
Abstract
Single-atom catalysts anchoring offers a desirable pathway for efficiency maximization and cost-saving for photocatalytic hydrogen evolution. However, the single-atoms loading amount is always within 0.5% in most of the reported due to the agglomeration at higher loading concentrations. In this work, the highly dispersed and large loading amount (>1 wt%) of copper single-atoms were achieved on TiO2, exhibiting the H2 evolution rate of 101.7 mmol g−1 h−1 under simulated solar light irradiation, which is higher than other photocatalysts reported, in addition to the excellent stability as proved after storing 380 days. More importantly, it exhibits an apparent quantum efficiency of 56% at 365 nm, a significant breakthrough in this field. The highly dispersed and large amount of Cu single-atoms incorporation on TiO2 enables the efficient electron transfer via Cu2+-Cu+ process. The present approach paves the way to design advanced materials for remarkable photocatalytic activity and durability.
Similar content being viewed by others

Introduction
Hydrogen evolution from solar-driven water splitting is a promising alternative strategy for future energy convertion1,2,3,4. Titanium oxide (TiO2) has been vastly employed5 and is still used as a benchmark photocatalyst for water splitting6 as it is low cost and more importantly, extremely stable and high efficiency under UV. Even so, TiO2 still suffers from high charge-carrier recombination and sluggish proton reduction kinetics7. Hence, addressing these issues is essential to enhance the charge separation and to promote H2 fuel synthesis. Several strategies such as doping, defect engineering, heterojunction formation, morphology variation, etc., have been reported to reduce the charge-carrier recombination and improve the H2 evolution8. Amongst them, loading a cocatalyst on the surface of the TiO2 is proved as an appropriate approach to enhance the charge separation through the established metal-semiconductor Schottky junction, which not only extracts the photogenerated electrons but also dramatically reduces the energy barrier for proton reduction.
Noble metals such as Pt, Au, and Pd are commonly used as cocatalysts in photocatalysis due to their low activation energy and efficient charge separation. For example, Pt loading improved TiO2 for H2 production by a factor of 129. However, they are not only rare elements and hence high cost but also the efficiency achieved is still moderate. Recently, there were many studies on earth-abundant transition metals (e.g., Cu, Ni, Co, and Fe) to substitute these noble metals as a suitable alternative for photocatalysis10,11,12,13. On the other hand, all these cocatalyst-loaded photocatalysts still struggle to achieve a breakthrough inefficiency due to the low-atom utilization while the cocatalysts are in their bulk composition. Very recently, single-atom catalysts (SACs) have been highly focused due to maximizing the reaction active sites, resembling the homogeneous catalysis14,15. The isolated and active metal atoms anchored onto the photocatalysts offer more water-molecule adsorption and active sites. So far, SACs loaded TiO2 have been investigated for H2 evolution6,16, CO2 reduction17, and dye degradation18. However, the aggregation of SACs is inevitable during the catalytic reaction due to their high surface energy or leaching due to the unstable anchoring as the majority were synthesized by post-treatment (e.g., impregnation approach)19,20,21. More importantly, the larger the percentage of SACs, the higher the activity, whereas to load higher than 0.5 wt% of SACs is very challenging as the majority of the studies reported a limited amount of SACs (usually near 0.1–0.3 wt %)1,2 onto the high surface area of substrates and it is difficult to control and reproduce4,22,23. Hence, obtaining the highly dispersed and high concentration of SACs remains to be the main bottleneck in photocatalytic H2 production.
The easily-changed valence states of Cu nanoparticles have been a promising candidate for efficient charge separation and transfer, leading to higher catalytic performance compared to even noble metal loaded TiO2 samples24,25,26,27. A recent wrap-bake-peel process, using SiO2@M/TiO2@SiO2 as the intermediate following NaOH etching to produce Cu SACs, has achieved a benchmark apparent quantum efficiency (AQE) of 45.5% at 340 nm6. It is due to the reduction and regeneration of the active sites during the catalytic cycle. Such a success stimulates us to investigate a more efficient strategy to stabilize Cu SACs and more importantly to generate an in-situ self-heal approach for continuous H2 production from water, thus no need for the regeneration step. To achieve this, the intact interaction between the single atoms and the support is crucial to obtain atomically evenly-dispersed Cu28,29.
Here, we have developed a bottom-up approach, which is different from the post-treatment approach reported in the literature including the very recent report to obtain evenly dispersed SACs on the substrate6,30. The metal-organic framework (MOF) MIL-125 was first synthesized using it as a precursor. Then metal ions were anchored into the MOF MIL-125 to generate a metal-oxygen-titanium bond, which is the key to ensuring uniformly immobilized metal SAC on the final catalysts31,32,33. Finally, the metal-MIL-125 intermediates were calcined to synthesize the final photocatalysts. This new strategy ensures atomic dispersion of metal cocatalyst and enables to achieve a higher loading amount ~1.5 wt%. The optimised sample shows the photocatalytic H2 evolution rate of 101.7 mmol g−1 h−1 (or 2.03 mmol h−1). To make a straightforward comparison with the reported H2 evolution rate, we converted it to the widely used unit of mmol per unit mass per unit time under simulated solar light irradiation, somewhat higher than the best photocatalyst PtSA-TiO2 (95.3 mmol g−1 h−1). The CuSA-TiO2 exhibits AQE of 56% at 365 nm irradiation, exceeding all the state-of-the-art TiO2-based photocatalysts (AQE of 4.3–45.5%6,34,35).
Results
Photocatalytic properties of CuSA-TiO2
First, four sets of experiments were performed for AQE optimisation and the detailed conditions were presented in Supplementary Fig. 1. The AQE results of CuSA-TiO2 under different wavelength light irradiation (365 nm, 385 nm, 420 nm, and 520 nm) are shown in Supplementary Fig. 1a. It indicates a decrease with increasing the wavelength followed by a slight increase at 520 nm due to the Cu-induced defects absorption (Supplementary Fig. 14). Also, the AQE measurement using different amounts of the photocatalyst was carried out, indicating the CuSA-TiO2 mass can affect AQE (Supplementary Fig. 1b). The changing trend is likely due to the fact that the higher photocatalyst mass would scatter more light when it is over 50 mg. With the 2:1 methanol:water ratio, the AQE at different light intensities was also tested (Supplementary Fig. 1d). The AQE result collected from various ratios of methanol: water indicates that methanol facilitates the H2 evolution from water. When the light intensity increases from 200 W/m2 to 500 W/m2, the AQE shows a slight increase. Hence, The CuSA-TiO2 represents an efficient and low-cost photocatalyst for continuous renewable H2 production.
