndc80C binding to Mis12C is not essential for microtubule binding. Our data indicate that the KMN network usually forms KMN-N (one copy of Ndc80C binds to KM as a KMN unit and the other directly binds CENP-T) on CENP-T for its functions, but KM-N-N (two copies of Ndc80C directly bind to CENP-T and KM independently binds to CENP-T) is functional in the network. We also demonstrated that Ndc80C binding to Mis12C is not essential for microtubule binding in human cells. Since the Ndc80C-Mis12C interaction is thought to be critical for the kinetochore functions7,42,43, our observations provide new insights for kinetochore studies and explain how the KMN network plays an essential role in kinetochore functions via the CENP-T pathway.
Results
The CENP-T-Mis12C interaction is essential on the CENP-T pathway
Mis12C binds to both CENP-C and CENP-T, and we have previously shown that the CENP-T-Mis12C interaction is much more critical than the CENP-C-Mis12C interaction in chicken DT40 cells35. However, deletion of the Mis12C-binding region (amino acids: aa 121-240 region) in chicken CENP-T did not lead to complete lethality in DT40 cells, although it caused a growth delay. This might be because Mis12C on CENP-C has redundant roles in kinetochore functions. We initially generated a cell line in which Mis12C-binding regions in both CENP-C and CENP-T were deleted (Fig. 1a-d). Since the Mis12C-binding site (N-terminal 73 aa region) in chicken CENP-C is dispensable in DT40 cells, we introduced mScarlet-fused CENP-C∆73 into one endogenous β-actin allele with disrupting the endogenous CENP-C gene using CRISPR/Cas9-mediated genome editing (Supplementary Fig. 1a-e). We chose the β-actin locus to obtain stable expression of mScarlet-fused CENP-C∆73, since the β-actin is ubiquitously expressed. Using this cell line, we introduced a cDNA for 3X FLAG-tagged CENP-T lacking the Mis12C-binding region (CENP-TΔ121-240) into one CENP-T allele (Supplementary Fig. 1f, g), and another allele was replaced with a drug resistance gene, and a wild-type (WT) CENP-T transgene was expressed under the control of a tetracycline (Tet)-responsive promoter (Tet-CENP-T, Fig. 1d). We called this line conditional knockout (cKO)-CT (Tet-Off) -CCΔ73/CENP-TΔ121-240, and CENP-T and CENP-C were completely replaced with 3X FLAG-CENP-TΔ121-240 and mScarlet-CENP-CΔ73, respectively, after Tet addition in this cell line (Fig. 1d). Using different combinations of CENP-T and CENP-C, we also generated various cell lines, such as cKO-CT-CCΔ73/CENP-TΔ90 or cKO-CT-CCWT/CENP-TΔ121-240 cells (Supplementary Fig. 1h). All cell lines generated in this study are summarized in Supplementary Table 1. We note that the CENP-T N-terminal region (aa 1-90) contains an Ndc80C-binding region (Fig. 1b), and CENP-TΔ90 does not directly bind to Ndc80C35,39.

a Recruiting pathway of KMN network via CENP-C or CENP-T. b Schematic representation of chicken CENP-T. Ndc80C- and Mis12C-binding domains were deleted in CENP-T∆90 and CENP-T∆121-240, respectively. c Schematic representation of chicken CENP-C. The Mis12C-binding domain was deleted in CENP-C∆73. d Immunoblot analyses of various CENP-C or CENP-T in cKO-CENP-T (Tet-Off) cells. Expression of the CENP-T transgene (Tet-CENP-T) was terminated by 24 h after Tet addition. mScarlet-CENP-C∆73 was expressed in CENP-C knockout cells. The 3X FLAG-fused CENP-T mutants were stably expressed. CENP-C migration was slightly slow, due to CENP-C phosphorylation in mitosis. Alpha-tubulin (tubulin) was used as the loading control. The asterisks indicate nonspecific bands. None depicts parental cKO-CENP-T cells. e The growth of cKO-CENP-T cells expressing 3X FLAG-CENP-T∆121-240 in the absence or presence of Tet (–Tet or +Tet) with CENP-C WT (left) or mScarlet-CENP-C∆73 (right). f Schematic representation of each CENP-T mutant. g GFP-Dsn1 levels at kinetochores in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and various CENP-T mutants in the presence of Tet for 30 h. None indicates parental cKO-CENP-T/CENP-C∆73/GFP-Dsn1 cells. Subtract values are presented using signal intensities of None. DNA was stained with DAPI. Error bars show the mean and standard deviation. p values were calculated by one-way ANOVA followed by Tukey’s test. h The growth of cKO-CENP-T cells expressing CENP-CΔ73 and either CENP-TΔ121-160, CENP-TΔ161-200, CENP-TΔ201-216, CENP-TΔ217-230, or CENP-TΔ231-240 in the absence or presence of Tet. Some time points (N.D.) were missing because of no viable cells. i Cell cycle distribution of cKO-CENP-T/CENP-C∆73 (None), cKO-CENP-T/CENP-C∆73 expressing CENP-TWT or CENP-T∆121-240 in the presence of Tet for 30 h. j Chromosome alignment values in cKO-CENP-T/CENP-C∆73 (None), cKO-CENP-T/CENP-C∆73 expressing CENP-TWT, or CENP-T∆121-240. The cells were cultured in the presence of Tet for 30 h and MG132 for last 3 h. Error bars indicate the mean and standard deviation. p values were calculated as in g. k Numbers of spindle pole stained by anti-γ-tubulin in cKO-CENP-T/CENP-C∆73 (None), cKO-CENP-T/CENP-C∆73 expressing CENP-TWT, or CENP-T∆121-240 in the presence of Tet for 30 h.
After confirming the expression of each transgene using immunoblot analysis (Fig. 1d), we analyzed the growth of each cell line in the absence or presence of Tet. In the absence of Tet (expression of Tet-CENP-T), all cell lines grew well. After Tet addition, cKO-CT-CCWT/CENP-TΔ90 cells died by ~96 h (Supplementary Fig. 1i). However, cKO-CT-CCWT/CENP-TΔ121-240 cells were still viable in the presence of Tet (Fig. 1e), although they showed a growth delay, which is consistent with the results of our previous study35. We further tested the growth of cKO-CT-CCΔ73/CENP-TΔ121-240 cells in the presence of Tet and found that these cells completely died by ~120 h after Tet addition (Fig. 1e). These results indicate that Mis12C on CENP-C has redundant roles in the kinetochore functions, although it is not sufficient to completely compensate for the lack of CENP-T-Mis12C interaction. Therefore, to precisely evaluate the Mis12C function on the CENP-T pathway in native kinetochores, we must analyze it in the absence of CENP-C-KMN network interaction (expression of CENP-CΔ73 instead of CENP-CWT).
Although we defined the aa 121-240 region as the Mis12C-binding region of chicken CENP-T, we attempted to define a more precise region of CENP-T required for Mis12C binding, using cKO-CT-CCΔ73 cells (Fig. 1f). In addition to centromeric signals, some mScarlet-fused CENP-CΔ73 localized to the non-centromeric chromosome region in these cells (Fig. 1g). This may be due to overexpression of mScarlet-fused CENP-CΔ73 under the control of β-actin promoter. Since we confirmed that cells expressing mScarlet-fused CENP-CΔ73 are viable, mScarlet-fused CENP-CΔ73 should be functional. We also introduced GFP-Dsn1 (a component of Mis12C) into endogenous Dsn1 alleles in these cells (Supplementary Fig. 1j-l). We note that cKO-CT-CCΔ73 cells (None in Fig. 1g) died before the complete depletion of CENP-T. This explains why GFP-Dsn1 is still visible in cKO-CT-CCΔ73 cells, and we also presented normalization data for GFP-Dsn1 based on data from cKO-CT-CCΔ73 cells. As shown in Fig. 1g, GFP-Dsn1 levels were significantly reduced in either cKO-CT-CCΔ73/CENP-TΔ161-200 or cKO-CT-CCΔ73/CENP-TΔ201-216 cells, suggesting that either aa 161–200 or 201–216 regions is essential for Mis12C recruitment to the CENP-T pathway. Consistent with the Dsn1 levels in these cell lines, both cKO-CT-CCΔ73/CENP-TΔ161-200 and cKO-CT-CCΔ73/CENP-TΔ201-216 cells died by ~72 h after Tet addition (Fig. 1h). These results suggest that Mis12C binds to the region of aa 161-216 of chicken CENP-T.
cKO-CT-CCΔ73/CENP-TΔ121-240 cells died in the presence of Tet (Fig. 1e), which might be due to mitotic defects. To test this prediction, we examined the cell cycle distribution (Fig. 1i), rate of chromosome alignment (Fig. 1j, Supplementary Fig. 1m-o), and the number of spindle poles (Fig. 1k) in these cells in the presence of Tet. Strong accumulation of G2/M fractions was observed (Fig. 1i), and chromosomes were not properly aligned (Fig. 1j, Supplementary Fig. 1o) in cKO-CT-CCΔ73/CENP-TΔ121-240 cells. Abnormal numbers of spindle poles were observed (Fig. 1k). These phenotypes were similar to those observed in cKO-CT-CCΔ73 cells. We concluded that cKO-CT-CCΔ73/CENP-TΔ121-240 cells died due to mitotic defects.
Ndc80C binding to CENP-T facilitates the Mis12C recruitment to CENP-T
To examine the functional roles of the KMN network on the CENP-T pathway, it is also important to know how each component of the network is recruited to CENP-T. Previous studies have suggested that CDK1-mediated phosphorylation of CENP-T facilitates recruitment of Ndc80C or Mis12C onto the kinetochore37,39,41. We further evaluated the effect of CENP-T phosphorylation on the recruitment of Ndc80C or Mis12C onto CENP-T in cKO-CT-CCΔ73 cells. We mutated potential CDK1 sites in CENP-T (CENP-TT72A-S88A) responsible for Ndc80C binding39 and introduced a mutant into cKO-CT-CCΔ73 cells (Fig. 2a, Supplementary Fig. 1f). To evaluate the levels of Ndc80C at kinetochores in these cell lines, we introduced Nuf2-GFP (a component of Ndc80C) into endogenous Nuf2 alleles (Supplementary Fig. 2a-c). We measured Nuf2-GFP levels at kinetochores. The data indicated that Nuf2 levels were significantly reduced in cKO-CT-CCΔ73/CENP-TT72A-S88A cells, compared to that in cKO-CT-CCΔ73/CENP-TWT cells (Fig. 2b). These results support those of our previous study39.

a Amino acids alignment of CENP-T between various species. Potential phosphorylation sites (T72 and S88) are marked. b Nuf2-GFP levels at kinetochores in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and either CENP-TWT, CENP-TΔ90, or CENP-TT72A-S88A in the presence of Tet for 30 h. None indicates parental cKO-CENP-T/CENP-C∆73/Nuf2-GFP cells. Subtract values are presented using signal intensities of None. DNA was stained with DAPI. Error bars indicate the mean and standard deviation. p values were calculated by one-way ANOVA followed by Tukey’s test. c Amino acids alignment of CENP-T between various species. Mutated S/T residues are marked. d GFP-Dsn1 levels at kinetochores in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and CENP-T8A in the presence of Tet for 48 h. DNA was stained with DAPI. Error bars indicate the mean and standard deviation. p values were calculated by two-tailed Welch’s t-test. e The growth of cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and either CENP-T8A or CENP-TWT in the absence or presence of Tet. f GFP-Dsn1 levels at kinetochores in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and either CENP-TWT, CENP-TΔ121-240, or CENP-TΔ90 in the presence of Tet for 30 h. None indicates parental cKO-CENP-T/CENP-C∆73/GFP-Dsn1 cells. Subtract values with None are presented. DNA was stained with DAPI. Error bars indicate the mean and standard deviation. p values were calculated as in b. g GFP-Dsn1 levels at kinetochores in cKO-Spc25 cells expressing mScarlet-CENP-C∆73 in the absence or presence of Tet for 18 h. DNA was stained with DAPI. Error bars indicate the mean and standard deviation. p values were calculated as in d. h GFP-Dsn1 levels at kinetochores in CENP-C KO cells expressing HA-CENP-C∆73 (CENP-C∆73 cells) in the presence of nocodazole or MG132. CENP-T was immuno-stained. DNA was stained with DAPI. Error bars indicate the mean and standard deviation. p values were calculated as in d. i Nuf2-GFP levels at kinetochores in CENP-C∆73 cells in the presence of nocodazole or MG132. CENP-T was immuno-stained. DNA was stained with DAPI. Error bars indicate the mean and standard deviation. p values were calculated as in d.
We also examined the effects of CENP-T phosphorylation on Mis12C recruitment. We further observed eight serine/threonine residues in the aa 161-216 region of chicken CENP-T, and a mutant CENP-T cDNA, in which all serine/threonine residues were replaced with alanine (CENP-T8A), was introduced into cKO-CT-CCΔ73 cells (Fig. 2c, Supplementary Fig. 2d). GFP-Dsn1 was also introduced into the endogenous Dsn1 alleles in these cells to measure Mis12C levels at the kinetochores (Supplementary Fig. 2d). As shown in Fig. 2d, Mis12C levels in cKO-CT-CCΔ73/CENP-T8A cells were not significantly different from those in cKO-CT-CCΔ73/CENP-TWT cells. Consistent with the data of Dsn1 levels, the growth of cKO-CT-CCΔ73/CENP-T8A cells in the presence of Tet was comparable to that of these cells in the absence of Tet (Fig. 2e). Considering these results, we conclude that phosphorylation of the aa 161–216 region of CENP-T is not essential for the recruitment of Mis12C onto the CENP-T pathway.
