
Abstract
Doping has been widely used to control the charge carrier concentration in organic semiconductors. However, in conjugated polymers, n-doping is often limited by the tradeoff between doping efficiency and charge carrier mobilities, since dopants often randomly distribute within polymers, leading to significant structural and energetic disorder. Here, we screen a large number of polymer building block combinations and explore the possibility of designing n-type conjugated polymers with good tolerance to dopant-induced disorder. We show that a carefully designed conjugated polymer with a single dominant planar backbone conformation, high torsional barrier at each dihedral angle, and zigzag backbone curvature is highly dopable and can tolerate dopant-induced disorder. With these features, the designed diketopyrrolopyrrole (DPP)-based polymer can be efficiently n-doped and exhibit high n-type electrical conductivities over 120 S cm−1, much higher than the reference polymers with similar chemical structures. This work provides a polymer design concept for highly dopable and highly conductive polymeric semiconductors.
Similar content being viewed by others
Introduction
Polymeric semiconductors have been intensively studied for next-generation flexible or stretchable optoelectronic devices because of their unique mechanical properties and good solution processability1,2. To tune the charge-carrier concentration, Fermi level, and electrical conductivity of conjugated polymers, molecular doping is widely used3,4,5,6,7. However, the introduced dopants tend to randomly distribute within polymer film and bring large structural and energetic disorder8. P-type dopants can be small-size Lewis acids (e.g., ferric chloride), small molecules (e.g., 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane), and polymers (e.g., poly(4-styrene sulfonate) (PSS)). These p-dopants are strong oxidants or acids, and can be incorporated into the polymer matrix without significantly disrupting the ordered molecular packings and thus leading to high p-type electrical conductivities9. High charge-carrier concentration over 1021 cm−3 and high electrical conductivities over 1000 S cm−1 have been achieved in p-doped polymers, such as poly(2,5-bis(3-hexadecylthiophen-2-yl)thieno[3,2-b]thiophene)7 and poly(3,4-ethylenedioxythiophene):PSS)10. In contrast, the commonly used n-type dopants usually have weak reducing ability and relatively large size to achieve good air stability11, resulting in a significant increase of disorder after doping. Therefore, the charge-transport properties of n-type polymers are strongly hindered by the undesirable doping-induced disorder. Only a few n-doped conjugated polymers are reported to achieve conductivities approaching or over 10 S cm−1 12,13,14,15,16,17.
The low electrical conductivities of n-doped conjugated polymers are mainly restricted by low doping efficiency and dopant-induced structural and energetic disorder. Many efforts have been devoted to improving the n-doping efficiency of conjugated polymers through the introduction of electron-deficient building blocks in the past decades18. However, the synthesis of strong electron-deficient building blocks is challenging17, as organic molecules with the lowest unoccupied molecular orbital (LUMO) energy levels lower than −4.4 eV might become unstable19. Furthermore, the n-doping efficiency is also limited by the solid-state miscibility between polymer and n-dopants, as efficient charge transfer between polymer and dopant requires close contact20,21. To enhance the solid-state miscibility, several polymer design strategies have been developed. For example, polar ethylene glycol side chains were employed to replace conventional alkyl side chains to improve the n-doping efficiency22. Twisted and “kinked” donor moieties were used to decrease the crystallinity of the conjugated polymers, constructing percolation space for dopants to improve doping efficiency23,24. However, these strategies always result in significant structural and energetic disorder, bringing about declined charge-carrier mobilities and poor electrical conductivities22,23,24.
Here we explore the design of n-type conjugated polymers with high tolerance to dopant-induced disorder. With a computer-aided molecular design approach, we screened currently available polymer building blocks and their possible combinations. We found that the pyrazine (Pz) and 3,4-difluorothiophene (2FT) combination exhibited the highest torsional barrier, best planarity, and single dominant conformation, providing a highly rigid polymer backbone (Fig. 1). Therefore, we synthesized the polymer P(PzDPP-2FT) (Fig. 1b) with a zigzag backbone and relatively large Urbach energy, which are usually thought to result in low charge-carrier mobilities25,26. However, P(PzDPP-2FT) exhibits high electron mobility due to its narrow conformation distribution, low energetic disorder, and strong interchain interactions. The zigzag backbone provides the polymer with a dopant-binding cavity compared with polymers with linear backbones and enhances doping efficiency. High mobility and enhanced doping efficiency synergistically contribute to a high n-type electrical conductivity over 120 S cm−1.

a Computer-aided polymer building block screening process: (i) Collecting representative high-performance D–A copolymers used for OFETs; (ii) extracting building blocks; (iii) further extracting the segments for simplifying the screening process; (iv) PES of the dihedral angles of different segment combinations. (b) Chemical structures of P(PzDPP-2FT), P(PzDPP-4F2T), and P(PzDPP-T). c PES results of the torsional angles φ2, φ5, and φ7 in these three polymers (ωB97X-D/6-311G(d,p)). The energy is relative to the minimal energy conformer. Planarity indices 〈cos2φ〉 of these torsional angles were calculated based on PES results. d The DFT-optimized geometries of the tetramers of P(PzDPP-2FT) and P(PzDPP-4F2T). Long alkyl chains were replaced with methyl groups.