First, the reference MIL-125 derived TiO2 was tested for photocatalytic H2 evolution under Xe lamp using methanol as a hole-scavenger. Fig. 1a shows the H2 evolution activities of TiO2 and other photocatalysts loaded with different metals such as Co, Ni, Fe, Mn, Zn, and Pt (0.75 wt% metal to precursor MIL-125(Tiv) before sintering). The samples were further analysed to determine the real amount of metal on TiO2 by an inductively coupled plasma test (Supplementary Table 1). All of them produce H2 higher than that of pristine TiO2 (4.2 mmol g−1 h−1) except Zn and Mn-TiO2, revealing that the metal single-atoms introduction plays a crucial role in the H2 evolution reaction. The activity order is CuSA-TiO2 (101.7 mmol g−1 h−1) > PtSA-TiO2 (95.3 mmol g−1 h−1) > FeSA-TiO2 (19.1 mmol g−1 h−1) > NiSA-TiO2 (12.0 mmol g−1 h−1) > CoSA-TiO2 (8.2 mmol g−1 h−1) > TiO2 (4.2 mmol g−1 h−1) > MnSA-TiO2 (2.3 mmol g−1 h−1) > ZnSA-TiO2 (2.2 mmol g−1 h−1). Interestingly, the CuSA-TiO2 evolve higher H2 than the benchmark Pt-loaded TiO2. The higher H2 evolution rate observed for the CuSA-TiO2 is due to the highly dispersed Cu SACs as proved later and their charge separation and catalytic effect as discussed below. Moreover, the larger loading amount of Cu SACs (1.5 wt%) compared with Pt SACs (0.64%) allows maximum utilization of the active sites to realize such an amazing activity, which might be ascribed to the easier coordination of dissociated Cu2+ with oxygen compared with [PtCl4]2−. Furthermore, the weight percentage of the Cu attached to the TiO2 was optimized as shown in Fig. 1b, and the highest H2 evolution activity is obtained on ca. 1.5 wt% CuSA-TiO2 (Supplementary Table 1). More importantly, an unprecedented AQE of 56% at 365 nm has been achieved on the ca. 1.5 wt% CuSA-TiO2, a new record. These results indicate that both the large atomic weight percentage and interaction of Cu with TiO2 are crucial for the extraordinary H2 evolution.

The photocatalytic H2 evolution rate of (a) TiO2 (non) and M-TiO2 derived from M-MiL-125(Tiv). b TiO2 and TiO2 loaded with different ratios of Cu SACs. c The photocatalytic activity of the ca. 1.5 wt% CuSA-TiO2 for six cyclic water splitting experiments and the last run is the activity of the sample after storing in the lab for 380 days. d The photocatalytic H2 evolution mechanism on 1.5 wt% CuSA-TiO2. e The corresponding schematic representation of the formation of copper SAC in the Ti lattice of TiO2, together with the related images.
While increasing the Cu atoms amount beyond ca. 1.5 wt%, the decreased photocatalytic activity was observed. It is perhaps due to the limited pores available for higher Cu precursor anchoring, leading to screening the irradiated light. To show the experimental evidence of excessive Cu accumulation, we measured the HRTEM for 2.57 wt% CuSA-TiO2 and 1.5 wt% CuSA-TiO2, compared with pristine TiO2. Supplementary Figure 10 shows the patterns of 1.5 wt% CuSA-TiO2, which is similar to the pristine TiO2. While clear patterns of Cu nanoparticles (about 2–5 nm) observed in 2.57 wt% CuSA-TiO2 (Supplementary Fig. 2) are marked with arrows and circles. Hence, the poor activity observed for the Cu atom amount beyond ca. 1.5 wt% might be ascribed to the Cu accumulation, which inhibits the incident light from reaching the photocatalyst surface.
The long-term stability and reproducibility of the H2 evolution using the optimised CuSA-TiO2 were analyzed by the six consecutive photocatalytic water-splitting experiments under simulated solar light irradiation (Fig. 1c). In addition, the long-term experiment i.e., 20 days were also carried out for the stability test and results are shown in Supplementary Fig. 3. The sample was kept in the solution for each cycle, overall lasting for 20 days. The sample exhibits similar activity. Moreover, the photocatalyst after the long-term test was characterized by the ICP-AES, listed in Supplementary Table 1. The Cu amount of CuSA-TiO2 after the long-term test was estimated to be 1.54 wt%, which is similar to that of the fresh sample. The long-term activity and ICP-AES result further confirm the long-term stability and reproducibility of CuSA-TiO2.
It can be seen that no noticeable decrease was observed in the H2 evolution rate, suggesting that the prepared sample is highly stable and the results are reproducible. Furthermore, the photocatalytic activity of the 1.5 wt% CuSA-TiO2 after 380-day-storage in the lab was tested and we found that it remains the same as of the freshly prepared sample (Fig. 1c). Supplementary Table 2 lists the very recent progress on TiO2-based photocatalysts for water splitting, indicating TiO2 is still a highly attractive photocatalyst. Compared with these reports, one can see that our CuSA-TiO2 sample is two times more active than the reported ca 1 wt% Pt atom-TiO2 and six times better than the recently reported Cu-TiO2 in terms of specific mass evolution rate. Another finding is that it presents a record AQE of 56%, which is also more stable (>30 h). Such enhancement is believed to be due to the unique Cu states on TiO2 prepared by our pre-encapsulation synthesis approach.
Morphology and structure characterization of CuSA-TiO2
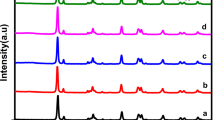
To explore the science behind the outstanding photocatalytic performance of 1.5 wt% CuSA-TiO2 (named CuSA-TiO2 subsequently), a series of studies were performed. Firstly, the X-ray diffraction (XRD) patterns of the as-prepared MIL-125(Tiv) were observed as shown in Fig. 2a, which agrees well with the earlier report36, suggesting that the MOF precursor was prepared successfully. The precursor MOF (MIL-125(Tiv)) was also observed with a regular cake-like morphology, the high specific surface area (SSA, 1361 m2 g−1, Supplementary Fig. 4a–b) and favorable pores31, which facilitates the Cu ions anchoring, leading to the formation of CuSA-TiO2 with the SSA of 294 m2 g−1 (Supplementary Fig. 4c–d). After incorporating the Cu in MIL-125(Tiv), the XRD pattern is nearly identical to that of the pristine MIL-125(Tiv), indicating that Cu2+ is encapsulated into the framework of MIL-125(Tiv) with high dispersity38. The same conclusion can be drawn from the sintered samples (TiO2 and CuSA-TiO2). Furthermore, the addition of Cu does not change the phase of TiO237, so that there is only an anatase crystal structure existing in the CuSA-TiO2. Furthermore, the Zeta potential test of MIL-125(Tiv) indicates a potential value of −40.6 mV (Supplementary Fig. 5), that means the dispersion of MIL-125(Tiv) in water is stable with such large negative charges. More importantly, the positive Cu2+ ions can be readily electrostatically bonded to the Ti vacancies of MIL-125(Tiv) via M-O-Ti structure after adding the CuCl2 precursor aqueous solution. The Cu-encapsulated MIL-125(Tiv) (Cu-MIL-125(Tiv)) was then sintered at 450 °C to form Cu SACs on TiO2 (CuSA-TiO2). Such catalyst synthesis temperature was derived from the thermogravimetric differential thermal (TG-DTA) measurement (Supplementary Fig. 6). When the temperature is higher than 450 °C (black curve), the weight loss tends to be stable, which might be explained by the removal of organic ligands and stabilisation of Cu species at that temperature. Moreover, the actual ratio of Cu SACs on TiO2 is up to 1.5wt%, indicating the proposed strategy not only ensures the highly dispersed Cu anchoring but also achieves a rather large loading amount of Cu SACs.