Previous CENP-T tethering experiments into a non-centromere locus suggested that the extreme N-terminal region of CENP-T required for Ndc80C binding might be involved in the CENP-T-Mis12C interaction34,37. However, it is unclear whether the same regulation occurs in native kinetochores. To examine whether the CENP-T N-terminal region facilitates Mis12C recruitment to CENP-T in native kinetochores, we examined Mis12C (Dsn1) levels in cKO-CT-CCΔ73/CENP-TΔ90 cells (Fig. 2f, Supplementary Fig. 2e) and observed that Dsn1 levels in these cells were significantly lower than those in cKO-CT-CCΔ73/CENP-TWT cells (Fig. 2f).
Since the CENP-T N-terminal region is critical for Ndc80C binding, we attempted to demonstrate whether direct binding of Ndc80C to the CENP-T N-terminal region facilitates Mis12C recruitment. We generated cKO-Spc25 (a direct CENP-T-binding protein in Ndc80C) (Tet-Off) cells in the absence of the CENP-C-KMN interaction (cKO-Spc25-CCΔ73 cells) (Supplementary Fig. 2f-h) in which GFP-Dsn1 was introduced into endogenous Dsn1 alleles. As shown in Fig. 2g, Dsn1 levels in kinetochores were significantly reduced in cKO-Spc25-CCΔ73 cells after Tet addition, compared with those in these cells before Tet addition. Since these cells contained intact CENP-T, including the Mis12C-binding region, data indicate that direct binding of Ndc80C to CENP-T facilitates Mis12C recruitment onto CENP-T. As Mis12C on CENP-T binds to Knl1C and additional Ndc80C to form a Knl1C-Mis12C-Ndc80C unit (KMN unit), we concluded that direct binding of Ndc80C to CENP-T facilitates the formation of KMN-N (one copy of Ndc80C binds to KM as a KMN unit, and another copy of Ndc80C directly binds CENP-T) on the CENP-T pathway in chicken native kinetochores.
Since Ndc80C-CENP-T direct binding facilitates the Mis12C-CENP-T interaction to form KMN-N, it is possible that the tension caused by kinetochore-microtubule interactions might facilitate Mis12C recruitment onto CENP-T. Using nocodazole (for the absence of tension) or MG132 (for the presence of tension), we prepared chromosome spreads after confirming that chromosomes were aligned in the presence of MG132 (Fig. 2h, i, Supplementary Fig. 2i-m) in CENP-C-deficient cells expressing CENP-CΔ73 (CENP-CΔ73 cells). Further, we compared the levels of Ndc80C and Mis12C at kinetochores in the presence or absence of tension. Both Ndc80C and Mis12C levels did not change in CENP-CΔ73 cells in either the presence or absence of tension (Fig. 2h, i, Supplementary Fig. 2i), suggesting that the tension from microtubules might not have facilitated Mis12C recruitment onto CENP-T. It would be an important further challenge to clarify the molecular mechanisms of how direct binding of Ndc80C to CENP-T facilitates Mis12C recruitment onto CENP-T in cells.
Knl1C on the CENP-T pathway is essential for sister chromatid cohesion
Since cKO-CT-CCΔ73/CENP-TΔ121-240 cells led to severe mitotic defects (Fig. 1), we examined the levels of Mis12C-interacting proteins in these cells. We measured Knl1C levels in cells lacking CENP-T-Mis12C and CENP-C-Mis12C interactions (Fig. 3a, Supplementary Fig. 3a). As shown in Fig. 3a, Knl1C levels were significantly reduced in cKO-CT-CCΔ73/CENP-TΔ121-240 cells, and the reduction levels in these cells were similar to those in cKO-CT-CCΔ73 cells.

a Knl1 levels at kinetochores in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and either CENP-TWT or CENP-TΔ121-240 in the presence of Tet for 30 h. None indicates parental cKO-CENP-T/CENP-C∆73 cells. Subtract values are presented using signal intensities of None. DNA was stained with DAPI. Error bars indicate the mean and standard deviation. p values were calculated by one-way ANOVA followed by Tukey’s test. b Bub1 levels at kinetochores in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and either CENP-TWT or CENP-TΔ121-240 in the presence of Tet for 30 h. None indicates parental cKO-CENP-T/CENP-C∆73 cells. Subtract values with None are presented. DNA was stained with DAPI. Error bars indicate the mean and standard deviation. p values were calculated as in a. c Immuno-localization of H3T3ph on chromosomes in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and either CENP-TWT or CENP-TΔ121-240 in the presence of Tet for 30 h and nocodazole for last 4 h. The insets show magnified views of the boxed regions. Graphs summarize H3T3ph localization profiles on centromeres or non-centromeres. Error bars show the mean and standard deviation. p values were calculated by two-tailed Welch’s t-test. d Immuno-localization of Aurora B on chromosomes in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and either CENP-TWT or CENP-TΔ121-240 in the presence of Tet for 30 h and nocodazole for last 4 h. The insets present magnified views. Graphs summarize Aurora B localization profiles on centromeres or non-centromeres. Error bars show the mean and standard deviation. p values were calculated as in c. e Kinetochore-kinetochore distances of mitotic chromosomes in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and either CENP-TWT or CENP-T∆121-240 in the presence of Tet for 30 h and nocodazole for last 4 h. Error bars indicate the mean and standard deviation. p values were calculated as in c. f Mis12C and Knl1C (KM) on the CENP-T pathway function for the maintenance of sister chromatid cohesion (left). When KM is absent from CENP-T (right), H3T3ph and Aurora B are diffused into non-centromere regions, causing defects in sister chromatid cohesion.
Since Knl1C recruits Bub1 to kinetochores10, we investigated Bub1 localization in cKO-CT-CCΔ73/CENP-TΔ121-240 cells, using an anti-Bub1 antibody (Fig. 3b, Supplementary Fig. 3a). Bub1 signals were almost invisible in cKO-CT-CCΔ73/CENP-TΔ121-240 cells, and these levels were similar to those in cKO-CT-CCΔ73 cells (Fig. 3b). Knl1C-Bub1 is a critical target of the checkpoint protein Mps111,12, and the checkpoint protein BubR1 also binds to Knl1C10. Therefore, localization data of Knl1C and Bub1 suggest that checkpoint functions were compromised in cKO-CT-CCΔ73/CENP-TΔ121-240 cells.
Additionally, Bub1 recruitment leads to the proper localization of the chromosome passenger complex (CPC) and Shugoshin. These complexes together with phosphorylated threonine 3 of histone H3 (H3T3ph) contribute to the maintenance of sister chromatid cohesion in centromeres10,34,44. We then examined the distribution of H3T3ph, which is governed by Haspin kinase, in cKO-CT-CCΔ73/CENP-TΔ121-240 or cKO-CT-CCΔ73/CENP-TWT cells. In cKO-CT-CCΔ73/CENP-TWT cells, H3T3ph signals were concentrated in the centromeric region, but these signals were diffused along the entire chromosome arm in cKO-CT-CCΔ73/CENP-TΔ121-240 cells (Fig. 3c). Consistent with this localization profile of H3T3ph in cKO-CT-CCΔ73/CENP-TΔ121-240 cells, Aurora B (a component of CPC) signals were also distributed in the non-centromeric region in these cells (Fig. 3d). We interpret that the reduction of Knl1C and Bub1 leads to a reduction in the phosphorylation levels of threonine 120 of histone H2A in centromeres governed by Bub144,45, which causes the mislocalization of H3T3ph and Aurora B46,47. Centromere localization of H3T3ph and Aurora B is related to each other, and the reduction of upstream components affects their centromeric localization (Fig. 3c, d). In addition to these localization profiles, we observed that the distance between sister kinetochores increased in cKO-CT-CCΔ73/CENP-TΔ121-240 cells, compared to that in cKO-CT-CCΔ73/CENP-TWT cells (Fig. 3e). These data suggest that Mis12C and Knl1C (KM) on the CENP-T pathway facilitate proper localization of CPC and H3T3ph to centromeric regions through Bub1 recruitment to maintain the cohesion of sister chromatids (Fig. 3f). KM is known to function on CENP-C20,48, and we emphasize that our data demonstrated KM functions on CENP-T in cells lacking CENP-C-KMN interaction.
Two copies of Ndc80C on CENP-T are essential for their proper functions
While KM (Knl1C and Mis12C) on the CENP-T pathway plays a critical role in the maintenance of sister chromatids cohesion and the recruitment of checkpoint proteins, the roles of Ndc80C must be clarified in the KMN network on CENP-T. CENP-T has two copies of Ndc80C (N-N): one copy directly binds to the CENP-T N-terminus and the second copy is recruited via Mis12C (Fig. 3f). Therefore, the network forms KMN-N. Therefore, it is important to address whether N-N is essential for its microtubule-binding function on the CENP-T pathway independent of KM functions or whether Ndc80C must bind to Mis12C for its function. To test whether N-N is necessary for chromosome segregation, we first examined Ndc80C levels in cells lacking CENP-T-Mis12C and CENP-C-Mis12C interactions. To evaluate Ndc80C levels, we introduced Nuf2-GFP into the endogenous Nuf2 alleles in cKO-CT-CCΔ73/CENP-TΔ121-240, cKO-CT-CCΔ73/CENP-TΔ161-200, or cKO-CT-CCΔ73/CENP-TΔ201-216 cells (Supplementary Fig. 2a, 3b). As shown in Fig. 4a, Ndc80C levels in these cells were significantly reduced, albeit these levels were higher than those in cKO-CT-CCΔ73 cells (None in Fig. 4a), suggesting that one copy of Ndc80C still binds to the N-terminal region of CENP-TΔ121-240 (Fig. 4a), which is consistent with our previous observation35.

a Nuf2-GFP levels at kinetochores in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and various CENP-T mutants in the presence of Tet for 30 h. None indicates parental cKO-CENP-T/CENP-C∆73/Nuf2-GFP cells. Subtract values were presented using signal intensities of None. DNA was stained with DAPI. Error bars show the mean and standard deviation. p values were calculated by one-way ANOVA followed by Tukey’s test. b Schematic representation of chicken Dsn1∆326-349. c Schematic representation explaining that chicken Dsn1∆326-349 does not bind to Ndc80C but binds to Knl1C via the formation of Mis12C. d Immunoprecipitation with anti-GFP in cKO-Dsn1 cells expressing either GFP-Dsn1WT or GFP-Dsn1∆326-349 in the presence of Tet. Samples were subjected to immunoblot analyses using anti-Dsn1, -Mis12, -Knl1, and -Ndc80 antibodies. e Nuf2-mScarlet levels at kinetochores in cKO-Dsn1 cells expressing either GFP-Dsn1WT or GFP-Dsn1∆326-349 in the presence of Tet for 30 h. DNA was stained with DAPI. Error bars indicate the mean and standard deviation. p values were calculated by two-tailed Welch’s t-test. f The growth of cKO-Dsn1 cells expressing either GFP-Dsn1WT or GFP-Dsn1∆326-349 in the absence or presence of Tet (–Tet or +Tet). None indicates parental cKO-Dsn1 cells. g Microtubule staining using an anti-tubulin antibody (red) for cKO-Dsn1 cells expressing either GFP-Dsn1WT or GFP-Dsn1∆326-349 in the presence of Tet for 30 h. The cells were incubated in ice-cold medium for 10 min prior to fixation. DNA was stained with DAPI. h mScarlet-fused vinculin head domain (VH-mScarlet) localization to kinetochore in cKO-Dsn1 cells expressing CENP-T-TR reporter and either GFP-Dsn1 WT or GFP-Dsn1∆326-349. Disordered region of CENP-T (aa 241-529) was exchanged with the chicken TR domain (aa 482-889) (CENP-T TR), was expressed from the CENP-T locus. Cells were treated with or without nocodazole (Noc + or –), and mScarlet signals were measured. DNA was stained with DAPI. Error bars indicate the mean and standard deviation. p values were calculated as in a.
Although we observed that one copy of Ndc80C per one CENP-T still exists in cells lacking Mis12C, we analyzed the impact of one copy of Ndc80C without disrupting other Mis12C functions. Previous studies have suggested that the Dsn1 C-terminal region in Mis12C is critical for Ndc80C binding (Fig. 4b, c)35,48. Although another study suggested that the PVIHL motif in human Nsl1 in Mis12C might be also involved in Ndc80C binding43, this region in Nsl1 was not essential in DT40 cells, based on analyses using cKO-Nsl1 (Auxin-Inducible-Degron: AID) cells (Supplementary Fig. 4a-e). Therefore, in this study, we focused on the Dsn1 C-terminal region. We generated cKO-Dsn1 (Tet-Off) cells, in which GFP-Dsn1WT (cKO-Dsn1/Dsn1WT) or GFP-Dsn1Δ326-349 lacking the Ndc80C-binding region (cKO-Dsn1/Dsn1Δ326-349) were expressed under the control of the endogenous Dsn1 promoter (Supplementary Fig. 5a). We confirmed that Dsn1Δ326-349 formed Mis12C and bound to Knl1C but not to Ndc80C based on immunoprecipitation experiments using an anti-GFP nanobody (Fig. 4d). Consistent with the biochemical analyses, Dsn1 and Knl1 levels at kinetochores in cKO-Dsn1/Dsn1Δ326-349 cells were more than 80%, compared with those in cKO-Dsn1/Dsn1WT cells (Supplementary Fig. 5a-c), and Bub1 levels were similar to those in control cells (Supplementary Fig. 5a, d). In addition, the localization profiles of H3T3ph and Aurora B were similar to those in control cells (Supplementary Fig. 5e, f); therefore, sister chromatid cohesion defects were not observed (Supplementary Fig. 5g). These results suggest that KM retains the checkpoint function and sister chromatid cohesion maintenance function, lacking KM and Ndc80C interaction in cKO-Dsn1/Dsn1Δ326-349 cells (KM-N).