Results
Computer-aided polymer design
Previous studies have suggested that to achieve efficient charge transport in conjugated polymers, intrachain charge transport and interchain short-range contacts are critical27,28. As the intrachain charge-transport behavior is closely associated with the conformational distribution of the polymer backbone, carefully tuning the torsion-angle distribution between the polymer building blocks from the level of molecular design is crucial for maintaining low conformation disorder29. Therefore, we propose that there are three critical rules of designing conjugated polymers to enhance the tolerance to dopant-induced disorder: (1) high coplanarity to obtain good intrachain and interchain charge transport, which can be quantified through a planarity index 〈cos2φ〉30; (2) single preferential planar backbone conformation; and (3) high and steep torsion barriers that are essential to restrain the polymer backbone to maintain a coplanar conformation and enhance the tolerance to dopant-induced disorders. Although Rule (1) (〈cos2φ〉 value) has considered all torsional conformations and their relative contribution to the overall structural disorder, it is still not enough to screen the desired polymers with minimal dihedral angle distributions. Therefore, we introduced Rules (2) and (3) (see Supplementary Information for more details).
Based on these concepts, we employed density functional theory (DFT) calculations to screen currently available high-performance polymer building blocks. We hope to find a combination with the best planarity, highest torsional barrier, and single dominant conformation (Fig. 1a and more details in Supplementary Information Section 2). After extracting donor and acceptor segments from various representative donor–acceptor (D-A) polymers, relaxed potential energy scans (PESs) were performed at the dihedral angles of adjacent units. After comparing the performance of several different computational methods, ωB97X-D/6-311G(d, p) was selected due to its reasonable accuracy and better efficiency in predicting the torsional barrier heights of conjugated polymers (Supplementary Fig. 4). We found that Pz and 2FT combination shows the highest 〈cos2φ〉 value of 0.9329, i.e., the highest planarization, which could be attributed to its highest torsional barrier as well as its lowest relative energy at 0°. Electronegative atom N on the ring increases the rotational barriers by enhancing the backbone conjugation31. The fluorine has little steric hindrance and provides attractive non-covalent interactions, which also plays a significant role in promoting the planarization of Pz-2FT (see detailed discussions in the Supplementary Information Section 2). Thus, based on these building blocks, P(PzDPP-2FT) was designed (Fig. 1b), in which the torsion angles are small (φ1 = φ2 = 0.002°). For comparison, reference polymers, P(PzDPP-4F2T) and P(PzDPP-T), with similar chemical structures were also synthesized (Fig. 1b). DFT calculations reveal that the three polymers exhibit coplanar backbones from the side view under the optimized conformations (Supplementary Fig. 8). PES results demonstrate that Pz-DPP tends to form planar connections for all the polymers (φ1, φ3, and φ6) due to the non-covalent O ∙ ∙ ∙ H bonding (Fig. 1c). However, the small torsional barrier between the two thiophene units (φ5) in P(PzDPP-4F2T) makes the polymer readily deviate from planarity. Even though the dihedral angle of the Pz-T (φ7) in P(PzDPP-T) exhibits a relatively high torsional barrier, the low torsional barriers at both 0° and 180° make P(PzDPP-T) have more than one preponderant conformation. Interestingly, the strong F ∙ ∙ ∙ H interaction at Pz-2FT brings about a steeper and higher torsional barrier, and only one preponderant conformation at 0°. These two crucial features promote P(PzDPP-2FT) to form narrower torsion-angle distributions and thus low conformation disorder. Compared with the pseudo-linear backbone of both P(PzDPP-4F2T) and P(PzDPP-T), P(PzDPP-2FT) has a zigzag backbone curvature (Fig. 1d and Supplementary Fig. 8). We will show that this structure feature allows P(PzDPP-2FT) to form unique binding sites with n-dopants and further reducing the energetic disorder after doping.
Polymer properties
According to the cyclic voltammetry measurement, the estimated LUMO energy levels of P(PzDPP-2FT) and P(PzDPP-4F2T) are −3.90 eV and −3.82 eV, respectively (Supplementary Fig. 12). As P(PzDPP-2FT) and P(PzDPP-4F2T) have very similar chemical structures and close LUMO energy levels, we will focus on the comparison of their properties and device performance. Previous study has shown that the absorption profiles of polymers can reflect the rigidity and coplanarity of polymer backbones32. Conjugated polymers with larger torsional angles will become more planar in solid state due to interchain π–π stacking, leading to a red-shift in the solid-state absorption spectra. The thin-film absorption spectra of P(PzDPP-2FT) do not exhibit a noticeable shift compared to its solution one (Fig. 2a), suggesting that P(PzDPP-2FT) has a rigid and coplanar backbone with similar molecular conformations in both solution and solid state. In contrast, the film absorption spectrum of P(PzDPP-4F2T) exhibited an obvious red-shift compared to solution one (Fig. 2b), suggesting that P(PzDPP-4F2T) has a relatively flexible backbone and it may adopt more planar backbone conformation in solid state. Moreover, it has been proved that conjugated polymers with more planar and rigid backbone usually exhibit smaller Stokes shift and smaller full width at half maxima (FWHM) of their absorption and emission peaks33,34. Figure 2c shows that the Stokes shift of P(PzDPP-2FT) is 17 nm, which is much smaller than that of P(PzDPP-4F2T) (35 nm). The FWHM of the 0-0 absorption and emission peaks of P(PzDPP-2FT) are 41.1 and 39.5 nm, respectively, which are also smaller than that of P(PzDPP-4F2T) (43.8 and 41.6 nm for the absorption and emission peaks, respectively) (Supplementary Fig. 15). These results indicate that P(PzDPP-2FT) has a more planar and more shape-persistent backbone than P(PzDPP-4F2T).