a The XRD images of MIL-125, Cu-MIL-125, TiO2, and CuSA-TiO2. b HAADF STEM raw image of CuSA-TiO2. (Insert: low magnification TEM images of CuSA-TiO2). c Filtered HAADF STEM image from the area highlighted with a red rectangle in b and the corresponding line scan profiles. d–f Line 1, Line 2, and Line 3 marked in Fig. c. g STEM-EDS mapping of Ti, O, and Cu of the fresh CuSA-TiO2.
Figure 1e shows the synthesis of the final catalyst from the intermediate MOF. The cake-shaped morphology of the intermediates (Supplementary Figure 7) was collapsed to form TiO2 or CuSA-TiO2 with clear lattice fringes, upon calcination of the MIL-125(Tiv), or Cu-MIL-125(Tiv). Supplementary Figs. 8 and 9 show the transmission electron microscopy (TEM) image of the MIL-125 derived TiO2 and the CuSA-TiO2, respectively. The collapsed morphology has significant merits as it can allow more Cu atoms exposure. The incorporation of Cu atoms on the Ti vacancy sites in the intermediate MOF is important as indicated in Fig. 1e, which not only stabilises Cu ions but also ensures single atom distribution of Cu in the prepared photocatalysts. This was investigated by the high-angle annular dark-field (HAADF) STEM (Fig. 2b–c, Supplementary Figs. 10–12). The bright contrast spots can be clearly seen only on the Ti atomic row. These bright spots are attributed to Cu as indicated in Supplementary Fig. 11f–i, confirming that Cu atoms are exclusively present in the Ti vacancies and other Cu configurations (e.g., clusters or nanoparticles) are not detected8. The line scan profiles marked with three blue lines randomly selected are shown in Fig. 2d–f, where lines 1 only contains Ti atoms while line 2 and 3 have both Ti and Cu atoms, confirming that there are Cu-O-Ti clusters.
The STEM-EDS mapping (Fig. 2g, Supplementary Fig. 12e) and EDS spectrum of CuSA-TiO2 (Supplementary Fig. 13) verify the existence of Ti, O, and Cu elements. Due to the porous nature and unsaturated bonds of MIL-125(Tiv), the Cu single atoms can be well stabilized. The stability of Cu atoms is further studied by measuring the HAADF-STEM of CuSA-TiO2 after 24-h photocatalytic reaction (Supplementary Figure 12). The line scan profile and STEM-EDS demonstrates that CuSA is still well dispersed on TiO2, confirming the excellent stability and strong anchoring of CuSA on TiO2, which is the key reason that our photocatalyst not only shows the record activity but also does not require regeneration as reported by others6.
Photo-electric characterization of CuSA-TiO2
UV-visible absorption spectroscopy (UV-vis) was used to explore the optical properties of TiO2 and CuSA-TiO2. The pristine TiO2 shows an absorption edge starting from 380 nm, corresponding to its wide bandgap transition (Supplementary Fig. 14). After loading CuSA, CuSA-TiO2 exhibits an obvious absorption in the visible region as well, a broad hump between 400 nm and 1050 nm, which is attributed to the d-d transition of Cu2+ state6. It is also evidenced from Supplementary Fig. 14 that the TiO2 absorption remains unchanged after loading CuSA. The transfer and separation efficiency of photogenerated charge carriers was studied by photoluminescence (PL) experiments. Figure 3a shows the PL spectra of TiO2 and CuSA-TiO2 excited at 375 nm. It is obvious that the pristine TiO2 shows high emission intensity due to the serious charge carrier recombination, which is significantly reduced after loading CuSA, indicating that the CuSA loading might effectively facilitate the photogenerated electrons extraction to the Cu active sites39,40. It is possible because the reduction potential of Cu2+/Cu+ (0.16 V vs. NHE) is more positive than the conduction band of TiO2 (−0.1 V vs. NHE)41,42.

a Photoluminescence spectra of TiO2 and CuSA-TiO2 under an excitation wavelength of 375 nm. b In-situ Cu 2p XPS of CuSA-TiO2. c In-situ electron paramagnetic resonance spectra of CuSA-TiO2 with various states. d Transient absorption spectra of TiO2 and CuSA-TiO2 in the absence and presence of AgNO3 (2 mM), monitored in argon atmosphere after 320 nm excitation. e Charge dynamics decay in TiO2 and CuSA-TiO2 with and without methanol (10%) monitored at 650 nm. f The spectra of H2, HD, and D2 evolution on CuSA-TiO2 from the solution of CD3OD in H2O. g The spectra of H2, HD, and D2 evolution on CuSA-TiO2 from the solution of CH3OH in D2O.
PL lifetime is the average time that the fluorophore stays in the excited state before emission occurs43. The instrument response function (IRF) was first measured to be 530 ps (Supplementary Fig. 15). The PL decay tests for TiO2 and CuSA-TiO2 were then performed with 375 nm excitation beam taking into account TiO2 absorption spectra (Supplementary Fig. 15) and observed at 430 nm. The decay kinetics were fitted with a biexponential function as shown in Supplementary Table 3 after considering this IRF: I(t) = B1 exp(−t/τ1) + B2 exp(−t/τ2), where τ1 and τ2 are the decay times for the faster and slower components, respectively, B1 and B2 are the contributions of each component. The carrier lifetime of TiO2 and CuSA-TiO2 were determined to be 3.82 ns and 2.04 ns, respectively. The CuSA-TiO2 shows a shorter PL lifetime compared to that of TiO2, revealing the faster electron transfer in CuSA-TiO222,44.
To further understand the charge carrier separation, photoelectrochemical experiments were performed and the photocurrents observed for TiO2 and CuSA-TiO2 are shown in Supplementary Fig. 16a. CuSA-TiO2 exhibits 3 times higher photocurrent than the pristine TiO2, indicating the more efficient carrier separation in CuSA-TiO2 following photoexcitation. This might be due to Cu-O as the bridge for more efficient carrier transfer to Cu active sites for H2 generation45. In addition, the photocurrent response of CuSA-TiO2 shows high reproducibility and stability for several on/off cycles. Moreover, the electrochemical impedance spectra (EIS) were used to study the transfer properties of charge carriers46, as indicated in Supplementary Fig. 16b. CuSA-TiO2 shows similar but dramatically decreased EIS Nyquist curves compared to pristine TiO2, indicating the CuSA species serve as electron acceptors, thus facilitating the interfacial charge separation. The efficient electron mobility is possibly ascribed to the reversible redox process between Cu2+ and Cu+ in CuSA-TiO2. Then, the electrochemical linear sweep voltammetry of CuSA-TiO2 was performed and shown in Supplementary Fig. 17, showing an overpotential of −0.72 V vs RHE for H2 production.