In contrast, Ndc80C (Nuf2-mScarlet) levels in cKO-Dsn1/Dsn1Δ326-349 cells were significantly reduced, albeit Nuf2 signals were still visible (Fig. 4e, Supplementary Fig. 5h), suggesting that this mutation does not affect the CENP-T-Ndc80C direct interaction. We further investigated the growth rate of cKO-Dsn1/Dsn1Δ326-349 cells and observed that these cells died by ~ 96 h after Tet addition (Fig. 4f). Although cKO-Dsn1/Dsn1Δ326-349 cells died, kinetochores in these cells may still be bound to microtubules, since some of the Ndc80C are still bound to CENP-T. We then stained microtubules and observed chromosome alignments in cKO-Dsn1/Dsn1Δ326-349 cells. As shown in Fig. 4g, unaligned chromosomes and abnormal spindles were observed in these cells, and microtubules did not appear to bind to kinetochores. These data indicate that proper kinetochore-microtubule attachment does not occur in cKO-Dsn1/Dsn1Δ326-349 cells. Consistent with microtubule staining, tension from microtubules was not applied to CENP-T in these cells, based on analyses using the tension sensor probe (Fig. 4h, Supplementary Fig. 4i, j). We used the talin rod (TR) tension sensor system49,50, similar to that used in our previous study35. We expressed vinculin head domain (VH)-mScarlet and CENP-T-TR, and VH-mScarlet localized to kinetochores via binding to CENP-T-TR. In cells with wild-type Dsn1, VH-mScarlet signals decreased upon nocodazole treatment. However, in cells expressing Dsn1Δ326-349 VH-mScarlet signals did not decrease upon nocodazole treatment (Fig. 4h). Considering these results, one copy of Ndc80C directly binding to CENP-T is not sufficient for its microtubule-binding function, suggesting that two copies of Ndc80C (N-N) on CENP-T are essential for the establishment of appropriate kinetochore-microtubule interaction.
KM-Ndc80C interaction is not essential, and they are separable
Since some KM roles were ensured without interaction with Ndc80C (Fig. 5a), KM and Ndc80C might not be necessary to interact with each other and are separable (Fig. 5a). Alternatively, it is still possible that a second copy of Ndc80C might be necessary to interact with KM for its functions. To distinguish between these possibilities, we engineered the CENP-T N-terminus. If we introduced an additional Ndc80C-binding domain (aa 1-90) in the CENP-T N-terminal region, extra Ndc80C should be recruited without Mis12C binding (Fig. 5a, b). Initially, we introduced CENP-T2x(1-90) into the endogenous CENP-T allele in cKO-CT-CCΔ73 cells and confirmed that CENP-T2x(1-90) functions as wild-type CENP-T by examining the growth of these cells in the presence of Tet (Supplementary Fig. 6a, b). We also observed an increase in Ndc80C (Nuf2-GFP) levels in cKO-CT-CCΔ73/CENP-T2x(1-90) cells owing to an additional Ndc80C-binding site on CENP-T (Fig. 5c).

a The artificial conversion of KMN-N to KM-N-N. b Various CENP-T constructs providing an extra Ndc80C-binding site. c Nuf2-GFP levels at kinetochores in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and either CENP-TWT or CENP-T2X(1-90) in the presence of Tet for 30 h. DNA was stained with DAPI. Error bars show the mean and standard deviation. p values were calculated by two-tailed Welch’s t-test. d Tubulin staining in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and either CENP-TWT, CENP-T∆121-240, or CENP-T2X(1-90)_∆121-240 in the presence of Tet for 30 h. None indicates CENP-T KO cells. e Number distributions of spindle poles stained with anti-γ-tubulin in cKO-CENP-T/CENP-C∆73 (None), cKO-CENP-T/CENP-C∆73 expressing CENP-TWT, CENP-T∆121-240, or CENP-T2X(1-90)_∆121-240. f Chromosome alignment values in cKO-CENP-T/CENP-C∆73 (None), cKO-CENP-T/CENP-C∆73 expressing CENP-TWT, CENP-T∆121-240, or CENP-T2X(1-90)_∆121-240 in the presence of Tet for 30 h and MG132 for last 3 h. Error bars indicate the mean and standard deviation. p values were calculated by one-way ANOVA followed by Tukey’s test. g Cell cycle distribution of cKO-CENP-T/CENP-C∆73 (None), cKO-CENP-T/CENP-C∆73 expressing CENP-TWT, CENP-T∆121-240, or CENP-T2X(1-90)_∆121-240. h Kinetochore-kinetochore distances in cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and either CENP-TWT or CENP-T2X(1-90)_∆121-240 in the presence of Tet for 30 h. Error bars indicate the mean and standard deviation. p values were calculated as in c. i The growth of cKO-CENP-T cells expressing mScarlet-CENP-C∆73 and CENP-T2X(1-90)_∆121-240 in the absence or presence of Tet. j Tubulin staining in cKO-Dsn1 cells expressing Dsn1WT, Dsn1∆326-349, or both Dsn1∆326-349 and CENP-T2X(1-90) in the presence of Tet for 30 h. k Number distributions of spindle poles in cKO-Dsn1 cells expressing Dsn1WT, Dsn1∆326-349 or both Dsn1∆326-349 and CENP-T2X(1-90). l Chromosome alignment values in cKO-Dsn1 cells expressing Dsn1WT, Dsn1∆326-349 or both Dsn1∆326-349 and CENP-T2X(1-90) in the presence of Tet for 30 h and MG132 for last 3 h. Error bars indicate the mean and standard deviation. p values were calculated as in f. m Cell cycle distribution of cKO-Dsn1 cells expressing Dsn1WT, Dsn1∆326-349 or both Dsn1∆326-349 and CENP-T2X(1-90). n The growth of cKO-Dsn1 cells expressing both GFP-Dsn1∆326-349 and 3XFLAG-CENP-T2X(1-90) in the absence or presence of Tet. None indicates no extra CENP-T expression.
As CENP-TΔ121-240 lacks the binding of Mis12C, which binds to Knl1C and Ndc80C, it loses KM and one Ndc80C copy. Therefore, in addition to defects due to a lack of KM roles (Fig. 3), microtubule-binding defects owing to Ndc80C reduction could occur in cKO-CT-CCΔ73/CENP-TΔ121-240 cells (Fig. 4a). We further stained microtubules and observed chromosome alignments in cKO-CT-CCΔ73/CENP-TΔ121-240, cKO-CT-CCΔ73/CENP-TWT, or cKO-CT-CCΔ73 cells (Fig. 5d). We frequently observed unaligned chromosomes, and the spindle shape was not normal in cKO-CT-CCΔ73/CENP-TΔ121-240 cells, as observed in cKO-CT-CCΔ73 cells (Fig. 5d). We also observed abnormal numbers of spindle poles (Fig. 5e), a higher rate of misalignment of chromosomes (Fig. 5f), and an increase in G2/M populations (Fig. 5g) in cKO-CT-CCΔ73/CENP-TΔ121-240 cells. These data suggest that proper kinetochore-microtubule attachment and spindle integrity were disrupted in cKO-CT-CCΔ73/CENP-TΔ121-240 cells. However, the addition of the Ndc80C-binding site to CENP-TΔ121-240 (CENP-T2x(1–90)_Δ121–240) might rescue defects in proper kinetochore-microtubule attachments. We further generated cKO-CT-CCΔ73/CENP-T2x(1–90)_Δ121–240 cells (Supplementary Fig. 6c) and analyzed the phenotype of these cells. Abnormal spindle shape, improper kinetochore-microtubule attachments, number of spindle poles, and defects in chromosome misalignment were largely suppressed in cKO-CT-CCΔ73/CENP-T2x(1–90)_Δ121–240 cells, although recovery of chromosome misalignment was not strong, compared with cKO-CT-CCΔ73/CENP-TWT cells (Fig. 5d–f, Supplementary Fig. 6d). These data suggest that some Ndc80C-related defects in cKO-CT-CCΔ73/CENP-TΔ121-240 cells were partially suppressed by the addition of extra Ndc80C-binding sites. However, significant cohesion defects were still observed in cKO-CT-CCΔ73/CENP-T2x(1–90)_Δ121–240 cells (Fig. 5h). In addition, cKO-CT-CCΔ73/CENP-T2x(1–90)_Δ121–240 cells still revealed G2/M accumulation (Fig. 5g) and ultimately died by ~ 96 h after Tet addition (Fig. 5i), implying that the addition of extra Ndc80C to CENP-T did not completely suppress mitotic defects in cKO-CT-CCΔ73/CENP-TΔ121-240 cells. One possible explanation is that KM mitotic functions are essential for cell growth. Another possible explanation is that the Mis12C-Ndc80C interaction is still critical for full mitotic functions and cell growth, even if the defects of microtubule-kinetochore attachment were rescued by extra Ndc80C.
To distinguish between these two possibilities, we expressed CENP-T2x(1-90) in cKO-Dsn1/Dsn1Δ326-349 cells (Supplementary Fig. 6e), which lost one copy of Ndc80C and retained KM functions (KM-N). Although the KMN network forms KMN-N (one copy of Ndc80C binds to Mis12C, and another Ndc80C directly binds CENP-T) in wild-type chicken cells, KM-N-N (two copies of Ndc80C directly bind to CENP-T distinct from KM) may be functional (Fig. 5a). Strikingly, expression of GFP-CENP-T2x(1-90) in cKO-Dsn1/Dsn1Δ326-349 cells completely compensated for mitotic defects in the cKO-Dsn1/Dsn1Δ326-349 cells in the presence of Tet (Fig. 5j-m, Supplementary Fig. 6f). Abnormal spindle shape, abnormal numbers of spindle poles, mitotic accumulation, and chromosome misalignment were almost suppressed. Additionally, growth defects in cKO-Dsn1/Dsn1Δ326-349 cells were completely rescued by the expression of GFP-CENP-T2x(1-90) (Fig. 5n). These data indicate that two copies of Ndc80C can be functional without direct binding to Mis12C. This result indicates that direct binding of Ndc80C to Mis12C is not required for the microtubule-binding function of Ndc80C. Comparing the phenotype of cKO-Dsn1/Dsn1Δ326-349 cells expressing CENP-T2x(1-90) (Fig. 5j-n) with that of cKO-CT-CCΔ73/CENP-T2x(1–90)_Δ121–240 cells (Fig. 5d-i), we demonstrated that KM has essential mitotic roles, distinct from the microtubule-binding functions of N-N. We conclude that KM and N-N (two copies of Ndc80C) are separable and have distinct essential functions for cell growth (Fig. 5a).
Mis12C-Ndc80C interaction is dispensable in human RPE-1 cells
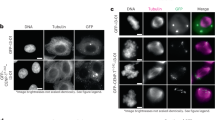
Our analyses in chicken DT40 cells demonstrated that KMN-N can be converted to KM-N-N in the KMN network on the CENP-T pathway (Fig. 6a). Interestingly, human CENP-T has three Ndc80C-binding sites (two are direct binding sites and one is via Mis12C: KMN-N-N) (Fig. 6a)35,41,51. If two copies of Ndc80C on CENP-T are sufficient for microtubule-kinetochore interactions in human cells, hDsn1 knockout cells expressing a mutant hDsn1 without the Ndc80C-binding site would be viable (KM-N-N), unlike cKO-Dsn1/Dsn1Δ326-349 DT40 cells. To test this hypothesis, we generated cKO-hDsn1 RPE-1 cells based on the AID system (cKO-hDsn1 (AID)) and expressed hDsn1Δ325-356 lacking the Ndc80C-binding region in these cells (Supplementary Fig. 7a-f). As observed in cKO-Dsn1/Dsn1Δ326-349 chicken DT40 cells, human Dsn1Δ325-356 was not associated with Ndc80C based on an immunoprecipitation experiment (Fig. 6b). In addition, Ndc80C levels were significantly decreased in cKO-hDsn1/hDsn1Δ325-356 cells after Indole-3-acetic acid (IAA) addition (Fig. 6c). Further, we examined the mitotic phenotype (Fig. 6d-f). In cKO-hDsn1 cells after IAA addition, cells with chromosome misalignment and lagging chromosomes significantly increased (Fig. 6e, f). However, the expression of Dsn1WT and Dsn1Δ325-356 suppressed these mitotic defects (Fig. 6d-f). In addition, the growth rate of cKO-hDsn1 cells expressing Dsn1Δ325-356 was comparable to those of cKO-hDsn1/hDsn1WT cells (Fig. 6g). These data support our hypothesis. Thus, we conclude that the Mis12C-Ndc80C interaction is dispensable in human cells.