a, b Normalized absorption spectra of both P(PzDPP-2FT) and P(PzDPP-4F2T) in solution, thin film, and annealed film. c Normalized absorption (solid lines) and photoluminescence (dash lines) (λex = 460 nm) spectra of the polymer solutions. d, e Schematic illustration of the possible microstructures of both polymers in solution. The red dot in the figure represents the crosslinking points or strong interchain short contacts. f Viscosity comparison of both polymer solutions as a function of concentration at 20 °C.
We observed that the P(PzDPP-2FT) solution can form a gel when aged at room temperature, whereas the P(PzDPP-4F2T) solution remains fluid after aging even though both polymers have similar solubility (Fig. 2d and Supplementary Fig. 28). As gel formation usually requires the presence of a cross-linked polymer chain network that percolates the system and confines the solvent35, we assume that P(PzDPP-2FT) may have strong interchain short contacts. To further prove this, viscosity was employed to evaluate properties of both polymers in solutions. As was expected, P(PzDPP-2FT) always exhibit higher viscosity and larger Martin constant KM (reflect the polymer–polymer interaction36) than P(PzDPP-4F2T). The intrinsic viscosity [η] of P(PzDPP-2FT) is over two times higher than that of P(PzDPP-4F2T), indicating the existence of larger macromolecules or aggregates37. These results confirm that P(PzDPP-2FT) has short interchain contacts or crosslinking points in solution to form such strong interactions, which will be further discussed and supported by the photothermal deflection spectroscopy (PDS) measurement.
Charge-transport properties and thin-film characterization
Various mass fractions of 4-(1,3-dimethyl-2,3-dihydro-1H-benzoimidazol-2-yl)phenyl)dimethylamine (N-DMBI) were mixed with P(PzDPP-2FT) and P(PzDPP-4F2T) to optimize their electrical conductivities. P(PzDPP-2FT) has a maximum conductivity of 43.3 S cm−1, which is almost 20 times higher than that of P(PzDPP-4F2T) (Fig. 3a). To eliminate the influence of different dopants and doping methods, the two polymers were also doped with tetrakis(dimethylamino)ethylene (TDAE) and bis(cyclopentadienyl)cobalt (CoCp2). In both cases, P(PzDPP-2FT) still showed higher n-type electrical conductivities than P(PzDPP-4F2T) (Fig. 3b, c). Notably, after being doped with CoCp2, P(PzDPP-2FT) exhibits the highest n-type electrical conductivity of 129 S cm−1. As both TDAE and CoCp2 are small and highly diffusible dopants (Supplementary Fig. 23), the conductivity enhancement is probably due to their better permeability and miscibility with the polymers, which can be supported by the uniform microstructures of the polymer films at higher doping concentrations (Supplementary Fig. 24). Besides, the temperature-dependent electrical conductivity of the doped P(PzDPP-2FT) shows weaker temperature dependence compared with that of P(PzDPP-4F2T) (Supplementary Fig. 17), suggesting that P(PzDPP-2FT) has lower charge-transport barriers after n-doping.

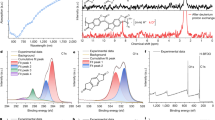
a Electrical conductivities at different N-DMBI/polymer ratios. b Electrical conductivities after different exposure times in TDAE vapor. c Electrical conductivities at different CoCp2/polymer ratios. The error bars are determined according to 2% measurement errors. d Absorption spectra of the pristine and N-DMBI-doped P(PzDPP-2FT) and P(PzDPP-4F2T) films. e UPS binding energy of the pristine and 15% N-DMBI-doped films. f EPR signals of the pristine and N-DMBI-doped films at different dopant/polymer ratios.
The doping efficiencies of the films were studied by absorption spectroscopy, ultraviolet photoelectron spectroscopy, and X-ray photoelectron spectroscopy (XPS). At each dopant/polymer ratio, P(PzDPP-2FT) showed stronger (bi)polaron absorptions than P(PzDPP-4F2T) (Fig. 3d). The larger shifts of the onset of the secondary electron cutoff for doped P(PzDPP-2FT) also imply that the Fermi level of P(PzDPP-2FT) shifts higher than that of P(PzDPP-4F2T) (Fig. 3e). The doping efficiency could also be evaluated by the newly formed signal of N-DMBI+ at 402 eV with XPS. For each dopant/polymer ratio, the relative intensity of the cationic N-DMBI+ to other N (1 s) signals in P(PzDPP-2FT) film is larger than that of P(PzDPP-4F2T) (Supplementary Fig. 18). The electron paramagnetic resonance (EPR) (Fig. 3f) and alternating current (AC) magnetic field Hall (Supplementary Fig. 19) measurements also showed that the N-DMBI-doped P(PzDPP-2FT) exhibited higher charge-carrier concentrations than P(PzDPP-4F2T). These results indicate that P(PzDPP-2FT) exhibited higher doping levels than P(PzDPP-4F2T) in film.