Valence state characterization of Cu SAs
To examine the real chemical states of the photocatalysts during the photocatalytic reaction, in-situ x-ray photoelectron spectroscopy (XPS) spectra of CuSA-TiO2 before and during the reaction were monitored (Fig. 3b). The Cu 2p before irradiation appears to be composed of four Gaussian peaks, where 952.9 eV and 932.9 eV are assigned to the Cu 2p1/2 and Cu 2p3/2 of Cu2+ (70.58%, Supplementary Table 4), respectively. Another set of peaks at 951.5 eV and 931.8 eV are likely associated with Cu+ species37 (29.42%), illustrating the Cu atoms bonded with TiO2 rather than physical adsorption on the surface. After 30 min irradiation, the Cu+ content increases to 61.68% and remains at 62–66% after that till the end of the reaction. Interestingly, after exposing the used catalyst in the air for 10 min, the ratio of Cu+ decreases to 53.45%, and the satellite peak of Cu2+ appears, which is ascribed to the Cu+ is partially oxidised to Cu2+ again. Also, Ti 2p and O 1 s of CuSA-TiO2 and reference PtSA-TiO2 before and after 30 min irradiation were compared in Supplementary Fig. 18. For CuSA-TiO2, the content of surface Ti3+ increases from 0.00% to 47.02% as well as oxygen vacancy (Vo) increases from 8.81% to 18.21% due to light irradiation (Supplementary Table 5), that is believed due to the photogenerated electrons accumulation on both Ti4+ and Cu2+ species47,48. For PtSA-TiO2, only 13.12% Ti3+ was formed by irradiation (Supplementary Fig. 18d), which can be explained by the stronger carrier exchange between Pt and TiO2 compared to Cu and TiO2. The Pt 4 f spectrum of PtSA-TiO2 shows two peaks located at 74.4 eV and 70.9 eV, corresponding to Pt2+. After irradiation, the new peak at 69.2 eV appears, corresponding to the metallic Pt, suggesting the partial reduction of Pt2+. The O 1 s spectrum of PtSA-TiO2 (Supplementary Fig. 18f) shows the peak at 530.8 eV before irradiation assigning to the trace oxidation of Pt, which vanishes after irradiation, and Vo increases from 11.20% to 22.00% (Supplementary Table 6), matching well with that of Pt 4f. Interestingly, although Vo change is very similar for both CuSA-TiO2 and reference PtSA-TiO2, the Ti3+ in the former is much higher than the latter, indicating more photoelectrons transfer from the excited TiO2 to Pt2+ than that to Cu2+, which is reasonable as the reduction potential of Pt2+/Pt0 (0.758 eV) is much more positive than that of Cu2+/Cu+ (0.153 eV)49. We think there is little Cu metal on the surface of TiO2 due to the strong interaction between Cu and substrate as mentioned above and one-electron reduction process (Cu2+ to Cu+) is easier than two-electron process (Cu2+ to Cu0), which is crucial for the Cu2+ and Cu+ cycle (or self-healing) as indicated in Fig. 1d. The valence states of Cu in CuSA-TiO2 before and after irradiation were further confirmed by in-situ electron paramagnetic resonance (EPR) (Fig. 3c). Before irradiation, the EPR spectrum of CuSA-TiO2 shows a strong signal of Cu2+, which decreases after 30 min irradiation and then increases after exposure to the air. This result indicates that EPR-silent Cu+ was formed during irradiation and then oxidized back to Cu2+ when exposed to air.
Charge dynamics analysis
To further confirm the charge dynamics of the excited TiO2 and CuSA-TiO2, the femto second transient absorption spectra (fs-TAS) were monitored and shown in Supplementary Fig. 19 and Fig. 3. Figure 3d shows the TAS-signals of TiO2 and CuSA-TiO2 with and without AgNO3 (2 mM) under 320 nm excitation. For TiO2, the monotonically strong background is ascribed to the excited photoelectrons either in the conduction band or the trap states50. After introducing Ag+ as an electron scavenger, the TAS signal of TiO2 exhibits the decreased signal intensity, indicating this wavelength range is the fingerprint of photoelectrons. When adding Cu SAC onto TiO2, the TAS is nearly identical to that of TiO2 in the presence of Ag+ ions, clearly indicating that photogenerated electrons are successfully captured by the Cu2+ or the efficiency of Cu2+ to abstract photoelectrons from TiO2 is close to that of Ag+ ions, which is believed due to the strong chemical bond interaction in the Cu-O-Ti clusters. For CuSA-TiO2 in the presence of Ag+, the TAS signal further decreases due to the electron scavenging by both Cu2+ and Ag+. On the other hand, when adding methanol as a hole scavenger to TiO2, the enhanced photoelectron signal is observed at long wavelengths compared with that in the absence of methanol (monitored at 650 nm in Fig. 3e) due to the hole scavenging by methanol. This photoelectron dynamics decay is likely due to the reaction of oxidized species of methanol with the electrons. As expected, CuSA-TiO2 shows similar features of photoelectron decay in the presence or absence of methanol, confirming that electrons can be efficiently trapped by Cu2+ when holes are scavenged by methanol. The TAS results are well consistent with in-situ XPS and in-situ EPR results.
Isotopic experiments
To differentiate the origin of the evolved H2, isotopic tracing experiments were performed on CuSA-TiO2. Figure 3f and Supplementary Fig. 20 exhibit the spectra of generated H2 from deuterated methanol and water (CD3OD/H2O). H2 is the major product, then HD, and finally deuterium (D2). This indicates that the source of protons is mainly derived from water and partially from methanol, consistent with others51. The same conclusion can be drawn from the other case study using deuterated water and methanol (D2O/CH3OH), as shown in Fig. 3g and Supplementary Fig. 21. Again, D2 shows the largest signal, then HD and finally H2, proving water is the major hydrogen source for H2 evolution.
Theoretic calculations of carrier transfer
The photogenerated electrons transfer to the CuSA and their dissipation was modelled by the DFT simulation. The CuSA-TiO2 sample was modelled using 1.5 wt% of Cu replacing Ti (Supplementary Fig. 22). The charge density diagrams of pristine TiO2 and CuSA-TiO2 under dark conditions are shown in Fig. 4a, b, respectively. It can be seen that when Ti is replaced by Cu (highlighted with a pink dotted square), the unbalanced charges lead to a slight accumulation of electrons on Cu. The gradual increase of electron density around Cu is obvious after irradiation for 100 ps (Fig. 4c), 200 ps (Fig. 4d), and 1 ns (Fig. 4e). While the irradiation stops, the charge density on Cu after 100 ps is obviously reduced (Fig. 4 f), indicating that the accumulated electrons reduce the Cu2+ to Cu+. At the same time, the Cu+ can be oxidized back to Cu2+ when no more photogenerated electrons are transferred to Cu while in contact with oxygen.