a Schematic representation of the KMN network on CENP-T in chicken and human cells. Chicken has KMN-N on CENP-T. While cKO-Dsn1/Dsn1∆326-349 cells (KMN-N) died, expression of CENP-T2X(1-90) in these cells (KMN-N-N) rescued growth deficiency. In human cells, KMN-N-N is on CENP-T. The phenotype of cKO-Dsn1 lacking the Mis12C-Ndc80C interaction (cKO-hDsn1/hDsn1∆325-356 cell: KMN-N-N) will be tested. The KMN network on the CENP-C pathway in chicken and human cells is also presented. Both gDsn1∆326-349 and hDsn1∆325-356 do not bind to Ndc80C on the CENP-C pathway. b Immunoprecipitation with anti-RFP in AID-based cKO-hDsn1 cells expressing either mScarlet-hDsn1WT or -hDsn1∆325-356 in the presence of nocodazole for 24 h and IAA for last 2 h. Samples were analyzed by anti-Dsn1, and -Ndc80 antibodies. c Ndc80 levels at kinetochores in cKO-hDsn1 cells expressing mScarlet-hDsn1WT or -hDsn1∆325-356 in the presence of IAA for 2 h. DNA was stained with DAPI. Ndc80 was stained with an anti-Ndc80 antibody (red). CENP-T was stained with an anti-CENP-T antibody (green). None indicates parental cKO-hDsn1 (AID) cells. Error bars indicate the mean and standard deviation. p values were calculated by one-way ANOVA followed by Tukey’s test. d Tubulin staining in cKO-hDsn1 cells expressing hDsn1WT or hDsn1∆325-356 in the presence of IAA for 2 h. Kinetochores were stained with an anti-human CENP-A antibody (red). The insets present the kinetochore-microtubule bindings. DNA was stained with DAPI. e Images of mitotic chromosomes stained with anti-CENP-A in cKO-hDsn1 cells (None) expressing hDsn1WT or hDsn1∆325-356 in the presence of IAA for 2 h. DNA was stained with DAPI. The graph summarizes the percentage of cells with misaligned chromosomes in each line. Error bars indicate the mean and standard deviation. p values were calculated as in c. f Images of anaphase for cKO-hDsn1 cells (None) expressing hDsn1WT or hDsn1∆325-356 in the presence of IAA for 2 h. DNA was stained with DAPI. The graph summarizes the percentage of cells with abnormal anaphase in each line. Error bars indicate the mean and standard deviation. p values were calculated as in c. g The growth of cKO-hDsn1 cells expressing mScarlet-hDsn1WT or -hDsn1∆325-356 in the absence or presence of IAA.
Discussion
We previously revealed that the CENP-T pathway is the main pathway for the recruitment of the KMN network onto kinetochores, compared to the CENP-C pathway, in chicken DT40 cells35. However, the specific roles of the KMN network on the CENP-T pathway remain unclear. The KMN network binds to both CENP-C and CENP-T. Therefore, to precisely evaluate the roles of the KMN network on the CENP-T pathway, we must remove the CENP-C-KMN interaction to eliminate some redundant functions of CENP-C. Moreover, since some proteins have multiple roles, we must use specific mutants of each protein, which do not disrupt their multiple functions to avoid misinterpretation of its protein function. Based on our analyses, we concluded that the KMN network forms KMN-N in chicken cells and that KM and N-N have distinct roles: Proper kinetochore-microtubule interaction via N-N (two copies of Ndc80C) and the maintenance of sister chromatid cohesion, recruitment of one copy of Ndc80C or checkpoint function via KM (Knl1C and Mis12C) (Fig. 7a, b). Importantly, we observed that the Mis12C-Ndc80C interaction was not necessary, and KM and N-N are separable (Fig. 7a, b). Since the Ndc80C-Mis12C interaction is thought to be critical for the kinetochore functions7,42,43, our observations provide new insights for kinetochore studies.

a Summary of numbers of K, M, and N in various mutants and their phenotypes in chicken and human cells. b A model of the essential roles of the KMN network on the CENP-T pathway. KM recruits Bub1, H3T3ph, and Aurora B to maintain sister chromatid cohesion and bind to checkpoint proteins. In addition, KM recruits one Ndc80C copy. However, two copies of Ndc80C (N-N) are critical for the establishment of kinetochore-microtubule interactions. As Ndc80C is not necessary to bind to Mis12C, KM and N-N are separable. In CENP-T2X(1-90)_∆121-240 cells (N-N lacking KM), cohesion defects were still observed, even if sufficient amounts of Ndc80C were bound to kinetochores. In Dsn1∆326-349 cells (KM-N lacking one N), only one copy of Ndc80 localized kinetochores, and proper chromosome alignments did not occur, while cohesion defects were not observed. c Amino acid sequence alignments of the CENP-T extreme N-terminal region, conserved second N-terminal region, and Dsn1 C-terminal end for Ndc80C binding between various species.
There are multiple recruitment pathways of the KMN network onto kinetochores in other organisms, in addition to the CENP-C and CENP-T-pathways52,53, and the pathway choice is variable among different species54. Although many species have more than two pathways, Drosophila melanogaster and Caenorhabditis elegans have only the CENP-C pathway55,56,57. In contrast, several Lepidoptera and Mucor have only the CENP-T pathway58,59. These observations suggest that each pathway can recruit the KMN network onto the kinetochore, and the CENP-T and CENP-C pathways might be redundant in organisms possessing more than two pathways. However, our previous study indicated that the CENP-T-KMN network interaction is essential, but the CENP-C-KMN network interaction is dispensable in chicken DT40 cells. Nevertheless, we hypothesized that the CENP-C-KMN network interaction plays a role, because cells lacking Mis12C binding to CENP-T in the presence of the CENP-C-KMN network interaction do not completely die. This study demonstrated that cells completely died in the absence of Mis12C interaction with both CENP-C and CENP-T, suggesting that Mis12C on CENP-C plays a role, which might contribute to sister chromatid cohesion or checkpoint function via KM or robust kinetochore-microtubule attachment via additional Ndc80C since Mis12C binds to Knl1C and Ndc80C. Therefore, to analyze the functional roles of KMN in each pathway, we must characterize it with the depletion of some redundancies. This strategy is essential for evaluating the function of each complex in different organisms with various recruitment pathways of the KMN network.
Two copies of Ndc80C (N-N) are necessary and sufficient for proper kinetochore-microtubule attachment in DT40 cells, suggesting that binding of a single copy of Ndc80C to spindle microtubules is not sufficient for full kinetochore functions. Single-molecule experiments demonstrated that the multivalency of Ndc80C efficiently tracked depolymerizing microtubules, whereas a single copy of Ndc80C did not51, which supports our observations. Other biophysics experiments suggest that microtubule pulling forces from kinetochores vary (0.4–8 pN)60,61, suggesting that the double increase of microtubule-binding proteins might not be sufficient for stable chromosome segregation. However, it is unclear how much the kinetochore with one copy of Ndc80C is unstable, and such kinetochores might not generate sufficient force for chromosome segregation. Thus, the extra copy of Ndc80C might have synergistic effects on kinetochore function in vivo. It is important to know how extra copies of Ndc80C contribute to kinetochore function.
The CENP-C-KMN network interaction is not sufficient in DT40 cells since Mis12C is easily dissociated from CENP-C35. However, forced binding of Mis12C to CENP-C stabilizes the CENP-C pathway. In this situation, there is only one copy of Ndc80C on the kinetochore, which possesses only the CENP-C pathway. However, kinetochores with one copy of the Ndc80C were functional in this case. This mechanism might be explained by the amount of protein at the kinetochore38. Suzuki et al.38 proposed ~ 215 molecules of CENP-C and ~ 72 molecules of CENP-T in a human kinetochore. Since only ~ 70 molecules of Mis12C bind to CENP-C, two-thirds of CENP-C are usually not used for the KMN network interaction. However, CENP-C has the capacity to accept more KMN components, and forced binding of Mis12C to CENP-C would cause a stable CENP-C pathway, even if the kinetochore with the CENP-C pathway has only one copy of Ndc80C. Additionally, this measurement may explain why two copies of Ndc80C are necessary for CENP-T. Compared with CENP-C amounts, CENP-T amounts are small at each kinetochore38. To maintain sufficient amounts of Ndc80C, two copies of Ndc80C per CENP-T may be critical for chromosome segregation.
Independent of the microtubule-binding function of two copies of Ndc80C (N-N), Knl1C and Mis12C (KM) have some essential roles, since even when cells had two copies of Ndc80C for proper kinetochore-microtubule interaction in cKO-CT-CCΔ73/CENP-T2x(1–90)_Δ121–240 cells, the cells died. Although it is still unclear which KM functions are critical for cell viability, KM has various roles, including the maintenance of sister chromatid cohesion or the recruitment of checkpoint proteins. KM binds to Bub1, which further recruits various factors required for the maintenance of sister chromatid cohesion, including the cohesin complex. The cohesin complex, H3T3ph, and CPC localize on entire chromosomes until prophase, but they localize to centromeric regions in metaphase cells. KM appears to play an essential role in proper centromere localization of H3T3ph or CPC in metaphase (Fig. 3f), since these localizations are dispersed into entire chromosomes in mutant cells lacking the Mis12C-CENP-T interaction. KM either directly or indirectly facilitates the removal of H3T3ph or CPC from chromosome arms and protects their removal from centromeres in metaphase for sister chromatid cohesion.
The functions of KM and N-N on the CENP-T pathway are relatively predictable based on previous observations2,6, however, it is surprising to find that KM and N-N are separable, which means that Mis12C is not necessary for binding to Ndc80C. This striking result contrasts with a previous yeast study62, which proposed that the microtubule-binding activity of yeast Ndc80C is regulated by its interaction with the MIND complex (yeast Mis12 complex).
For the CENP-C-KMN network interaction, Mis12C is the sole platform for Ndc80C; therefore, this interaction is critical. On the CENP-T-KMN network interaction, as one copy of Ndc80C is not sufficient, the Mis12C-Ndc80C interaction is critical in chicken cells. However, if Ndc80C is artificially supplied to CENP-T, this Ndc80C-recruitment function for Mis12C becomes dispensable (Fig. 5). Although this was observed in an artificial chicken situation, the Ndc80C-Mis12C interaction is dispensable in human cells, since humans have three copies of Ndc80C on CENP-T (KMN-N-N) (Fig. 7a)35,41. To bind to Ndc80C, CENP-T phosphorylation of the threonine residue of the TPR motif in its N-terminus is critical (Fig. 7c)39,41. Two TPR motifs exist in primate CENP-T, albeit a TPR motif in the extreme N-terminus of CENP-T is not clearly detected in CENP-Ts other than the primate CENP-T, while Chinese hamsters have two TPR motifs (Fig. 7c). This suggests that the TPR motif was acquired after the primate lineage to ensure robust kinetochore-microtubule interactions. Although we prefer to interpret that the copy number of Ndc80C binding sites on CENP-T is a critical factor for the increase in Ndc80C amounts in most organisms, it is also possible that the amount of Ndc80C would be increased in different ways. Therefore, this point is important and needs to be addressed.
While KM and N-N are separable, one copy of Ndc80C is usually supplied via Mis12C binding; however, the advantage of this system is unknown. This may be related to the Mis12C recruitment mechanism on the CENP-T pathway. Direct binding of Ndc80C to CENP-T facilitates the recruitment of Mis12C (Fig. 2), which further recruits additional copy of Ndc80C. This is an efficient way to recruit essential factors to the CENP-T pathway, and it is not necessary to have two Ndc80-binding sites in CENP-T. In in vitro experiments, Mis12C can bind to CENP-T, even if CENP-T does not directly bind to Ndc80C41. Therefore, the efficient Mis12C recruitment mechanism via direct Ndc80C binding to CENP-T cannot be explained only by performing in vitro studies. It would be an important further challenge to clarify the regulatory mechanisms of Mis12C recruitment onto the CENP-T pathway.
The kinetochore is a multi-protein complex with complicated protein-protein networks that often exhibit redundant functions. Therefore, it is necessary to dissect the functional roles of each kinetochore complex. In this study, using precise information on the binding sites of each complex, we defined the distinct functions of KM and N-N on the CENP-T pathway and demonstrated that they are separable. It is difficult to distinguish these functions using only a simple knockout approach for the components of the KMN network. Furthermore, functional kinetochores could be created by providing an artificial Ndc80C-binding site on CENP-T to explain the distinct KMN functions in the CENP-T pathway. Therefore, to control or manipulate kinetochore functions in the future, we must understand the precise roles of each complex in vivo, including redundant functions. We believe that this study provides a model for analyzing the precise function of each kinetochore complex in vivo and provides critical insights into kinetochore functions.
Methods
Cell culture
Chicken DT40 cells63 were cultured in Dulbecco’s modified Eagle’s medium (Nacalai tesque, 08459-64) containing 10% fetal bovine serum (Sigma, 172012-500 mL), 1% chicken serum (Thermo Fisher, 16110-082), 10 µM 2-Mercaptoethanol (Sigma, M3148), and penicillin (100 unit/mL)-streptomycin (100 µg/mL) (Thermo Fisher, 15140-122), at 38.5 °C with 5% CO2. For gene inactivation in tetracycline-dependent conditional knockout cell lines (cKO-CENP-T, cKO-Dsn1, and cKO-Spc25 cells), 2 µg/ml tetracycline (Sigma, T-7660) was added. For degradation of GFP-AID-Nsl1 in cKO-Nsl1 (AID) cells, 500 µM 3-Insole acetic acid (IAA; Wako, 090-07123) was added. For examination of the cell growth for each cKO cell line, tetracycline or IAA was added to the culture medium at 0 h and the cell numbers were measured at each time point. Cell numbers were normalized to that before tetracycline or IAA addition (0 h) for each cell line.
Human RPE-1 cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and penicillin (100 unit/mL)-streptomycin (100 µg/mL) at 37 °C with 5% CO2. For degradation of GFP-AID-hDsn1 in cKO-hDsn1 (AID) cells, 250 µM IAA was added. For examination of the cell growth for each cKO cell line, IAA was added to the culture medium at 0 h and the cell numbers were measured at each time point. Cell numbers were normalized to that before IAA addition (0 h) for each cell line.