Doping efficiency enhancement is usually attributed to the low-lying LUMO energy levels of the polymer or the enhanced miscibility between polymer and n-dopant. However, P(PzDPP-2FT) shows almost the same LUMO energy level as P(PzDPP-4F2T), which is relatively high among other n-type polymers reported in the literature (Supplementary Fig. 12 and Supplementary Table 4). Therefore, the slightly lower LUMO level of P(PzDPP-2FT) cannot explain its high doping levels. The microstructures of both the pristine and the N-DMBI-doped P(PzDPP-2FT) and P(PzDPP-4F2T) films were characterized by grazing incidence wide-angle X-ray scattering (GIWAXS). The two-dimensional (2D) GIWAXS patterns of the pristine polymer films are displayed in Supplementary Fig. 20. P(PzDPP-4F2T) exhibits stronger (h00) diffraction peaks than P(PzDPP-2FT) along the qz axis, indicating that P(PzDPP-4F2T) has a highly ordered polymer packing, whereas P(PzDPP-2FT) has lower crystallinity. P(PzDPP-2FT) mainly exhibits a face-on orientation, whereas P(PzDPP-4F2T) exhibits mixed face-on and edge-on orientations. The π–π stacking distances were estimated to be 3.48 Å and 3.37 Å for P(PzDPP-2FT) and P(PzDPP-4F2T), respectively. After N-DMBI doping, the lamellar packing and π–π stacking distances of P(PzDPP-4F2T) remain almost unchanged, whereas a slight increase of both the lamellar packing and π–π stacking distances was observed for P(PzDPP-2FT) (Supplementary Fig. 21). The paracrystallinity (g) change of both polymers is shown in Supplementary Fig. 2227. Usually, after heavily doping, polymer films showed increased paracrystallinity due to dopant-induced structural disorder. Interestingly, the paracrystallinity of P(PzDPP-2FT) does not change much even for dopant/polymer ratios of up to 60%, suggesting its tolerance to dopant-induced structural disorder. Atomic force microscope (AFM) combined with infrared-spectroscopy can be used to probe the dopant miscibility in polymer films38. When doped with 30% N-DMBI, the P(PzDPP-2FT) film exhibited a domain size of around 50 nm, much smaller than that of the doped P(PzDPP-4F2T) film (90–110 nm) (Supplementary Fig. 27). Moreover, the 30% N-DMBI doped P(PzDPP-4F2T) film has large doped polymer domains separated by a large amount of unreacted dopant (Supplementary Fig. 27b). In contrast, the doped P(PzDPP-2FT) film has smaller polymer domains and a tiny amount of unreacted dopant (Supplementary Fig. 27a). Therefore, the enhanced doping efficiency of P(PzDPP-2FT) is attributed to its good miscibility with dopants.
Disorder-tolerant features
The pristine P(PzDPP-2FT) exhibited comparable electron mobilities (1.30 ± 0.14 cm2 V−1 s−1) to that of P(PzDPP-4F2T) (1.28 ± 0.25 cm2 V−1 s−1) (Fig. 4a). The gate-voltage-dependent mobilities of P(PzDPP-2FT) and P(PzDPP-4F2T) were extracted from their transfer characteristics (Fig. 4b). For P(PzDPP-2FT), the mobility is nearly independent of the gate voltage (VG) when VG > 50 V, whereas the mobility of P(PzDPP-4F2T) increases as VG increases. The temperature-dependent drain current ID vs. VG in the saturation regime was tested (Supplementary Fig. 35). For P(PzDPP-2FT), the exponent γ, which reflects the temperature dependence, takes a nearly temperature-independent value of about 3.3 (Supplementary Fig. 36). In contrast, the exponent γ for P(PzDPP-4F2T) increases when decreasing temperature, where γ = T0/T + 2. This can be explained by the carrier hopping within an exponential density of states (DOSs)26,39. The temperature-dependent mobility measurement shows that P(PzDPP-2FT) has lower activation energy (Ea) than P(PzDPP-4F2T) (Supplementary Fig. 37). All these results suggest that the pristine P(PzDPP-2FT) has lower energetic disorder than P(PzDPP-4F2T)26. Previous studies demonstrated that disorder in semiconductors would result in the formation of the tail states of their DOSs, broadening of the absorption onset, and creating an exponential sub-bandgap absorption tail called the Urbach tail. The Urbach energy (Eu) is often used to evaluate the energetic disorder of a conjugated polymer film26,40. According to PDS characterization, P(PzDPP-2FT) exhibits a larger Urbach energy (48.8 meV) than P(PzDPP-4F2T) (36.6 meV) (Fig. 4d). This seems to conflict with our device results that P(PzDPP-2FT) has lower energetic disorder. In fact, the disorder in conjugated polymer film could come from the torsional disorder along the polymer backbone or different molecular packings. A conjugated polymer film usually contains crystalline, semi-crystalline, and amorphous regions. Compared with the crystalline region, the DOS broadening in the semi-crystalline and the amorphous regions is more significant (further discussed in the theoretical modeling part). The GIWAXS results