a Charge density distribution of pure TiO2. b CuSA-TiO2 under dark condition (Cu is in the dotted square). c CuSA-TiO2 after 100 ps irradiation. d CuSA-TiO2 after 200 ps irradiation. e CuSA-TiO2 after 1 ns irradiation. f CuSA-TiO2 after turning off the irradiation for 100 ps. In the middle panel, (a1–f1) is the magnified Cu atom highlighted by pink square in (a–f). Black and red colored circles indicate more electrons’ accumulation on Cu.
The DFT result for the pristine TiO2 after irradiation as shown in Supplementary Fig. 23. It can be seen that there is no increase of the charge density on Ti atoms until 1 ns (d1), which is back to the original state when irradiation stops. It suggests the lower charge density and slower charge mobility of pristine TiO2 compared with CuSA-TiO2. To make it clear, the selected areas in 4e and 4 f of Fig. 4 are magnified and compared in Supplementary Figure 24. The black circles and corresponding distances D1 and D2 (Supplementary Fig. 24) are used to present the charge density on the Cu atom after irradiation for 1 ns and after turning off the irradiation for 100 ps. Obviously, the D1 is larger than D2, indicating that the charge density on the Cu atom after irradiation for 1 ns is quite high while the obviously reduced density is observed when stopping the irradiation after 100 ps.
The surface photovoltage (SPV) was measured by the Kelvin probe (KP) to monitor the charge separation on CuSA-TiO2 and PtSA-TiO2 (Supplementary Fig. 25a–b). The ΔSPV comparison before and after irradiation for CuSA-TiO2 and PtSA-TiO2 has been shown in Supplementary Figure 25c. The average ΔSPV before and after irradiation for CuSA-TiO2 is 185 mV, which is higher than that of PtSA-TiO2 (144 mV), indicating the more enhanced charge separation rate in CuSA-TiO2.
Hydrogen evolution mechanism
The dramatic enhancement in photocatalytic H2 evolution activity of CuSA-TiO2 can be attributed to the following reasons. First of all, the synthesis strategy provides suitable sites (Ti vacancy) and a high specific surface area for Cu atoms stabilization (Fig. 1e). Secondly, the abundant Cu2+ effectively traps the photogenerated electrons, leading to the significantly reduced recombination of the photogenerated charges (step 1 in Fig. 1d). The Cu+ has a positive potential to reduce H2O to H2 (step 2 in Fig. 1d), then Cu+ returns to Cu2+. Such an interesting reversible process or in-situ self-healing enables CuSA-TiO2 to achieve higher photocatalytic activity than the conventional Pt/TiO251. It is also higher than other single atom sites decorated photocatalysts reported due to such a high concentration of Cu (1.5 wt%) anchored on the surface of TiO2. The interaction between Cu+ and H2O is also verified by density functional theory (DFT) calculation. The H2O reduction by Cu+ needs to overcome the energy barrier of 0.44 eV and is exothermic by 0.83 eV (Supplementary Fig. 26). To verify the oxidized products of methanol, the solution of D2O/CH3OH after the reaction was tested by nuclear magnetic resonance (NMR) and detected the by-product HCOOH (Supplementary Fig. 27), confirming that methanol was first oxidised to HCHO and further to HCOOH (Fig. 1d).
In summary, a reproducible and low-cost pre-encapsulation strategy was developed for stabilizing metal single atoms on TiO2 and was further demonstrated for solar fuel H2 synthesis. The strongly anchored Cu single atoms trigger a reversible/self-healing and continuous photocatalytic process, which has been proved by both experimental and theoretical studies. The synthesised CuSA-TiO2 shows the higher H2 evolution rate with the benchmark apparent quantum efficiency of 56% at 365 nm. The obtained higher activity is due to the advantage of MOF structure with extremely large surface area as the intermediate, which maximises the exposed sites for CuSA immobilization on TiO2, reaching ~1.5 wt%. After calcination, the strongly bonded CuSA on TiO2 effectively separates photoelectrons, and then electrons cascade to reduce water to H2, along with methanol oxidation. Both the diverse spectroscopic and in-situ experiments as well as DFT results reveal the most efficient charge separation by CuSA than any other cocatalysts and prove the significance of the in-situ self-healing effect of Cu species during the photocatalytic reaction. The present atomic-level photocatalytic material design strategy indeed paves the way towards a competitive H2 production for commercial application.
Methods
All commercially available chemicals, reagents, and solvents were used as received without further purification unless noted otherwise.
Preparation of MOF MIL-125(Tiv) precursor
The precursor of MIL-125 (MIL stands for Material from Institut Lavoisier) was prepared by following the reported procedure1. In a typical process, 3 g of terephthalic acid (1,4-benzenedicarboxylic acid) was added to the 54 ml of N,N dimethylacetamide (DMF) and magnetically stirred for 10 min. Then, 6 mL of methanol was added into the above mixture followed by the addition of 1.2 mL (to create Ti vacancies, we use 1.2 mL instead of 1.56 mL from the literature) of Ti(OC4H9)4 under the stirring condition for 5 min. After that, the solution was transferred into a 100 mL Schlenk tube and kept at 130 °C for 20 h. After cooling down to room temperature, the precipitate was separated by centrifugation and washed by DMF and methanol consecutively. The free solvent attached to the precipitate was removed by vacuum drying. The final obtained sample is MIL-125.
Synthesis of SAC-MIL and preparation of final photocatalysts
Typically, 0.5 g of MIL-125 particles were dispersed in 40 mL of DI water, then the metal-salt with different weight ratios (e.g., 6.8 mg CuCl2 for 0.75 wt% Cu) was added into the MIL-125 dispersion and stirred for 3 h for encapsulated precursor synthesis. The resulting materials were then centrifuged and washed with water, dried at 80 oC. Finally, the MSA-TiO2 particles (M = Cu, Co, Ni, Fe, Mn, Zn, and Pt) were obtained by annealing at 450 oC for 4 h in the air.