Plasmid constructions for cell transfection
Several CENP-T mutants (CENP-TΔ121–160, CENP-TΔ161–200, CENP-TΔ201–216, CENP-TΔ217–230, CENP-TΔ231–240, CENP-T8A, CENP-T2x(1-90), and CENP-T2x(1–90)_Δ121–240) were generated by PCR based mutagenesis or In-Fusion® HD Cloning Kit (Takara, 639648). To express 3X FLAG-fused CENP-T mutants, CENP-TWT, CENP-TΔ90, CENP-TT72A-S88A 39, CENP-TΔ121–24035, CENP-TΔ121–160, CENP-TΔ161–200, CENP-TΔ201–216, CENP-TΔ217–230, CENP-TΔ231–240, CENP-T8A, CENP-T2x(1-90), and CENP-T2x(1–90)_Δ121–240 were cloned into p3X FLAG-CMV-10 (Sigma, E7658).
To express 3X FLAG-fused CENP-T mutants under control of the endogenous CENP-T promoter in DT40 cells, 3X FLAG-fused CENP-T mutants and the Neomycin resistance gene expressed under the control of the SV40 promoter (SV40 NeoR) were cloned into the pBSKS_CT 2k vector, in which about 2 kb genome fragment (5′ and 3′ homology arm regions (approximately 1 kb each)) around the start codon of CENP-T is cloned35 by In-Fusion® HD Cloning Kit (pBSKS_CT KI_3X FLAG-CENP-TWT_SV40 NeoR, pBSKS_CT KI_3X FLAG-CENP-TΔ90_SV40 NeoR, pBSKS_ CT KI_3X FLAG-CENP-TT72A-S88A_SV40 NeoR, pBSKS_ CT KI_3X FLAG-CENP-TΔ121–240_SV40 NeoR, pBSKS_CT KI_3X FLAG-CENP-TΔ121–160_SV40 NeoR, pBSKS_ CT KI_3X FLAG-CENP-TΔ161–200_SV40 NeoR, pBSKS_ CT KI_3X FLAG-CENP-TΔ201–216_SV40 NeoR, pBSKS_ CT KI_3X FLAG-CENP-TΔ217–230_SV40 NeoR, pBSKS_CT KI_3X FLAG-CENP-TΔ231–240_SV40 NeoR, pBSKS_CT KI_3X FLAG-CENP-T8A_SV40 NeoR, pBSKS_ CT KI_3X FLAG-CENP-T2x(1-90)_SV40 NeoR, and pBSKS_CT KI_3X FLAG-CENP-T2x(1–90)_Δ121–240_SV40 NeoR). To disrupt the expression of the endogenous CENP-T, the Neomycin resistance cassette (SV40 NeoR) was cloned into the pBSKS_CT 2k vector (pBSKS_ CT KI_SV40 NeoR). To integrate these constructs into the endogenous CENP-T locus by using CRISPR/Cas9-mediated homologous recombination, the pX335_ggCENP-T containing single-guide RNA (sgRNA) against a genomic sequence around the start codon of CENP-T and SpCas9 nickase (D10A) gene35 was used.
The mScarlet-fused CENP-CΔ73 expression vector was generated by cloning of CENP-CΔ73 cDNA into the pmScarlet-C1 (Addgene, 85042) vector. To express mScarlet-fused CENP-CΔ73 under control of the endogenous β-actin (ACTB) promoter in DT40 cells, mScarlet-fused CENP-CΔ73 and the Hygromycin B resistance gene expressed under control of the PGK promoter (PGK HygroR) were cloned into the pBSKS_ACTB 2k vector, in which about 2 kb genome fragment (5′ and 3′ homology arm regions (approximately 1 kb each)) around the ACTB start codon is cloned35, by In-Fusion® HD Cloning Kit (pBSKS_ACTB KI_mScarlet-CENP-CΔ73_PGK HygroR). To express the HA-fused CENP-CΔ73 under control of the endogenous ACTB promoter in DT40 cells, mScarlet region of the pBSKS_ACTB KI_mScarlet-CENP-CΔ73_PGK HygroR was replaced with the HA-tag (pBSKS_ACTB KI_HA-CENP-CΔ73_PGK HygroR). To integrate these constructs into the endogenous ACTB locus by using CRISPR/Cas9-mediated homologous recombination, pX335_ggACTB containing sgRNA against a genomic sequence around the start codon of ACTB and SpCas9 nickase (D10A) gene35 was used.
To express the GFP-fused Dsn1WT or Dsn1Δ326–349 35, under control of the Dsn1 promoter in DT40 cells, the GFP-fused Dsn1WT or Dsn1Δ326–349 with puromycin resistance gene expressed by the PGK promoter (PGK PuroR), EcoGPT expressed by the PGK promoter (PGK EcoGPT), or the blasticidin S resistance gene expressed by ACTB promoter (ACTB BSR) were cloned into the pBSKS_Dsn1 2k vector, in which about 2 kb genome fragment (5′ and 3′ homology arm regions (approximately 1 kb each)) around the Dsn1 start codon is cloned, by In-Fusion® HD Cloning Kit (pBSKS_Dsn1 KI_GFP-Dsn1WT_PGK PuroR, pBSKS_Dsn1 KI_GFP-Dsn1WT_PGK EcoGPT, pBSKS_Dsn1 KI_GFP-DsnWT_ACTB BSR, and pBSKS_Dsn1 KI_GFP-DsnΔ326–349_ACTB BSR). To integrate these constructs into the endogenous Dsn1 locus by using CRISPR/Cas9-mediated homologous recombination, we designed sgRNA against a DNA sequence (GCGCGCGTAGCCGCCATGGA) around the start codon of Dsn1 using CRISPOR64, and cloned it into the BbsI site of the pX330 plasmid (Addgene, 42230) containing SpCas9 gene (pX330_ggDsn1).
To express the GFP-fused Nuf2 under control of the Nuf2 promoter in DT40 cells, GFP-fused Nuf2 and PGK PuroR or PGK EcoGPT were cloned into pBSKS_Nuf2 2k vector, in which about 2 kb genome fragment (5′ and 3′ homology arm regions (approximately 1 kb each)) around the end of the exon1 of Nuf2 genomic sequence is cloned, by In-Fusion® HD Cloning Kit (pBSKS_Nuf2 KI_Nuf2-GFP_PGK PuroR and pBSKS_Nuf2 KI_Nuf2-GFP_PGK EcoGPT). To express the mScarlet-fused Nuf2 under control of the Nuf2 promoter in DT40 cells, mScarlet-fused Nuf2 and ACTB BSR or PGK EcoGPT were cloned into pBSKS_Nuf2 2k vector by In-Fusion® HD Cloning Kit (pBSKS_Nuf2 KI_Nuf2-mScarlet_ACTB BSR and pBSKS_Nuf2 KI_Nuf2-mScarlet_PGK EcoGPT). To integrate these constructs into the endogenous Nuf2 locus by using CRISPR/Cas9-mediated homologous recombination, we designed sgRNA against a DNA sequence (GCCACCCTTAAGGTGTGTG) in the intron 1 of the Nuf2 genomic sequence using CRISPOR64, and cloned it into the BbsI site of the pX330 (pX330_ggNuf2).
To disrupt expression from the endogenous CENP-C gene by using CRISPR/Cas9 system, sgRNA against a DNA sequence in the exon2 of the CENP-C genomic sequence (CGAGCAAGATTCTGCGGGCA) was designed using CRISPOR64, and cloned it into the BbsI site of the pX330 (pX330_ggCENP-C).
To express the GFP-fused CENP-T2x(1-90), pEGFP-C3_CENP-T2x(1-90) was generated from the pEGFP-C3_CENP-TWT by In-Fusion® HD Cloning Kit.
To generate cKO-Nsl1 (AID) cells, we used CRISPR/Cas9-meidated genome editing combined with the auxin degron-based cKO system65,66,67. To disrupt the expression of the endogenous Nsl1 by using CRISPR/Cas9 system, sgRNA against a DNA sequence (TAAGAAATCATTACCTTGAG) across the boundary between the exon 4 and the intron 4 of the Nsl1 gene was designed using CRISPOR64, and cloned it into the Bbs1 site of pX330 (pX330_ggNsl1). To express the OsTIR1-T2A-BSR and GFP-mAID-fused Nsl1 simultaneously under control of the CMV promoter, Nsl1 cDNA was cloned into the pAID1.2-NEGFP containing CMV promoter_OsTIR1-T2A-BSR_IRES_GFP-mAID (pAID1.2-NEGFP-Nsl1). To linearize the pAID1.2-NEGFP-Nsl1, the pX330_pAID1.2 containing sgRNA against a back bone sequence of the pAID1.2-NEGFP-Nsl1 and SpCas9 gene was used67.
To express the GFP-fused Nsl1WT, the pEGFP-C3_Nsl1WT was used, and leucine 220 and leucine 222 residues of pEGFP-C3_Nsl1WT were substituted with glutamic acid (pEGFP-C3_Nsl1EE) by PCR based mutagenesis for the expression of GFP-fused Nsl1EE.
To express the VH-mScarlet under control of the endogenous ACTB promoter in DT40 cells for the tension sensor assay, GFP-sequence of VH-GFP (a gift from T. Maresca, University of Massachusetts Amherst, Amherst, MA, USA) was replaced with mScarlet (VH-mScarlet). Then, VH-mScarlet and PGK EcoGPT were cloned into pBSKS_ACTB 2k vector (pBSKS_ACTB 2k_VH-mScarlet).
To express the OsTIR1-T2A-BSR and GFP-mAID-fused hDsn1 simultaneously under control of the CMV promoter, hDsn1 cDNA was cloned into the pAID1.2-NEGFP (pAID1.2-NEGFP-hDsn1). To express the OsTIR1-T2A-BSR and GFP-mAID-hDsn1 under control of the CMV promoter from the AAVS1 locus in RPE-1 cells, CMV promoter_OsTIR1-T2A-BSR_IRES_GFP-mAID-hDsn1 fragment was cloned into the pBSKS_AAVS1 1.6k vector, in which about 1.6 kb genome fragment (5′ and 3′ homology arm regions (approximately 0.8 kb each)) around the AAVS1 intron 1 is cloned, by In-Fusion® HD Cloning Kit. To integrate this construct into the endogenous AAVS1 locus by using CRISPR/Cas9-mediated homologous recombination, we designed sgRNA against a DNA sequence (TAGTGGCCCCACTGTGGGGT) in the intron 1 of AAVS1 using CRISPOR64, and cloned it into the Bbs1 site of the pX330 (pX330_hsAAVS1).
To express the mScarlet-fused hDsn1 expressed under control of the endogenous hDsn1 promoter in RPE-1 cells, mScarlet-fused hDsn1WT or hDsn1Δ325-356 and NeoR or PuroR expressed by ACTB promoter (ACTB NeoR, ACTB PuroR) were cloned into pBSKS_hDsn1 2k vector, in which about 2 kb genome fragment (5′ and 3′ homology arm regions (approximately 1 kb each)) around the hDsn1 start codon is cloned, by In-Fusion® HD Cloning Kit (pBSKS_hDsn1 KI_mScarlet-hDsn1WT_ACTB NeoR, pBSKS_hDsn1 KI_mScarlet-hDsn1WT_ACTB PuroR, pBSKS_hDsn1 KI_mScarlet-hDsn1Δ325-356_ACTB NeoR, and pBSKS_hDsn1 KI_mScarlet-hDsn1Δ325-356_ACTB PuroR). To disrupt the expression of endogenous hDsn1 in RPE-1 cells, ACTB NeoR or ACTB PuroR were cloned into pBSKS_hDsn1 2k vector by In-Fusion® HD Cloning Kit (pBSKS_hDsn1 KI_ ACTB NeoR, pBSKS_hDsn1 KI_ACTB PuroR). To integrate these constructs into the endogenous hDsn1 locus by using CRISPR/Cas9-mediated homologous recombination, we designed sgRNA against a DNA sequence (CTTACCTTGGGTTCAGGCTT) in the intron1 of the hDsn1 genomic sequence using CRISPOR64, and cloned it into the Bbs1 site of pX330 (pX330_hsDsn1).
Generation of cell lines
The cell lines established in this study are listed in Supplementary Table 1.
To express the mScarlet-fused CENP-CΔ73 or HA-fused CENP-CΔ73 under control of the endogenous ACTB promoter, pBSKS_ACTB KI_mScarlet-CENP-CΔ73_PGK HygroR or pBSKS_ACTB KI_HA-CENP-CΔ73_PGK HygroR were transfected with pX335_ggACTB using Neon Transfection System (Thermo Fisher, MPK5000) with 6 times of pulse (1400 V, 5 msec) in Tet-Off CENP-T/CENP-TΔ/+ cells5 (mScarlet-CENP-CΔ73), cKO-Spc25 cells39 (mScarlet-CENP-CΔ73), and CL18 cells (HA-CENP-CΔ73). The transfected cells were selected in the medium containing 2.5 mg/mL Hygromycin B (Wako, 087-06152) in 96 well plates to isolate single-cell clones.
To knockout the expression of endogenous CENP-C, pX330_ggCENP-C was transfected with pBSKS_ACTB-BSR using Neon Transfection System with six times of pulse (1400 V, 5 msec) in Tet-OFF CENP-T/CENP-TΔ/+ expressing mScarlet-CENP-CΔ73 cells, cKO-Spc25 cells expressing mScarlet-CENP-CΔ73, and CL18 cells expressing HA-CENP-CΔ73. The transfected cells were selected in the medium containing 25 µg/mL Blasticidin S hydrochloride (Wako, 029-18701) for 48 h, and seeded in 96 well plates to isolate single-cell clones (Tet-Off CENP-T/CENP-TΔ/+/CENP-CΔ73 cells, cKO-Spc25-CCΔ73 cells, and CENP-CΔ73 cells).