suggest that P(PzDPP-2FT) is less crystalline with more disordered polymer packing. As P(PzDPP-2FT) has a rigid and planar backbone, its larger Urbach energy does not come from the torsional disorder along the polymer backbone. Therefore, the larger Urbach energy of P(PzDPP-2FT) can be attributed to its low crystallinity and less ordered molecular packing. However, charge-transport properties in polymer film are mostly influenced by the DOS along the charge-transport pathways. We also compared the charge-carrier mobilities and Eu data of different types of polymers reported in the literature. It turns out that there is no clear relationship between charge-carrier mobility and Eu (Fig. 4e). Interestingly, for the polymers with high mobilities over 1 cm2 V−1 s−1, P(PzDPP-2FT) has the largest Eu. This result can be explained by a previous assumption that fast intrachain charge transport and a few interchain short-range contacts are sufficient to allow high charge-carrier mobilities in conjugated polymers27. Recent studies have indicated that a stronger sub-bandgap absorption coefficient area in PDS suggests the existence of stronger interchain short contacts within materials’ amorphous domains41. The larger sub-bandgap area of P(PzDPP-2FT) in PDS measurements also suggests it has stronger interchain short-range interactions (Supplementary Fig. 39), consistent with the gel formation experiment and the higher viscosity of P(PzDPP-2FT) solution. In addition, the DFT calculations, absorption profiles, and Stokes shifts all support that P(PzDPP-2FT) has a rigid and single preferential planar conformation, which guarantees its fast intrachain charge transport. Therefore, the high electron mobility of P(PzDPP-2FT) can be attributed to its efficient intrachain charge transport and good interchain short-range interactions.

a Transfer characteristics of the pristine P(PzDPP-2FT) and P(PzDPP-4F2T) films. b Gate-voltage-dependent saturation mobilities. c Extracted activation energies of the doped films at different dopant/polymer ratios. d Absorption coefficients of both the pristine polymer films measured by PDS. Dash lines represent exponential tail fits for extraction of the Urbach energies. e Mobility vs. Urbach energy of ours and other reported polymers. f Electrical conductivity comparison of reported n-doped semiconducting polymers.
The activation energies of the polymer films at different doping concentrations were extracted (Supplementary Fig. 40). P(PzDPP-2FT) exhibited constantly lower activation energies than P(PzDPP-4F2T) (Fig. 4c). The activation energies of both P(PzDPP-2FT) and P(PzDPP-4F2T) first decrease and then increase as the dopant concentration increasing. Molecular doping of a disorder organic semiconductor has two counteracting effects. On the one hand, it increases the charge-carrier concentration and fills the traps, leading to a reduced activation energy for charge transport42. On the other hand, the insertion of the ionized dopants causes structural and energetic disorder because of film microstructure damage and the randomly distributed Coulombic interactions between the dopant ions and the charge carriers. These effects result in increased activation energy, especially at high dopant concentrations43. Therefore, the decrease of the activation energy might be due to the trap-filling mechanism and the increase of the activation energy could be attributed to the excessive dopant-induced structural and energetic disorder44. Compared with P(PzDPP-4F2T), P(PzDPP-2FT) shows a smaller activation energy fluctuation with the change of the doping concentration and the excessive dopants (>20%) did not obviously increase the activation energy. These results suggest that P(PzDPP-2FT) has a stronger tolerance to the dopant-induced energetic disorder27,45. Therefore, the high miscibility with dopants and disorder-tolerant feature of P(PzDPP-2FT) allows it to show high n-type electrical conductivities (Fig. 4f).
P(PzDPP-T) has a coplanar and linear backbone with relatively high torsional barriers but multiple coplanar conformations (i.e., at 0° and 180°). It showed low electrical conductivity of 0.01 S cm−1 when doped with TDAE or N-DMBI (Supplementary Fig. 34). The EPR measurement indicated that the doped P(PzDPP-T) films had a charge-carrier concentration of ca. 1019 cm−3, comparable to that of P(PzDPP-2FT) and P(PzDPP-4F2T) (Supplementary Fig. 33 and Supplementary Table 1). Therefore, the low electrical conductivity of the doped P(PzDPP-T) film is primarily attributed to its low charge-carrier mobility or significant structural and energetic disorder. This result suggests that apart from a planar backbone, a single dominant conformation is also important to realize the disorder-tolerant feature.