Procedure of photocatalysis
The photocatalytic water-splitting experiments were performed on the full glass automatic on-line trace gas analysis system (Fig. 28a, Labsolar-6A, Perfect Light Ltd.) and Multichannel photochemical reactor (Fig. 28b, PCX-50C, Perfect Light Ltd.). With Labsolar-6A, a Xe lamp (Perfect light PLS-SXE300C) equipped with filters was used as the simulated solar spectral source. (The data in Fig. 1(a–c) was collected with the light intensity of 500 W/m2, and that in Supplementary Fig. 3 was collected with the slightly reduced light intensity of 325 W/m2 due to the safety issue for a very long-time experiment.) The as-prepared catalyst (20 mg) was uniformly dispersed in 120 mL of H2O/methanol aqueous solution by using a magnetic stirrer (containing H2O/methanol with a ratio of = 1:2). The system was vacuum-treated several times to remove the dissolved air, and the amount of produced H2 was measured by an on-line gas chromatograph (GC7900). During the reaction, the temperature was maintained at 40 °C using water circulation. For cyclic experiments, the sample was collected through centrifugation and drying after the photocatalytic reaction, without any other treatment. The sample storage for 380 days was stored in a glass bottle in a normal lab cabinet.
The PCX-50C, a 1 W UV LED (Wavelength range: 365 nm, light intensity: 34.5 mW/cm2) was used as the simulated light source for AQE calculation. The as-prepared catalyst (50 mg) was uniformly dispersed in 30 mL aqueous solution under magnetic stirring (containing H2O/methanol, v/v = 1:2). The rest of the conditions were similar as stated above.
The AQE is calculated by using the following equation and the photocatalytic H2 evolution (PHE) rate obtained from PCX-50C (Supplementary Fig.28b):
where M is the molar amount of hydrogen, NA is the Avogadro’s constant, h is the Planck constant, c is the light velocity, I is the intensity of the light, A is the irradiation area measured by the reactor window with a diameter of 3 cm, t is the reaction time, and λ is the wavelength of light (365 nm)1.
The amount of hydrogen via 50 mg photocatalyst in the reactor was measured to be 0.753 mmol within 1 h at 40 °C.
Characterization
The phase structures of the prepared samples were determined by the X-Ray diffraction (XRD) measurements using an X-ray diffractometer (Rigaku, Japan) with CuKa irradiation. The accelerating voltage and applied current were 40 kV and 80 mA, respectively. The morphology and microstructure of the samples were examined by emission scanning electron microscope (FE-SEM, Nova nanoSEM 450) and transmission electron microscope (TEM, JEM-2100). The high-angle annular dark-field (HAADF) STEM for CuSA-TiO2 was obtained using JEM-ARM300F equipment. The Brunauer–Emmett–Teller (BET) specific surface area of the prepared powders was analyzed by a 3H-2000PS2 sorption analyzer, and the porosity of the samples was evaluated based on nitrogen adsorption isotherms at 77 K. UV-vis diffused reflectance spectra of the samples were obtained using a Metash UV-9000S spectrophotometer. X-ray photoelectron spectroscopy (XPS) measurements were accomplished via a photoelectron spectrometer (Thermo ESCALAB 250Xi) with an Al Kα radiation source. The excitation wavelength was 320 nm, the scanning speed was 1200 nm min−1, and the PMT voltage was 700 V. The widths of the excitation slit and emission slit were both 5.0 nm. The Fourier transform infrared spectroscopy (FT-IR) spectra of starting materials and the as-synthesized samples were obtained using IR2000 equipment. Surface photovoltage (SPV) experiments were performed using the SKP5050 Kelvin probe. Electron spin resonance (EPR) spectroscopy was performed on Bruker EMXnano to detect the unpaired electrons of Cu2+ in CuSA-TiO2 powder at room temperature. Nuclear magnetic resonance (NMR, DRX500) was adopted to identify the product in the solution after the photocatalytic reaction.
Photoluminescence (PL) spectra and time-resolved fluorescence decay spectroscopy were obtained by an FLS 1000 fluorescence spectrophotometer (UK), where the sample powder was placed on a copper support. When testing the steady-state PL, 375 nm was selected for excitation. For PL decay testing, 375 nm and 430 nm were respectively selected for excitation and detection.
Isotopic experiment
The isotopic experiment was performed using a multichannel photochemical reactor under 365 nm LED irradiation. 10 mg of CuSA-TiO2 was added into the solution of 22.5 mL of water and 7.5 mL of methanol, and irradiated for 24 h. Two separate experiments were carried out under the same conditions except for the deuterated part (CD3OD/H2O or D2O/CH3OH). Finally, the products were identified by Mass spectrum (MS, Hiden HPR-40).
Inductively coupled plasma optical emission spectrometer (ICP) test
The ICP test was performed by PlasmaQuant PQ9000. The sample was dissolved in 5 mL freshly-made nitrohydrochloric acid at room temperature for 30 min and then heated to 136 oC for another 20 min. After cooling down to room temperature, the solid residue was filtered followed by dilution to 50 mL for the test.
Transient absorption spectroscopy (TAS) measurement
The fs-TAS measurement was carried out using a commercial transient absorption spectrometer (Newport TAS pump-probe system) that includes a 1 kHz Solstice (Newport Corp.) Ti:sapphire regenerative amplifier outputting 800 nm, 100 fs pulses. This laser light was split into two parts to generate the pump and the probe pulses. The tunable pump pulse was generated in a TOPAS-Prime (Light Conversion Ltd.) optical parametric amplifier and used to excite the sample at 320 nm. Broadband probe light (420–780 nm) was generated by focusing the Solstice output in a 2 mm sapphire crystal. Both the pump and probe beam overlapped spatially in the sample and the time delay between the pump and probe pulse was scanned by controlling the stage. 1 mg mL−1 sample was dispersed in an aqueous solution and transferred to 1 mm path length cuvettes. Samples were measured after purging with argon.
Density functional theory (DFT) study
All calculations are performed with the DMOL3 module of Materials Studio 5.0. The ultrasoft pseudopotential was used in the calculation because of its several advantages in efficiency and veracity in the reciprocal space. The electronic exchange-correlation energy is treated within the framework of the generalized gradient approximation (GGA) with Perdew-Burke-Ernzerhof (PBE). The plan-wave expansion is truncated by the cutoff energy of 450 eV. The Monkhorst–Park scheme K-point grid sampling is set as 3 × 3 × 9 for the model. Mulliken population analysis is used to analyze the average net charge. The Mulliken population is defined as the electronic charge assigned to the atoms and atomic orbitals. The convergence value is set as 0.01 nm for the maximum displacement tolerances, 0.01 eV Å−1 for the maximum force in the geometrical optimization. The convergence accuracy of SCF is 10−6 eV/atom. The crystal structures and atom coordinates were also optimized firstly under the principle of energy minimization to obtain the appropriate cell parameters with stable structures for the model. Based on this principle, the electronic structures and optical properties could be calculated. After geometrical optimization, the lattice parameters of the pure anatase TiO2 are given as follows: a = b = 3.79 Å, c = 9.51 Å. The supercell built as 8 × 8 × 1, thus the crystal parameters of the optimized supercell are a = b = 30.28 Å, c = 9.51 Å. The simulation of irradiation was realized by adding an electromagnetic field to simulate the UV irradiation. The charge density of the model was recorded in a certain time interval, i.e., 100 ps, 200 ps, and 1 ns.