To express 3X FLAG-fused CENP-T constructs under the control of the endogenous CENP-T promoter, each 3X FLAG-fused CENP-T construct (pBSKS_CT KI_3X FLAG-CENP-Tmutant_SV40 NeoR) was transfected with pX335_ggCENP-T using Neon Transfection System with 6 times of pulse (1400 V, 5 msec) in Tet-Off CENP-T/CENP-TΔ/+ cells or Tet-Off CENP-T/CENP-TΔ/+/CENP-CΔ73 cells. The transfected cells were selected in the medium containing 2 mg/mL G418 (Santa Cruz Biotechnology, SC-29065B) in 96 well plates to isolate single-cell clones (cKO-CT-CCWT/CENP-Tmutant cells, cKO-CT-CCΔ73/CENP-Tmutant cells).
To express the GFP-fused Dsn1 under control of the endogenous Dsn1 promoter, pBSKS_Dsn1 KI_GFP-Dsn1WT_PGK PuroR and pBSKS_Dsn1 KI_GFP-Dsn1WT_PGK EcoGPT were transfected with pX330_ggDsn1 using Neon Transfection System with 6 times of pulse (1400 V, 5 msec) in cKO-CT-CCΔ73 cells, cKO-CT-CCΔ73/CENP-TWT cells, cKO-CT-CCΔ73/CENP-TΔ90 cells, cKO-CT-CCΔ73/CENP-TΔ121–240 cells, cKO-CT-CCΔ73/CENP-TΔ121–160 cells, cKO-CT-CCΔ73/CENP-TΔ161–200 cells, cKO-CT-CCΔ73/CENP-TΔ201–216 cells, cKO-CT-CCΔ73/CENP-TΔ217–230 cells, cKO-CT-CCΔ73/CENP-TΔ231–240 cells, cKO-CT-CCΔ73/CENP-T8A cells, cKO-Spc25-CCΔ73 cells, and CENP-CΔ73 cells. The transfected cells were selected in the medium containing 0.5 µg/mL puromycin (Takara, Z1305N), 25 µg/mL mycophenolic acid (Wako, 138-11003), and 125 µg/mL xanthine (Sigma, 1002581797) in 96 well plates to isolate single-cell clones. pBSKS_Dsn1 KI_GFP-Dsn1WT_ACTB BSR or pBSKS_Dsn1 KI_GFP-Dsn1Δ326–349_ ACTB BSR transfected with the pX330_ggDsn1 using Neon Transfection System with 6 times of pulse (1400 V, 5 msec) in Tet-Off Dsn1/Dsn1Δ/+ cells, and then the transfected cells were selected in the medium containing 25 µg/mL Blasticidin S hydrochloride in 96 well plates to isolate single-cell clones (cKO-Dsn1/Dsn1WT cells and cKO-Dsn1/Dsn1Δ326-349 cells).
To express the GFP-fused Nuf2 under control of the endogenous Nuf2 promoter, pBSKS_Nuf2 KI_GFP-Nuf2_PGK PuroR and pBSKS_Nuf2 KI_GFP-Nuf2_PGK EcoGPT were transfected with the pX330_ggNuf2 using Neon Transfection System with 6 times of pulse (1400 V, 5 msec) in cKO-CT-CCΔ73 cells, cKO-CT-CCΔ73/CENP-TWT cells, cKO-CT-CCΔ73/CENP-TΔ90 cells, cKO-CT-CCΔ73/CENP-TT72A-S88A cells, cKO-CT-CCΔ73/CENP-TΔ121–240 cells, cKO-CT-CCΔ73/CENP-TΔ161–200 cells, cKO-CT-CCΔ73/CENP-TΔ201–216 cells, cKO-CT-CCΔ73/CENP-T2x(1-90) cells, and CENP-CΔ73 cells. The transfected cells were selected in the medium containing 0.5 µg/mL puromycin, 25 µg/mL mycophenolic acid, and 125 µg/mL xanthine in 96 well plates to isolate single-cell clones.
To express the mScarlet-fused Nuf2 under control of the endogenous Nuf2 promoter, pBSKS_Nuf2 KI_Nuf2-mScarlet_ACTB BSR and pBSKS_Nuf2 KI_Nuf2-mScarlet_PGK EcoGPT were transfected with the pX330_ggNuf2 using Neon Transfection System with 6 times of pulse (1400 V, 5 msec) in cKO-Dsn1/GFP-Dsn1WT cells and cKO-Dsn1/GFP-Dsn1Δ326-349 cells. The transfected cells were selected in the medium containing 25 µg/mL Blasticidin S hydrochloride in 96 well plates to isolate single-cell clones.
To generate cKO-Nsl1 (AID) cells, the pAID1.2-NEGFP-Nsl1 was transfected with the pX330_ggNsl1 and pX330_pAID1.2 using Neon Transfection System with 6 times of pulse (1400 V, 5 msec) in CL18 cells. The transfected cells were selected in the medium containing 25 µg/mL Blasticidin S hydrochloride in 96 well plates to isolate single-cell clones.
To express the GFP-fused Nsl1WT and Nsl1EE, the linearized pEGFP-C3_Nsl1WT or pEGFP-C3_Nsl1EE was transfected using Gene Pulser II electroporator (Bio-Rad, 165-2105) in cKO-Nsl1 (AID) cells. The transfected cells were selected in the medium containing 2 mg/mL G418 in 96 well plates to isolate single-cell clones.
To express the GFP-fused CENP-T2x(1-90), the linearized pEGFP-C3_CENP-T2x(1-90) was transfected using Gene Pulser II electroporator in cKO-Dsn1/GFP-Dsn1Δ326-349 cells. The transfected cells were selected in the medium containing 2 mg/mL G418 in 96 well plates to isolate single-cell clones.
For the tension sensor assay, pBSKS_CT 2k_TagRFP-CENP-T-TR (241-529;TR)35 was transfected with the pX335_ggCT using Neon Transfection System with 6 times of pulse (1400 V, 5 msec) in cKO-Dsn1/Dsn1WT or cKO-Dsn1/Dsn1Δ326-349 cells. The transfected cells were selected in the medium containing 2 mg/mL G418 in 96 well plates to isolate single-cell clones. To express the VH-mScarlet, the pBSKS_ACTB 2k_VH-mScarlet was transfected with the pX335_ggACTB using Neon Transfection System with 6 times of pulse (1400 V, 5 msec) in cKO-Dsn1/Dsn1WT or cKO-Dsn1/Dsn1Δ326-349 cells expressing the TagRFP-CENP-T-TR. The transfected cells were selected in the medium containing 25 µg/mL mycophenolic acid and 125 µg/mL xanthine in 96 well plates to isolate single-cell clones.
To express the OsTIR1-T2A-BSR and GFP-mAID-fused hDsn1, pAID1.2-NEGFP-hDsn1 was transfected with the pX330_hsAAVS1 using Neon Transfection System with 6 times of pulse (1400 V, 5 msec) in RPE-1 cells. The transfected cells were selected in the medium containing 10 µg/mL Blasticidin S hydrochloride. To disrupt the expression of endogenous hDsn1 in RPE-1 cells expressing the OsTIR1-T2A-BSR and GFP-mAID-hDsn1, pBSKS_hDsn1 KI_ACTB NeoR and pBSKS_hDsn1 KI_ACTB PuroR were transfected with the pX330_hDsn1 using Neon Transfection System with 6 times of pulse (1400 V, 5 msec). To replace endogenous hDsn1 with mScarlet-hDsn1WT or mScarlet-hDsn1Δ325-356 in RPE1 cells expressing OsTIR1-T2A-BSR and GFP-mAID-hDsn1, the pBSKS_hDsn1 KI_mScarlet-hDsn1WT_ACTB NeoR and pBSKS_hDsn1 KI_ mScarlet-hDsn1WT_ACTB PuroR or pBSKS_hDsn1 KI_mScarlet-hDsn1Δ325-356_ACTB NeoR and pBSKS_hDsn1 KI_ mScarlet-hDsn1Δ325-356_ACTB PuroR were transfected with the pX330_hDsn1 using Neon Transfection System with 6 times of pulse (1400 V, 5 msec). The transfected cells were selected in the medium containing 0.5 mg/ml G418 and 2 µg/ml puromycin.
Genotyping
For the genomic DNA extraction from DT40 cells, cells were collected, washed with PBS (Nissui Pharmaceutical Co., Ltd., 05913), and suspended in 0.2 mg/mL Proteinase K (Sigma, P2308) in 0.1% PBST (PBS, 0.1% Tween 20 (Nacalai tesque, 28353-85)). The suspended cells were incubated at 55 °C for 90 min and heated at 96 °C for 15 min.
For the genomic DNA extraction of RPE-1 cells, cells were collected after trypsinizing, washed with PBS, and suspended in 0.2 mg/mL Proteinase K in 0.1% PBST (PBS, 0.1% Tween 20). The suspended cells were incubated at 55 °C for 90 min and heated at 96 °C for 15 min.
To investigate the mutation sequence in the chicken CENP-C gene, the genomic regions flanking the sgRNA target site were amplified by PCR using the following primers: Fw: 5′- ATGCATCAACCAGGAGGCTGTC -3′, Rv: 5′- CTAAGGCACACCATTAGTTTTGG -3′. The PCR amplicons were cloned into pBluescript II SK (-) and sequenced.
The target integration of several constructs was confirmed by PCR. The primers used for PCRs were listed in Supplementary Table 2.
Immunoblot
DT40 cells were collected, washed with cold PBS, and suspended in 1xLaemmli Sample Buffer (LSB; 62.5 mM Tris (Sigma, T1503) –HCl (Nacalai tesque, 18321-05) (pH 6.8), 10% Glycerol (Nacalai tesque, 17018-83), 2% SDS (Nacalai tesque, 02873-75), 5% 2-mercaptoethanol (Sigma, M3148), bromophenol blue (Wako, 101123)) (final concentration 1 × 104 cells/µL). Following the sonication, the lysate was heated at 96 °C for 5 min.
RPE-1 cells were collected after trypsinization, washed with cold PBS, and suspended in 1xLSB (final concentration 1 × 104 cells/µL). Following the sonication, the lysate was heated at 96 °C for 5 min.
The collected samples (5 × 104 cells) were separated by SDS-PAGE (SuperSep Ace, 5–20% (Wako, 194-15021) or handmade 7.5%) and transferred onto PVDF membranes (Immobilon®-P (Merck, IPVH00010)). The membrane was probed with primary antibody diluted with Signal Enhancer Hikari (Nacalai Tesque, 02270-81) at 4 °C for overnight. After washing with 0.1% TBST (TBS: 50 mM Tris-HCl, 150 mM NaCl; 0.1% Tween 20) for 15 min, the membrane was probed with secondary antibody diluted with Signal Enhancer Hikari at room temperature for 1 h. After washing with 0.1% TBST for 15 min, the signals were detected using ECL Prime Western Blotting Detection Reagent (cytiva, RPN2232) and visualized by ChemiDoc Touch imaging system (Bio-Rad). The Image processing was performed using Image Lab 5.2.1 (Bio-Rad) and Adobe Photoshop v23.1.0 (Adobe).
Primary antibodies used in immunoblot analyses were rabbit anti-chicken CENP-T5 (1:10000), rabbit anti-chicken CENP-C18 (in this study) (1:10000), rabbit anti-chicken Dsn135 (1:10000), rabbit anti-chicken Nuf268 (1:5000), rabbit anti-chicken Ndc8068 (1:2000), rabbit anti-chicken Spc25 (in this study using recombinant chicken full-length Spc25 as an immunogen) (1:2000), rabbit anti-chicken Knl134 (1:2000), rabbit anti-chicken Mis1242 (1:2000), rabbit anti-human Dsn1(Bio Academia, 70-101) (1:5000), rabbit anti-GFP (MBL, 598) (1:2000), mouse anti-FLAG M2 (Sigma, F1804) (1:1000), rat anti-RFP (Chromotek, 5f8) (1:2000), and mouse anti-α-tubulin (Sigma, T9026) (1:5000). Secondary antibodies used in immunoblot analysis were horseradish peroxidase-conjugated (HRP)-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch, 111-035-144) (1:10000), HRP-conjugated goat anti-rat IgG (Jackson ImmunoResearch, 112-035-003) (1:10000), and HRP-conjugated rabbit anti-mouse IgG (Jackson ImmunoResearch, 115-035-003) (1:10000).
Immunoprecipitation
For GFP-Dsn1 immunoprecipitation, cKO-Dsn1/Dsn1WT or cKO-Dsn1/Dsn1∆326–349 cells were cultured with 2 µg/mL tetracycline for 36 h. For cKO-Dsn1/GFP-Dsn1WT cells cultured with 2 µg/mL tetracycline for 36 h, 100 ng/mL nocodazole (Sigma, M1404) were also added for last 10 h. For mScarlet-hDsn1 immunoprecipitation, cKO-Dsn1/hDsn1WT or cKO-Dsn1/hDsn1∆325–356 cells were cultured with 100 ng/mL nocodazole for 24 h and treated with IAA for last 2 h. These cells were collected, washed with cold PBS twice, suspended in Lysis buffer (50 mM NaPi (Disodium Hydrogenphosphate, 197-02865; Sodium Dihydrogenphosphate Dihydrate, 192-02815) (pH 8.0), 300 mM NaCl (Nacalai tesque, 31320-05), 0.1% NP40 (Nacalai tesque, 25223-004), 5 mM 2-mercaptoethanol, 1xcOmplete EDTA-free proteinase inhibitor (Roche, 11873580001), and 1xphosphatase inhibitor (10 mM Sodium Pyrophosphate (Sigma, 221368), 5 mM Sodium Azide (Wako, 195-11092), 10 mM NaF (Wako, 192-01972), 0.4 mM Sodium orthovanadate (Sigma, S6508), 20 mM β-Glycerophosphate (Wako, 048-34332))) (final: 1 × 108 cells/mL for DT40 cells, 2 × 107 cells/mL for RPE1 cells) and sonicated. The lysate was clarified by centrifugation and the supernatant was incubated with GFP-Trap Magnetic agarose (Chromotek, gtma) or RFP-Trap Magnetic agarose (Chromotek, rtma) at 4 °C for 1 h. Proteins precipitated with GFP-Trap Magnetic agarose or RFP-Trap Magnetic agarose were washed with Lysis buffer five times, eluted with 2 × LSB. The eluted samples were heated at 96 °C for 5 min and subjected to immunoblot analysis.