Theoretical modeling
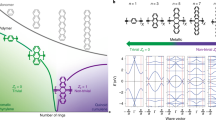
To further understand the difference among the three polymers in molecular scale and the importance of our design rules, we performed atomistic molecular dynamics simulations (see Supplementary Information for more details). Crystalline and disordered regions in polymer films are critical for interchain and intrachain charge transport27. Figure 5a, b show that both P(PzDPP-2FT) and P(PzDPP-4F2T) exhibit narrower torsion-angle distributions and more planar backbone in the crystalline region compared with P(PzDPP-T). This result is consistent with the better planarity of the fluorine-substituted polymers according to design Rule (1). Disorder regions may become more critical in doped conjugated polymers, as previous studies have shown that dopants tend to insert into the side chain or/and π–π stacking zones, perturbing the polymer packing and cause large structural disorder46. We then expanded the π–π stacking and lamellar distances of the polymers to simulate the disorder regions or the dopant-induced disorder regions. Without the restrain of tight π–π stacking, P(PzDPP-4F2T) shows obviously broadened torsion-angle distributions and its backbone severely deviates from planarity, whereas P(PzDPP-2FT) can still maintain planar conformation, thanks to its high and steep torsional barrier in each dihedral angle. The shape-persistent backbone and narrower torsion-angle distributions in P(PzDPP-2FT) provide the polymer with good interchain interactions, as demonstrated in the gelation experiment. As for P(PzDPP-T), it not only displays the widest dihedral angle distribution due to its low torsional potentials but also twists partly on account of its non-unique preponderant conformation at 0° or 180°, further emphasizing the importance of Rule (2) and Rule (3). To quantify the degree of disorder, we calculated the DOSs via DFT and compared the characteristic depth of the trap states (Eb) of the two fluorinated polymers. In crystalline regions, both polymers exhibit similar Eb of the DOS of LUMO energy level. When turned to disorder regions, the Eb of P(PzDPP-2FT) increased slightly from 18 to 23 meV, compared to P(PzDPP-4F2T) from 19 to 31 meV, outlining that the former owns a better disorder-tolerant ability. Thus, charge carriers in P(PzDPP-2FT) can transport more efficiently with less energetic disorder after dopants are added47. Considering the large proportion of disorder regions in polymer films, the intrachain charge transport in these disorder regions may strongly affect the whole charge-transport characteristics. All these results allow us to conclude that P(PzDPP-2FT) undoubtedly has excellent transport properties and superior disorder-tolerant ability.

a MD simulations of the polymers in crystalline and disorder regions. b Torsion-angle distributions for dihedral angles along polymer backbone (φ1 ~ φ7). The torsion-angle distribution width (W) based on the Gaussian fitting could be considered as the SD from planarity. c Calculated DOS distributions for dodecamers of both polymers. d, e δginter-isosurface and δginter-sign(λ2)ρ 2D fingerprint plots for illustrating the different intermolecular interactions between d P(PzDPP-2FT)−-TDAE+ and e P(PzDPP-4F2T)−-TDAE+.
We also performed DFT calculations to explore the intermolecular interactions between the polymers and the dopants. Using TDAE as an example, we found that P(PzDPP-2FT) has a stronger binding energy with TDAE+, because the cationic TDAE+ tends to dock into the “cavity” formed by the zigzag backbone (Supplementary Fig. 44 and Supplementary Table 3). Independent Gradient Model (IGM) analysis in Fig. 5d suggests that the major interactions between TDAE+ and P(PzDPP-2FT)− are hydrogen bonding and Van der Waals interactions, whereas only weaker Van der Waals interactions are found for P(PzDPP-4F2T)−-TDAE+ fragment. According to the IGM analysis, non-zero values of δginter exclusively correspond to interaction situations: the larger the δginter value, the stronger the interactions48. Therefore, larger δginter in the 2D fingerprint plot of P(PzDPP-2FT)−-TDAE+ supports the stronger intermolecular interaction and thus larger binding energy, compared to that of P(PzDPP-4F2T)−-TDAE+ (Fig. 5d and Supplementary Table 3). During the geometry-optimization process, we found that TDAE+ was easily captured in the “cavity” formed by the zigzag backbone of P(PzDPP-2FT), whereas it tended to stay on the top of the P(PzDPP-4F2T) backbone. This could result in serious disruption of the polymer packing in P(PzDPP-4F2T) or refraining the TDAE from doping. These results indicate that the zigzag backbone curvature can effectively capture TDAE dopants without much destruction to polymer packing and lead to enhanced miscibility and higher doping efficiency in P(PzDPP-2FT) films.
Discussion
In conclusion, with three design rules and a computer-aided building block screening process, we have successfully designed and synthesized a disorder-tolerant n-type polymer, P(PzDPP-2FT). Our results indicate that a highly dopable and disorder-tolerant polymer might need the following features: (i) high and steep torsion-angle barriers to prevent any distortions caused by dopants; (ii) single preferential planar conformation; (iii) strong interchain interactions (as indicated by the gel formation experiment) that can endure dopant interference and provide efficient interchain charge transport; and (iv) strong dopant-binding sites that do not disturb polymer packing. We envisage that our disorder-tolerant design could provide a valuable strategy for highly dopable and highly conductive conjugated polymers.
Data availability
The source data generated in this study have been deposited in the materials cloud database (https://archive.materialscloud.org/record/2021.127) and are also available from the corresponding author upon reasonable request.
Code availability
The codes that support the findings of this study are available from the corresponding author upon reasonable request.
References
Wang, S. et al. Skin electronics from scalable fabrication of an intrinsically stretchable transistor array. Nature 555, 83–88 (2018).
Someya, T., Bao, Z. & Malliaras, G. G. The rise of plastic bioelectronics. Nature 540, 379–385 (2016).
Xu, K. et al. Ground-state electron transfer in all-polymer donor-acceptor heterojunctions. Nat. Mater. 19, 738–744 (2020).