Photoelectrochemical measurements
The photoelectrodes were prepared on fluorine-doped tin oxide (FTO) glass slides, which were cleaned with ethanol, rinsed with DI water, and dried before use. A 10 mg of the prepared samples were added into 200 μL of ethanol and 800 μL water with 10 μL of Nafion (5wt.%) and then carefully ground for a little while to form a homogenous slurry. Subsequently, the obtained slurry was evenly distributed onto the conductive side of FTO glass. After drying in the air, the photoelectron chemical properties of the obtained electrodes were tested in a three-electrode system using a CHI-760E electrochemical workstation. The prepared electrode, Pt wire, and Ag/AgCl electrode were used as the working, counter, and reference electrodes, respectively. The 0.2 M Na2SO4 aqueous solution was used as an electrolyte, and the photoelectrodes were irradiated using a 150 W xenon lamp with a light density of 95 mW/cm2. The photocurrents of the electrodes were measured using the amperometric (I–t curves) technique under repeatedly interrupted light irradiation. Electrochemical impedance spectroscopy (EIS) measurements were performed at an applied voltage of 5 mV with a frequency in the range of 105–0.1 Hz.
Statistical analysis
All data were presented as means with standard deviations (SD).
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Change history
11 April 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41467-022-29799-z
References
Zhao, Y. et al. Simultaneous oxidative and reductive reactions in one system by atomic design. Nat. Catal. 4, 1–10 (2021).
Cao, L. et al. Identification of single-atom active sites in carbon-based cobalt catalysts during electrocatalytic hydrogen evolution. Nat. Catal. 2, 134–141 (2019).
Wang, Y. et al. Current understanding and challenges of solar-driven hydrogen generation using polymeric photocatalysts. Nat. Energy 4, 746–760 (2019).
Dan, K. et al. Recent advances in visible-light driven water oxidation and reduction in suspensions systems. Mater. Today 21, 897–922 (2018).
Moniz, S. J. A. et al. Visible-Light Driven Heterojunction Photocatalysts for Water Splitting– A Critical Review. Energy Environ. Sci. 8, 731–759 (2015).
Lee, B. H. et al. Reversible and cooperative photoactivation of single-atom Cu/TiO2 photocatalysts. Nat. Mater. 18, 620–626 (2019).
Tang, J. et al. Mechanism of photocatalytic water splitting in TiO2. Reaction of water with photoholes, importance of charge carrier dynamics, and evidence for four-hole chemistry. J. Am. Chem. Soc. 130, 13885–13891 (2008).
Linsebigler, A. L., Lu, G. & Yates, J. T. Photocatalysis on TiO2 Surfaces: Principles, Mechanisms, and Selected Results. Chem. Rev. 95, 735–758 (1995).
Cao, S. et al. Photocatalytic pure water splitting with high efficiency and value by Pt/porous brookite TiO2 nanoflutes. Nano Energy 67, 104287 (2020).
Barroso, M. et al. The role of cobalt phosphate in enhancing the photocatalytic activity of α-Fe2O3 toward water oxidation. J. Am. Chem. Soc. 133, 14868–14871 (2011).
Wang, G. et al. Photoinduction of Cu single atoms decorated on UiO-66-NH2 for enhanced photocatalytic reduction of CO2 to liquid fuels. J. Am. Chem. Soc. 142, 19339–19345 (2020).
Xiao, M. et al. Molten‐Salt‐Mediated Synthesis of an Atomic Nickel Co‐catalyst on TiO2 for Improved Photocatalytic H2. Evolution. Angew. Chem. 132, 7297–7301 (2020).
Xie, J. et al. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species. Nat. Catal. 1, 889–896 (2018).
Liu, L. & Corma, A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 118, 4981–5079 (2018).
Qiao, B. et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 3, 634–641 (2011).
Hejazi, S. et al. On the controlled loading of single platinum atoms as a co-catalyst on TiO2 anatase for optimized photocatalytic H2 generation. Adv. Mater. 32, 1908505 (2020).
Lam, E. et al. Isolated Zr surface sites on silica promote hydrogenation of CO2 to CH3OH in supported Cu catalysts. J. Am. Chem. Soc. 140, 10530–10535 (2018).
Yin, Y. et al. Boosting Fenton-like reactions via single atom Fe catalysis. Environ. Sci. Technol. 53, 11391–11400 (2019).
Wang, Y. et al. Catalysis with two-dimensional materials confining single atoms: concept, design, and applications. Chem. Rev. 119, 1806–1854 (2019).
Guo, J. et al. Nitrogen-doped porous carbon supported nonprecious metal single-atom electrocatalysts: from synthesis to application. Small Meth. 3, 1–33 (2019).
Zhang, H., Liu, G., Shi, L. & Ye, J. Single-atom catalysts: emerging multifunctional materials in heterogeneous catalysis. Adv. Energy Mater. 8, 1–24 (2018).
Jiang, X. H. et al. Silver Single atom in carbon nitride catalyst for highly efficient photocatalytic hydrogen evolution. Angew. Chem. - Int. Ed. 59, 23112–23116 (2020).
Trofimovaite, R. et al. Single atom Cu(I) promoted mesoporous titanias for photocatalytic Methyl Orange depollution and H2 production. Appl. Catal. B Environ. 232, 501–511 (2018).
Seadira, T. W. P., Sadanandam, G., Ntho, T., Masuku, C. M. & Scurrell, M. S. Preparation and characterization of metals supported on nanostructured TiO2 hollow spheres for production of hydrogen via photocatalytic reforming of glycerol. Appl. Catal. B Environ. 222, 133–145 (2018).
Yu, J. & Ran, J. Facile preparation and enhanced photocatalytic H2-production activity of Cu(OH)2 cluster modified TiO2. Energy Environ. Sci. 4, 1364–1371 (2011).
Foo, W. J., Zhang, C. & Ho, G. W. Non-noble metal Cu-loaded TiO2 for enhanced photocatalytic H2 production. Nanoscale 5, 759–764 (2013).
Xiao, S. et al. Copper nanowires: A substitute for noble metals to enhance photocatalytic H2 generation. Nano Lett. 15, 4853–4858 (2015).
Saffman, M. The next step in making arrays of single atoms. Nature 567, 468–470 (2019).
Fang, X. et al. Single Pt atoms confined into a metal–organic framework for efficient photocatalysis. Adv. Mater. 30, 1–7 (2018).
Chen, L. N. et al. Efficient hydrogen production from methanol using a single-site Pt1/CeO2 catalyst. J. Am. Chem. Soc. 141, 17995–17999 (2019).
Song, Y. et al. Metal-organic framework nodes support single-site nickel(II) hydride catalysts for the hydrogenolysis of Aryl Ethers. ACS Catal. 9, 1578–1583 (2019).