Immunofluorescence analysis
The cells were cytospan onto slide glasses or coverslips by the Cytospin3 centrifuge (Shandon, TH-CYTO3), fixed with 3% paraformaldehyde (PFA; Nacalai tesque, 26126-25) in 250 mM HEPES (Nacalai tesque, 17514-15) -NaOH (Nacalai tesque, 31511-05) (pH 7.4) at RT for 15 min, and permeabilized in 0.5% NP-40 in PBS at RT for 10 min. For the γ-tubulin staining, the cells were cultured on the Concanavalin A (Sigma, L7647) coated chamber slides for 30 min to promote cell attachment and fixed with 3% PFA in 250 mM HEPES-NaOH (pH 7.4) at RT for 10 s and Methanol at –25 °C for 10 min. After blocking with 0.5% BSA (Equitech-Bio Inc, BAC62) in PBS for 5 min, the cells were incubated with primary antibodies diluted in 0.5% BSA in PBS at 37 °C for 1 h. The cells were washed three times with 0.5% BSA in PBS, incubated with secondary antibodies diluted in 0.5% BSA in PBS at 37 °C for 1 h, and washed three times with 0.5% BSA in PBS. The cells were post-fixed for 10 min. DNA was stained with 100 ng/mL DAPI in PBS for 20 min. The stained cells were mounted with VECTASHIELD Mounting Medium (Vector Laboratories, H-1000).
Primary antibodies used in immunofluorescence analysis were rabbit anti-chicken Knl134 (1:5000), rabbit anti-chicken CENP-T5 (1:2000), rabbit anti-chicken Bub134 (1:1000), rabbit anti-chicken Aurora B34 (1:5000), mouse anti-H3T3ph34 (1:10000), mouse anti-human Ndc80 (Abcam, ab3613) (1:1000), mouse anti-FLAG-M2 (Sigma, F1804) (1:1000), mouse anti-α-tubulin (Sigma T9026) (1:5000), rabbit anti-γ-tubulin (Sigma, T5192) (1:1000), FITC-conjugated mouse anti-α-tubulin (Sigma, F2168) (1:1000), mouse anti-human CENP-A69 (1:200), and rat anti-human CENP-T (immunogen: HNPDSDSTPRTLLRRVLDTC, a gift from Kinya Yoda, Nagoya University, Nagoya, Japan) (1:1000). Secondary antibodies used in immunofluorescence analysis were FITC-conjugated goat anti-rabbit IgG F(ab’)2 (1:1000), FITC-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, 115-095-003) (1:1000), FITC-conjugated goat anti-rat IgG (Jackson ImmunoResearch, 112-095-003) (1:1000), Cy3-conjugated mouse anti-rabbit IgG (Jackson ImmunoResearch, 211-165-109) (1:1000), Cy3-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, 115-165-003) (1:1000), and Alexa647-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, 115-605-003) (1:1000).
For counterstaining of DNA, 100 ng/mL DAPI (Roche, 10236276001) in PBS was used for 20 min incubation. The stained cells were mounted with VECTASHIELD Mounting Medium.
Chicken CENP-C antibody generation
The chicken CENP-C fragment (amino acids 1-302) was cloned into pET30b (Merk, 69910). The His-tag fused chicken CENP-C (1-302) protein was expressed in an Escherichia coli cell line, Rosetta2 (DE3) (Merk, C2527I). Cells expressing His-tag fused chicken CENP-C (1-302) were harvested by centrifugation, resuspended in binding buffer (20 mM Tris-HCl (pH 7.5), 500 mM NaCl, 5 mM Imidazole (Wako, 095-00015), and 1xcomplete EDTA-free proteinase inhibitor), and lysed by sonication. The lysate was clarified by centrifugation and the supernatant was incubated with Ni-NTA beads (Qiagen, 30210). After washing Ni-NTA beads with wash buffer (20 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 40 mM Imidazole), bound proteins were eluted with elute buffer (20 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 800 mM Imidazole). The elution was dialyzed against PBS, concentrated using Amicon-Ultra-15 10 K (Merk, UFC901008), frozen in liquid nitrogen and stored at −80 °C. The purified protein was sent to Cosmo bio to raise antibodies against chicken CENP-C in rabbits.
Chromosome spread
For the preparation of nocodazole treated samples, the cells were cultured with 100 ng/mL nocodazole for 14 h or 500 ng/mL nocodazole for 4 h. For the preparation of MG132 (Sigma, C2211) treated samples, the cells with cultured with 100 ng/mL nocodazole for 10 h, washed with 1xPBS three times, and treated with 10 µM MG132 for 4 h. Nocodazole or MG132 treated cells were collected and suspended in 0.56% hypotonic buffer (40 mM KCl (Nacalai tesque, 28514-75), 10 mM HEPES/NaOH (pH 7.4), 0.5 mM EDTA (Nacalai tesque, 15111-45)) (Fig. 2h, 2i, 3c, 3d, 3e, 5h, Supplementary Fig. 5e, 5f, 5g). The cells (3.5–4 × 105 cells) were cytospan onto slide glasses or coverslips by the Cytospin3 centrifuge, and fixed with 3% PFA in 250 mM HEPES-NaOH (pH 7.4) at RT for 15 min. The samples were subjected to immunofluorescence analysis.
Microtubule staining
For the microtubule staining of DT40 cells, the cells were cultured on the Concanavalin A coated chamber slides for 30 min to promote cell attachment. Then, the cells were incubated with ice-cold medium for 10 min. After fixed with 3% PFA in 250 mM HEPES-NaOH (pH 7.4) at RT for 15 min, the samples were subjected to immunofluorescence analysis with an anti-α-tubulin antibody.
For the microtubule staining of RPE1 cells, the cells were cultured on the coverslips and incubated with the ice-cold medium for 10 min. After extracted with 0.2% triton X-100 (Sigma, T8787) in PHEM buffer (60 mM PIPES (Sigma, P1851), 25 mM HEPES, 10 mM EGTA (Sigma, E3889), and 2 mM MgCl2 (Nacalai tesque, 20909-55), pH 7.4) for 1 min, equal amount of 4% PFA in PHEM was added (final 2% PFA) and fixed for 5 min. After removed the solution, the cells were fixed with 4% PFA in PHEM for 5 min, washed with 0.1% Triton X-100 in PBS (PBSTX) for 5 min, blocked with 3% BSA in PBSTX for 5 min, and incubated with primary antibodies diluted in 3% BSA in PBSTX at 37 °C for 1 h. The cells were washed three times with PBSTX, incubated with secondary antibodies diluted with 3% BSA in PBSTX at 37 °C for 1 h, and washed three times with PBSTX. DNA was stained with 100 ng/mL DAPI in PBS for 20 min. The stained cells were mounted with VECTASHIELD Mounting Medium.
Image acquisition
Immunofluorescence images and DAPI staining images were acquired every 0.2 μm intervals of z-slice using a Zyla 4.2 sCMOS camera (Andor) mounted on a Nikon Eclipse Ti inverted microscope with an objective lens (Nikon; Plan Apo lambda 100×/1.45 NA) with a spinning disk confocal scanner unit (CSU-W1; Yokogawa) controlled with NIS-elements v4.60 (Nikon). The images in the Figures are the maximum intensity projection or sum intensity projection (chromosome spread samples) of the Z-stack generated with Fiji (v1.53)70 and were processed using Fiji (v1.53) and Adobe Photoshop v23.1.0 (Adobe).
Evaluation of chromosome alignment in DT40 cells
The cells were cultured on the Concanavalin A-coated chamber slides for 30 min to promote cell attachment. After fixed with 3% PFA in 250 mM HEPES-NaOH (pH 7.4) at RT for 15 min, the cells were subjected to immunofluorescence analysis. For the calculation of chromosome alignment value, positions of each kinetochore signal were plotted and linear approximation straight-line was drawn. Absolute values of a correlation coefficient were defined as a chromosome alignment value.
Flow cytometry analysis
The cells were treated with tetracycline (2 μg/mL) for 30 h and incubated with 20 μM BrdU (5-bromo-2-deoxyuridine; Sigma, 10280879001) for 20 min and harvested. The harvested cells were washed with the ice-cold PBS, fixed with ice-cold 70% ethanol and stored at –20 °C. The fixed cells were washed with 1% BSA/PBS, incubated in 4 N HCl with 0.5% Triton X-100 at RT for 30 min, and washed with 1% BSA/PBS three times. The cells were incubated with an anti-BrdU antibody (BD, 347580) at RT for 1 h and washed with 1% BSA/PBS three times. Then, the cells were incubated with FITC-conjugated antimouse IgG (Jackson ImmunoResearch, 115-095-003; at 1:20 dilution in 1% BSA/PBS) at RT for 30 min, washed with 1% BSA/PBS and stained DNA with propidium iodide (Sigma, P4170) (10 μg/mL) in 1% BSA/PBS at 4 °C overnight or RT for 4 h. The stained cells were applied to a Flow cytometry (Guava easyCyte: Merck). Obtained data were analyzed with an InCyte software (guava soft 3.1.1; Merck). The cell cycle gates were manually adjusted and the percentages of each cell cycle stage (G1-, S-, G2/M-phase or dead cell) were calculated.
Quantification, statistics, and reproducibility
The fluorescence signal intensities of GFP-Dsn1, Nuf2-GFP, Nuf2-mScarlet, anti-Bub1, anti-Knl1, VH-mScarlet, and anti-human Ndc80 on more than 60 kinetochores in each of 10 cells (Figs. 1g, 2b, 2d, 2f, 2g-i, 3a, 3b, 4a, 4e, 4h, 5c, 6c and Supplementary Fig. 5b-d) and background signals in nonkinetochore region for each sample were quantified using Imaris v9.0.2. (Bitplane). The mean of fluorescence signal intensities on kinetochores in each cell were subtracted with mean of background signals in non-kinetochore region. p values were calculated by two-tailed Welch’s t-test (Figs. 2d, 2g-I, 4e, 5c and Supplementary Fig. 5b-d) or one-way analysis of variance (ANOVA) followed by Tukey’s test (Fig. 1g, 2b, 2f, 3a, 3b, 4a, 4h and 6c).
The fluorescence signal intensities of H3T3ph and Aurora B in the centromere region and the noncentromere region were quantified using Fiji. The centromere region was selected by the 10 × 15 pixels (length × width) region along the chromosome axis (the region of interest: ROI-A). This region was enlarged with the 10 × 20 pixels (length × width) region (ROI-B) for background subtraction. To determine the background signal intensity, the sum of fluorescence signal intensity of ROI-B was subtracted with the sum of fluorescence signal intensity of ROI-A (ROI-B - ROI-A). The mean of fluorescence signal intensity on the centromere region (ROI-A) was subtracted with the mean of the background signal intensity. The noncentromere region was selected by the length size (pixel length size depends on length of chromosome arm) × 15 pixels (length × width) region along with chromosome axis. Background subtraction was performed in the same way as the centromere region. The fluorescence signals on 4 chromosomes in each cell (Fig. 3c: n = 14 (WT) and 13 (∆121–240) cells; Fig. 3d, Supplementary Fig. 5e, 5f: n = 13 cells) were quantified. p values were calculated by two-tailed Welch’s t-test.
The kinetochore-kinetochore distances of more than 10 mitotic chromosome in each of 10 cells (Figs. 3e, 5h, Supplementary Fig. 5g) in chromosome spread samples were measured using Imaris v9.0.2. (Bitplane). p values were calculated by two-tailed Welch’s t-test.
The chromosome alignment values were calculated as described in a section for “Evaluation of chromosome alignment in DT40 cells”. p values were calculated by one-way ANOVA followed by Tukey’s test (Figs.1j, 5f, 5l) or two-tailed Welch’s t-test (Supplementary Fig. 1n) (Fig. 1j: n = 41 (None), 46 (WT), and 46 (∆121-240) cells; 5 f: n = 42 (None), 41 (WT), 41 (∆121-240) and 41 (2X(1-90)_∆121-240) cells; 5 l: n = 37 (Dsn1WT), 35 (Dsn1∆326-349) and 42 (Dsn1∆326-349 + CENP-T2X(1-90)) cells; Supplementary Fig. 1n: n = 44 (Nocodazole), and 42 (MG132) cells).
Each experiment was repeated at least two (Figs. 1d, 1e, 1g-j, 2e, 2h, 2i, 3a, 3d, 3e, 4f, 4g, 5c, 5d, 5f-n, 6b-d, 6g and Supplementary Figs. 1c, 1e, 1g, 1i, 1k-o, 2b-e, 2h, 2k-m, 3a, 3b, 4c-e, 5a, 5e-h, 5j, 6a-f, 7c, 7e and 7f) or three times (Figs. 1k, 2b, 2d, 2f, 2g, 3b, 3c, 4a, 4d-e, 4h, 5e and Supplementary Fig. 5b-d). Representative data of technical replicates are presented.
The percentage of cells with misaligned chromosomes (Fig. 6e) or cells with abnormal chromosome (Fig. 6f) were calculated. Each experiment was repeated three times (Fig. 6e: n = 24 or 25 cells per experiment) or four times (Fig. 6f: n = 24 or 25 cells per experiment), and mean and standard deviation of three (Fig. 6e) or four (Fig. 6f) experiments are presented. p values were calculated by one-way ANOVA followed by Tukey’s test.