Yurash, B. et al. Towards understanding the doping mechanism of organic semiconductors by Lewis acids. Nat. Mater. 18, 1327–1334 (2019).
Schwarze, M. et al. Molecular parameters responsible for thermally activated transport in doped organic semiconductors. Nat. Mater. 18, 242–248 (2019).
Kolesov, V. A. et al. Solution-based electrical doping of semiconducting polymer films over a limited depth. Nat. Mater. 16, 474–480 (2017).
Yamashita, Y. et al. Efficient molecular doping of polymeric semiconductors driven by anion exchange. Nature 572, 634–638 (2019).
Kang, K. et al. 2D coherent charge transport in highly ordered conducting polymers doped by solid state diffusion. Nat. Mater. 15, 896–902 (2016).
Patel, S. N. et al. Morphology controls the thermoelectric power factor of a doped semiconducting polymer. Sci. Adv. 3, e1700434 (2017).
Kim, G.-H., Shao, L., Zhang, K. & Pipe, K. P. Engineered doping of organic semiconductors for enhanced thermoelectric efficiency. Nat. Mater. 12, 719–723 (2013).
Lussem, B. et al. Doped organic transistors. Chem. Rev. 116, 13714–13751 (2016).
Wang, S. H. et al. Thermoelectric properties of solution-processed n-doped ladder-type conducting polymers. Adv. Mater. 28, 10764–10771 (2016).
Liu, J. et al. N-type organic thermoelectrics of donor-acceptor copolymers: improved power factor by molecular tailoring of the density of states. Adv. Mater. 30, 1804290 (2018).
Liu, J. et al. Overcoming coulomb interaction improves free-charge generation and thermoelectric properties for n‑doped conjugated polymers. ACS Energy Lett. 4, 1556–1564 (2019).
Lu, Y. et al. Rigid coplanar polymers for stable n-type polymer thermoelectrics. Angew. Chem. Int. Ed. 58, 11390–11394 (2019).
Shi, K. et al. Toward high performance n-type thermoelectric materials by rational modification of BDPPV backbones. J. Am. Chem. Soc. 137, 6979–6982 (2015).
Lu, Y. et al. Persistent conjugated backbone and disordered lamellar packing impart polymers with efficient n-doping and high conductivities. Adv. Mater. 33, 2005946 (2021).
Wang, H. & Yu, C. Organic thermoelectrics: materials preparation, performance optimization, and device integration. Joule 3, 53–80 (2019).
Ye, Q. et al. Cyanated diazatetracene diimides with ultrahigh electron affinity for n-channel field effect transistors. Org. Lett. 15, 1194–1197 (2013).
Lu, Y., Wang, J. & Pei, J. Strategies to enhance the conductivity of n-type polymer thermoelectric materials. Chem. Mater. 31, 6412–6423 (2019).
Naab, B. D. et al. Mechanistic study on the solution-phase n-doping of 1,3-dimethyl-2-aryl-2,3-dihydro-1H-benzoimidazole derivatives. J. Am. Chem. Soc. 135, 15018–15025 (2013).
Liu, J. et al. Enhancing molecular n-type doping of donor-acceptor copolymers by tailoring side chains. Adv. Mater. 30, 1704630 (2018).
Perry, E. E. et al. High conductivity in a nonplanar n-doped ambipolar semiconducting polymer. Chem. Mater. 29, 9742–9750 (2017).
Shin, Y. et al. Improving miscibility of a naphthalene diimide-bithiophene copolymer with n-type dopants through the incorporation of “kinked” monomers. Adv. Electron. Mater. 4, 1700581 (2018).
Mei, J., Diao, Y., Appleton, A. L., Fang, L. & Bao, Z. Integrated materials design of organic semiconductors for field-effect transistors. J. Am. Chem. Soc. 135, 6724–6746 (2013).
Venkateshvaran, D. et al. Approaching disorder-free transport in high-mobility conjugated polymers. Nature 515, 384–388 (2014).
Noriega, R. et al. A general relationship between disorder, aggregation and charge transport in conjugated polymers. Nat. Mater. 12, 1038–1044 (2013).
Fratini, S., Nikolka, M., Salleo, A., Schweicher, G. & Sirringhaus, H. Charge transport in high-mobility conjugated polymers and molecular semiconductors. Nat. Mater. 19, 491–502 (2020).
Gumyusenge, A. et al. Semiconducting polymer blends that exhibit stable charge transport at high temperatures. Science 362, 1131–1134 (2018).
Che, Y. & Perepichka, D. F. Quantifying planarity in the design of organic electronic materials. Angew. Chem. Int. Ed. 60, 1364–1373 (2021).
Lin, T.-J. & Lin, S.-T. Theoretical study on the torsional potential of alkyl, donor, and acceptor substituted bithiophene: the hidden role of non-covalent interaction and backbone conjugation. Phys. Chem. Chem. Phys. 17, 4127–4136 (2015).
Mei, J., Kim, D. H., Ayzner, A. L., Toney, M. F. & Bao, Z. Siloxane-terminated solubilizing side chains: bringing conjugated polymer backbones closer and boosting hole mobilities in thin-film transistors. J. Am. Chem. Soc. 133, 20130–20133 (2011).