Remiro-Buenamañana, S. et al. Influence of co-catalysts on the photocatalytic activity of MIL-125(Ti)-NH2 in the overall water splitting. Appl. Catal. B Environ. 254, 677–684 (2019).
Ji, P. et al. Titanium(III)-oxo clusters in a metal-organic framework support single-site Co(II)-hydride catalysts for arene hydrogenation. J. Am. Chem. Soc. 140, 433–440 (2018).
Xiang, Q., Yu, J. & Jaroniec, M. Synergetic effect of MoS2 and graphene as cocatalysts for enhanced photocatalytic H2 production activity of TiO2 nanoparticles. J. Am. Chem. Soc. 134, 6575–6578 (2012).
Liu, Y. et al. TiO2/Cu2O core/ultrathin shell nanorods as efficient and stable photocatalysts for water reduction. Angew. Chem. - Int. Ed. 54, 15260–15265 (2015).
Dan-Hardi, M. et al. A new photoactive crystalline highly porous titanium(IV) dicarboxylate. J. Am. Chem. Soc. 131, 10857–10859 (2009).
Kampouri, S. et al. Photocatalytic hydrogen generation from a visible-light responsive metal-organic framework system: The impact of nickel phosphide nanoparticles. J. Mater. Chem. A 6, 2476–2481 (2018).
Wang, H. et al. Facile synthesis of Sb2S3/ultrathin g-C3N4 sheets heterostructures embedded with g-C3N4 quantum dots with enhanced NIR-light photocatalytic performance. Appl. Catal. B Environ. 193, 36–46 (2016).
Wolff, C. M. et al. All-in-one visible-light-driven water splitting by combining nanoparticulate and molecular co-catalysts on CdS nanorods. Nat. Energy 3, 862–869 (2018).
Liu, H., Zhang, J. & Ao, D. Construction of heterostructured ZnIn2S4@NH2-MIL-125(Ti) nanocomposites for visible-light-driven H2 production. Appl. Catal. B Environ. 221, 433–442 (2018).
Ao, D., Zhang, J. & Liu, H. Visible-light-driven photocatalytic degradation of pollutants over Cu-doped NH2-MIL-125(Ti). J. Photochem. Photobiol. A Chem. 364, 524–533 (2018).
Scanlon, D. O. et al. Band alignment of rutile and anatase TiO2. Nat. Mater. 12, 798–801 (2013).
Berezin, M. Y. & Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 110, 2641–2684 (2010).
Luo, H. et al. Pt single-atoms supported on nitrogen-doped carbon dots for highly efficient photocatalytic hydrogen generation. J. Mater. Chem. A 8, 14690–14696 (2020).
Zhu, S. et al. Cooperation between inside and outside of TiO2: Lattice Cu+ accelerates carrier migration to the surface of metal copper for photocatalytic CO2 reduction. Appl. Catal. B Environ. 264, 118515 (2020).
Ahmed, S. A. Structural, optical, and magnetic properties of Cu-doped TiO2 samples. Cryst. Res. Technol. 52, 1–8 (2017).
Jiang, Z. et al. Living atomically dispersed Cu ultrathin TiO2 nanosheet CO2 reduction photocatalyst. Adv. Sci. 6, 3–7 (2019).
Zhang, Y. et al. Covalent organic framework-supported Fe-TiO2 nanoparticles as ambient-light-active photocatalysts. J. Mater. Chem. A 7, 16364–16371 (2019).
Kim, M. R., Lee, D. K. & Jang, D. J. Facile fabrication of hollow Pt/Ag nanocomposites having enhanced catalytic properties. Appl. Catal. B Environ. 103, 253–260 (2011).
Sachs, M., Pastor, E., Kafizas, A. & Durrant, J. R. Evaluation of surface state mediated charge recombination in anatase and rutile TiO2. J. Phys. Chem. Lett. 7, 3742–3746 (2016).
Kandiel, T. A., Ivanova, I. & Bahnemann, D. W. Long-term investigation of the photocatalytic hydrogen production on platinized TiO2: An isotopic study. Energy Environ. Sci. 7, 1420–1425 (2014).
Acknowledgements
This work was mainly supported by the National Natural Science Foundation of China (no.51562038), Yunnan Yunling Scholars Project, the Key Project of Natural Science Foundation of Yunnan (2018FY001(−011)) received by QL, and Yunnan basic applied research project (No. 202101AT070013) receive by YZ. WZ and JM are thankful for the Shannxi Key Research Grant (No. 2020GY-244). HW, MT, and JT are thankful for financial support from the UK EPSRC (EP/S018204/2), Royal Society Newton Advanced Fellowship grant (NAF\R1\191163 and NA170422), and Leverhulme Trust (RPG-2017-122).
Author information
Authors and Affiliations
Contributions
QL and JT conceived and supervised the progress of the entire project. YZ designed the experiments, prepared the materials, and performed the MS, in-situ XPS, isotopic, ICP characterizations, photocatalytic performances test (H2 evolution test and AQE test) JZ prepared the materials and performed the BET, TG-DTA, in-situ XPS, SPV characterizations, and photocatalytic performances test (long-term stability test). HW repeated activity tests and AQE analysis and discussion of XPS, TAS, PL, AQE, and photochemical results. X. Z. carried out the first-principle DFT modulations and SEM, TEM characterizations. WZ. and YG carried out the TAS measurement and analysis. BX performed the in-situ EPR, XRD characterizations and analyzed the HADDF, STEM-EDS data. TL performed the electrochemical test, PL and PL decay test. MT heavily contributed to the discussion on the fundamental understanding. JZ performed Zeta, UV-vis, and NMR characterizations. JM supervised TAS experiments and heavily contributed to the TAS analysis. LL and RH performed the HADDF characterization. The paper was written through collective contributions from all authors. All authors approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review information
Nature Communications thanks Moussab Harb, Hiroyuki Matsuzaki, Ki Tae Nam and Rosendo Valero for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Zhao, J., Wang, H. et al. Single-atom Cu anchored catalysts for photocatalytic renewable H2 production with a quantum efficiency of 56%. Nat Commun 13, 58 (2022). https://doi.org/10.1038/s41467-021-27698-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-27698-3
This article is cited by
-
Photochemical tuning of dynamic defects for high-performance atomically dispersed catalysts
Nature Materials (2024)
-
Pythium oligandrum-derived carbon/graphene oxide as sensitized substrate: large-scale preparation and enhancing photocatalytic hydrogen evolution
Biomass Conversion and Biorefinery (2024)
-
Coupled Cu doping and Z-scheme heterojunction for synergistically enhanced tetracycline photodegradation
Nano Research (2024)
-
High quantum efficiency of hydrogen production from methanol aqueous solution with PtCu–TiO2 photocatalysts
Nature Materials (2023)
-
Non-oxidative Propane Dehydrogenation over Vanadium Doped Graphitic Carbon Nitride Catalysts
Catalysis Letters (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