Statistical analyses were performed using GraphPad Prism7(GraphPad Software).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. All data supporting the findings of this study are available within the paper and supplementary information. Source data are provided with this paper.
References
Fukagawa, T. & Earnshaw, W. C. The centromere: chromatin foundation for the kinetochore machinery. Developmental cell 30, 496–508 (2014).
McKinley, K. L. & Cheeseman, I. M. The molecular basis for centromere identity and function. Nat. Rev. Mol. cell Biol. 17, 16–29 (2016).
Hara, M. & Fukagawa, T. Critical Foundation of the Kinetochore: The Constitutive Centromere-Associated Network (CCAN). Prog. Mol. Subcell. Biol. 56, 29–57 (2017).
Hara, M. & Fukagawa, T. Kinetochore assembly and disassembly during mitotic entry and exit. Curr. Opin. cell Biol. 52, 73–81 (2018).
Hori, T. et al. CCAN makes multiple contacts with centromeric DNA to provide distinct pathways to the outer kinetochore. Cell 135, 1039–1052 (2008).
Musacchio, A. & Desai, A. A Molecular View of Kinetochore Assembly and Function. Biology (Basel) 6, 5 (2017).
Cheeseman, I. M., Chappie, J. S., Wilson-Kubalek, E. M. & Desai, A. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127, 983–997 (2006).
DeLuca, J. G. et al. Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 127, 969–982 (2006).
Alushin, G. M. et al. The Ndc80 kinetochore complex forms oligomeric arrays along microtubules. Nature 467, 805–810 (2010).
Kiyomitsu, T., Obuse, C. & Yanagida, M. Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Developmental cell 13, 663–676 (2007).
Yamagishi, Y., Yang, C. H., Tanno, Y. & Watanabe, Y. MPS1/Mph1 phosphorylates the kinetochore protein KNL1/Spc7 to recruit SAC components. Nat. cell Biol. 14, 746–752 (2012).
Vleugel, M. et al. Sequential multisite phospho-regulation of KNL1-BUB3 interfaces at mitotic kinetochores. Mol. cell 57, 824–835 (2015).
Foltz, D. R. et al. The human CENP-A centromeric nucleosome-associated complex. Nat. cell Biol. 8, 458–469 (2006).
Okada, M. et al. The CENP-H-I complex is required for the efficient incorporation of newly synthesized CENP-A into centromeres. Nat. cell Biol. 8, 446–457 (2006).
Weir, J. R. et al. Insights from biochemical reconstitution into the architecture of human kinetochores. Nature 537, 249–253 (2016).
Pesenti, M. E. et al. Reconstitution of a 26-Subunit Human Kinetochore Reveals Cooperative Microtubule Binding by CENP-OPQUR and NDC80. Mol. cell 71, 923–939 e910 (2018).
Saitoh, H. et al. CENP-C, an autoantigen in scleroderma, is a component of the human inner kinetochore plate. Cell 70, 115–125 (1992).
Fukagawa, T., Pendon, C., Morris, J. & Brown, W. CENP-C is necessary but not sufficient to induce formation of a functional centromere. EMBO J. 18, 4196–4209 (1999).
Kwon, M. S., Hori, T., Okada, M. & Fukagawa, T. CENP-C is involved in chromosome segregation, mitotic checkpoint function, and kinetochore assembly. Mol. Biol. cell 18, 2155–2168 (2007).
Klare, K. et al. CENP-C is a blueprint for constitutive centromere-associated network assembly within human kinetochores. J. cell Biol. 210, 11–22 (2015).
Watanabe, R. et al. CDK1-mediated CENP-C phosphorylation modulates CENP-A binding and mitotic kinetochore localization. J. cell Biol. 218, 4042–4062 (2019).
Ariyoshi, M. et al. Cryo-EM structure of the CENP-A nucleosome in complex with phosphorylated CENP-C. EMBO J. 40, e105671 (2021).
Nishino, T. et al. CENP-T-W-S-X forms a unique centromeric chromatin structure with a histone-like fold. Cell 148, 487–501 (2012).
Takeuchi, K. et al. The centromeric nucleosome-like CENP-T-W-S-X complex induces positive supercoils into DNA. Nucleic acids Res. 42, 1644–1655 (2014).
Nagpal, H. et al. Dynamic changes in CCAN organization through CENP-C during cell-cycle progression. Mol. Biol. cell 26, 3768–3776 (2015).
McKinley, K. L. et al. The CENP-L-N Complex Forms a Critical Node in an Integrated Meshwork of Interactions at the Centromere-Kinetochore Interface. Mol. cell 60, 886–898 (2015).
Pentakota, S. et al. Decoding the centromeric nucleosome through CENP-N. eLife 6, e33442 (2017).
Chittori, S. et al. Structural mechanisms of centromeric nucleosome recognition by the kinetochore protein CENP-N. Science 359, 339–343 (2018).
Tian, T. et al. Molecular basis for CENP-N recognition of CENP-A nucleosome on the human kinetochore. Cell Res. 28, 374–378 (2018).
Cheeseman, I. M., Hori, T., Fukagawa, T. & Desai, A. KNL1 and the CENP-H/I/K complex coordinately direct kinetochore assembly in vertebrates. Mol. Biol. cell 19, 587–594 (2008).
Basilico, F. et al. The pseudo GTPase CENP-M drives human kinetochore assembly. eLife 3, e02978 (2014).
Hori, T., Okada, M., Maenaka, K. & Fukagawa, T. CENP-O class proteins form a stable complex and are required for proper kinetochore function. Mol. Biol. cell 19, 843–854 (2008).
Gascoigne, K. E. et al. Induced ectopic kinetochore assembly bypasses the requirement for CENP-A nucleosomes. Cell 145, 410–422 (2011).
Hori, T., Shang, W. H., Takeuchi, K. & Fukagawa, T. The CCAN recruits CENP-A to the centromere and forms the structural core for kinetochore assembly. J. cell Biol. 200, 45–60 (2013).
Hara, M., Ariyoshi, M., Okumura, E. I., Hori, T. & Fukagawa, T. Multiple phosphorylations control recruitment of the KMN network onto kinetochores. Nat. cell Biol. 20, 1378–1388 (2018).
Kim, S. & Yu, H. Multiple assembly mechanisms anchor the KMN spindle checkpoint platform at human mitotic kinetochores. J. cell Biol. 208, 181–196 (2015).
Rago, F., Gascoigne, K. E. & Cheeseman, I. M. Distinct organization and regulation of the outer kinetochore KMN network downstream of CENP-C and CENP-T. Curr. Biol. 25, 671–677 (2015).
Suzuki, A., Badger, B. L. & Salmon, E. D. A quantitative description of Ndc80 complex linkage to human kinetochores. Nat. Commun. 6, 8161 (2015).
Nishino, T. et al. CENP-T provides a structural platform for outer kinetochore assembly. EMBO J. 32, 424–436 (2013).
Malvezzi, F. et al. A structural basis for kinetochore recruitment of the Ndc80 complex via two distinct centromere receptors. EMBO J. 32, 409–423 (2013).
Huis In ‘t Veld, P. J. et al. Molecular basis of outer kinetochore assembly on CENP-T. eLife 5, e21007 (2016).
Kline, S. L., Cheeseman, I. M., Hori, T., Fukagawa, T. & Desai, A. The human Mis12 complex is required for kinetochore assembly and proper chromosome segregation. J. cell Biol. 173, 9–17 (2006).
Petrovic, A. et al. The MIS12 complex is a protein interaction hub for outer kinetochore assembly. J. cell Biol. 190, 835–852 (2010).
Yamagishi, Y., Honda, T., Tanno, Y. & Watanabe, Y. Two histone marks establish the inner centromere and chromosome bi-orientation. Science 330, 239–243 (2010).
Kawashima, S. A., Yamagishi, Y., Honda, T., Ishiguro, K. & Watanabe, Y. Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science 327, 172–177 (2010).
Kelly, A. E. et al. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science 330, 235–239 (2010).
Wang, F. et al. Histone H3 Thr-3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science 330, 231–235 (2010).
Petrovic, A. et al. Structure of the MIS12 Complex and Molecular Basis of Its Interaction with CENP-C at Human Kinetochores. Cell 167, 1028–1040 e1015 (2016).
del Rio, A. et al. Stretching single talin rod molecules activates vinculin binding. Science 323, 638–641 (2009).
Ye, A. A., Cane, S. & Maresca, T. J. Chromosome biorientation produces hundreds of piconewtons at a metazoan kinetochore. Nat. Commun. 7, 13221 (2016).
Volkov, V. A., Huis In ‘t Veld, P. J., Dogterom, M. & Musacchio, A. Multivalency of NDC80 in the outer kinetochore is essential to track shortening microtubules and generate forces. eLife 7, e36764 (2018).
Hara, M. & Fukagawa, T. Where is the right path heading from the centromere to spindle microtubules? Cell cycle 18, 1199–1211 (2019).
Sridhar, S., Hori, T., Nakagawa, R., Fukagawa, T. & Sanyal, K. Bridgin connects the outer kinetochore to centromeric chromatin. Nat. Commun. 12, 146 (2021).
Hamilton, G. E. & Davis, T. N. Biochemical evidence for diverse strategies in the inner kinetochore. Open Biol. 10, 200284 (2020).
Moore, L. L. & Roth, M. B. HCP-4, a CENP-C-like protein in Caenorhabditis elegans, is required for resolution of sister centromeres. J. cell Biol. 153, 1199–1208 (2001).
Orr, B. & Sunkel, C. E. Drosophila CENP-C is essential for centromere identity. Chromosoma 120, 83–96 (2011).
Drinnenberg, I. A., Henikoff, S. & Malik, H. S. Evolutionary Turnover of Kinetochore Proteins: A Ship of Theseus? Trends cell Biol. 26, 498–510 (2016).
Navarro-Mendoza, M. I. et al. Early Diverging Fungus Mucor circinelloides Lacks Centromeric Histone CENP-A and Displays a Mosaic of Point and Regional Centromeres. Curr. Biol. 29, 3791–3802 e3796 (2019).
Cortes-Silva, N. et al. CenH3-Independent Kinetochore Assembly in Lepidoptera Requires CCAN, Including CENP-T. Curr. Biol. 30, 561–572 e510 (2020).
Nicklas, R. B. The forces that move chromosomes in mitosis. Annu Rev. Biophys. Biophys. Chem. 17, 431–449 (1988).
Powers, A. F. et al. The Ndc80 kinetochore complex forms load-bearing attachments to dynamic microtubule tips via biased diffusion. Cell 136, 865–875 (2009).
Kudalkar, E. M. et al. Regulation of outer kinetochore Ndc80 complex-based microtubule attachments by the central kinetochore Mis12/MIND complex. Proc. Natl Acad. Sci. USA 112, E5583–E5589 (2015).
Buerstedde, J. M. et al. Light chain gene conversion continues at high rate in an ALV-induced cell line. EMBO J. 9, 921–927 (1990).
Concordet, J. P. & Haeussler, M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic acids Res. 46, W242–W245 (2018).
Nishimura, K., Fukagawa, T., Takisawa, H., Kakimoto, T. & Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. methods 6, 917–922 (2009).
Nishimura, K. & Fukagawa, T. An efficient method to generate conditional knockout cell lines for essential genes by combination of auxin-inducible degron tag and CRISPR/Cas9. Chromosome Res. 25, 253–260 (2017).
Nishimura, K. et al. A super-sensitive auxin-inducible degron system with an engineered auxin-TIR1 pair. Nucleic acids Res. 48, e108 (2020).
Hori, T., Haraguchi, T., Hiraoka, Y., Kimura, H. & Fukagawa, T. Dynamic behavior of Nuf2-Hec1 complex that localizes to the centrosome and centromere and is essential for mitotic progression in vertebrate cells. J. cell Sci. 116, 3347–3362 (2003).
Obuse, C. et al. Proteomics analysis of the centromere complex from HeLa interphase cells: UV-damaged DNA binding protein 1 (DDB-1) is a component of the CEN-complex, while BMI-1 is transiently co-localized with the centromeric region in interphase. Genes Cells 9, 105–120 (2004).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. methods 9, 676–682 (2012).
Acknowledgements
The authors are very grateful to Yuko Fukagawa, Kaori Oshimo, Yumiko Takeshita and Reika Fukuoka for their technical assistance. We also thank Thomas Maresca for providing plasmid constructs. This work was supported by CREST of JST (JPMJCR21E6), JSPS KAKENHI Grant Numbers 16H06279, and 17H06167, 20H05389, 21H05752 to TF, JSPS KAKENHI Grant Numbers 16K18491, and 21H02461 to MH.
Author information
Authors and Affiliations
Contributions
Y.T. designed and performed all experiments in this study under the supervision of M.H. and T.F. T.F. wrote the manuscript in collaboration with Y.T., discussing it with M.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ajit Joglekar and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Takenoshita, Y., Hara, M. & Fukagawa, T. Recruitment of two Ndc80 complexes via the CENP-T pathway is sufficient for kinetochore functions. Nat Commun 13, 851 (2022). https://doi.org/10.1038/s41467-022-28403-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28403-8
This article is cited by
-
Molecular cloning, subcellular localization, and rapid recruitment to DNA damage sites of chicken Ku70
Scientific Reports (2024)
-
Higher-order protein assembly controls kinetochore formation
Nature Cell Biology (2024)
-
Structure of the human KMN complex and implications for regulation of its assembly
Nature Structural & Molecular Biology (2024)
-
Structure of the human outer kinetochore KMN network complex
Nature Structural & Molecular Biology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