Scherf, U. & Mullen, K. Polyarylenes and poly(arylenevinylenes), 7. A soluble ladder polymer via bridging of functionalized poly(p-phenylene)-precursors. Macromol. Rapid Comm. 26, 1362–1370 (2005).
Köhler, A., Hoffmann, S. T. & Bässler, H. An order-disorder transition in the conjugated polymer MEH-PPV. J. Am. Chem. Soc. 134, 11594–11601 (2012).
Yao, S., Beginn, U., Gress, T., Lysetska, M. & Würthner, F. Supramolecular polymerization and gel formation of bis(merocyanine) dyes driven by dipolar aggregation. J. Am. Chem. Soc. 126, 8336–8348 (2004).
Clulow, A. J., Gilbert, E. P., Wolfer, P., Burn, P. L. & Gentle, I. R. Clustering of high molecular weight PCDTBT in bulk-heterojunction casting solutions. Macromolecules 48, 8331–8336 (2015).
Durand, A. Aqueous solutions of amphiphilic polysaccharides: concentration and temperature effect on viscosity. Eur. Polym. J. 43, 1744–1753 (2007).
Xiong, M. et al. Efficient n-doping of polymeric semiconductors through controlling the dynamics of solution-state polymer aggregates. Angew. Chem. Int. Ed. 60, 8189–8197 (2021).
Brondijk, J. J. et al. Two-dimensional charge transport in disordered organic semiconductors. Phys. Rev. Lett. 109, 056601–056601 (2012).
Nikolka, M. et al. High-mobility, trap-free charge transport in conjugated polymer diodes. Nat. Commun. 10, 2122 (2019).
Xiao, M. et al. Linking glass-transition behavior to photophysical and charge transport properties of high-mobility conjugated polymers. Adv. Funct. Mater. 30, 2007359 (2020).
Higgins, A., Mohapatra, S. K., Barlow, S., Marder, S. R. & Kahn, A. Dopant controlled trap-filling and conductivity enhancement in an electron-transport polymer. Appl. Phys. Lett. 106, 163301 (2015).
Arkhipov, V. I., Heremans, P., Emelianova, E. V. & Bässler, H. Effect of doping on the density-of-states distribution and carrier hopping in disordered organic semiconductors. Phys. Rev. B 71, 045214 (2005).
Salleo, A. et al. Intrinsic hole mobility and trapping in a regioregular poly(thiophene). Phys. Rev. B 70, 115311 (2004).
Liu, J. et al. N-type organic thermoelectrics: improved power factor by tailoring host-dopant miscibility. Adv. Mater. 29, 1701641 (2017).
Lu, Y. et al. The critical role of dopant cations in electrical conductivity and thermoelectric performance of n-doped polymers. J. Am. Chem. Soc. 142, 15340–15348 (2020).
Boyle, C. J. et al. Tuning charge transport dynamics via clustering of doping in organic semiconductor thin films. Nat. Commun. 10, 2827 (2019).
Lefebvre, C. et al. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 19, 17928–17936 (2017).
Acknowledgements
This work is supported by National Natural Science Foundation of China (22075001) and the Key-Area Research and Development Program of Guangdong Province (2019B010934001). X.Y. thanks the support of China Postdoctoral Science Foundation (8206200018 and 8206300146). W.H., Y.W., and J.Z. thank the financial support from National Natural Science Foundation of China Grant 21773016. S.Z., N.P., and X.G. thank the financial support from U.S. Department of Energy, Office of Science, Office of Basic Energy Science under the award number of DE-SC0019361, to make the X-ray scattering and AFM-IR measurements possible. We thank beamline BL14B1 (Shanghai Synchrotron Radiation Facility) for providing beamtime used for part of the XRD study. The computational part is supported by High-performance Computing Platform of Peking University. A portion of this research was conducted at the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility. A portion of this research used resources at the Spallation Neutron Source, a DOE Office of Science User Facility operated by the Oak Ridge National Laboratory.
Author information
Authors and Affiliations
Contributions
X.Y., M.X., and T.L. conceived the project. X.Y. synthesized the polymers and performed some characterization. K.-K.L. performed some characterization. M.X., J.-T.L., X.-Q.W., and J.-Y.W. performed device fabrication and characterization. J.-T.L. and X.-Y.D. performed theoretical modeling. S.Z., N.P., Z.C., and X.G. performed GIWAXS and AFM-IR measurements. Z.Z. and S.K.S. performed the PDS measurement. W.H., Y.W., and J.Z. performed part of the DFT calculations. X.Y., M.X., and T.L. wrote the manuscript. All the authors revised and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial or non-financial interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yan, X., Xiong, M., Deng, XY. et al. Approaching disorder-tolerant semiconducting polymers. Nat Commun 12, 5723 (2021). https://doi.org/10.1038/s41467-021-26043-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-26043-y
This article is cited by
-
Diketopyrrolopyrrole-based Conjugated Polymers as Representative Semiconductors for High-Performance Organic Thin-Film Transistors and Circuits
Chinese Journal of Polymer Science (2023)
-
Toward High Performance Ambipolar Transport from Super-exchange Perspective: Theoretical Insights for IID-based Copolymers
Chinese Journal of Polymer Science (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
