
Abstract
Metal/oxide interface is of fundamental significance to heterogeneous catalysis because the seemingly “inert” oxide support can modulate the morphology, atomic and electronic structures of the metal catalyst through the interface. The interfacial effects are well studied over a bulk oxide support but remain elusive for nanometer-sized systems like clusters, arising from the challenges associated with chemical synthesis and structural elucidation of such hybrid clusters. We hereby demonstrate the essential catalytic roles of a nanometer metal/oxide interface constructed by a hybrid Pd/Bi2O3 cluster ensemble, which is fabricated by a facile stepwise photochemical method. The Pd/Bi2O3 cluster, of which the hybrid structure is elucidated by combined electron microscopy and microanalysis, features a small Pd-Pd coordination number and more importantly a Pd-Bi spatial correlation ascribed to the heterografting between Pd and Bi terminated Bi2O3 clusters. The intra-cluster electron transfer towards Pd across the as-formed nanometer metal/oxide interface significantly weakens the ethylene adsorption without compromising the hydrogen activation. As a result, a 91% selectivity of ethylene and 90% conversion of acetylene can be achieved in a front-end hydrogenation process with a temperature as low as 44 °C.
Similar content being viewed by others


Introduction
Metal/oxide interface is of great fundamental and practical interest to heterogeneous catalysis, because it raises essential questions regarding the strong metal–support interaction1,2 and plays a pivotal role in several catalytic processes3,4. From a structural perspective, a metal/oxide interface is constructed by components that significantly differ from each other in terms of chemical compositions, bonding characters, lattice parameters, and electric and mechanical properties5,6, of which the adhesion structure and chemistry turn out to be a compelling research topic, while from a functional perspective, the chemical bonding and associated charge transfer7 at the metal/oxide interface allow the modulations of morphology, size, and electronic structures of metals to optimize the bonding strength of reaction intermediates for better catalytic performances3. During the past few decades, considerable progress has been achieved in the structural elucidation and tuning of well-defined metal/oxide interfaces that usually adopt a bulk oxide support to facilitate the nucleation, adsorption, or deposition of metals8. It is expected that a nanometer metal/oxide interface, perhaps formed by heterografting between metal and oxide clusters, would reinforce the structural and electronic effect to achieve better catalytic performance. However, due to the great challenges in the chemical synthesis and structural elucidation of such hybrid clusters, there are limited insights into the nanometer metal/oxide interface.
As a representative reaction where oxides supported metal catalysts are frequently used, the selective hydrogenation of acetylene to ethylene is subjected to an inherent trade-off between two essential requirements for both high catalytic activity and selectivity: the facile activation of hydrogen and the weak binding of ethylene9. Despite the significant progress achieved by Pd-based catalysts10, a simultaneous optimization of these two parameters is still challenging, especially in the front-end process where H2 and C2H4 are in large excess. To reach this goal requires a sophisticated tuning of the geometric and electronic structure of Pd, which motivates people to engineer the metal/oxide interface. In most Pd/oxide catalysts, only Pd nanoparticles or isolated Pd atoms are loaded. Unfortunately, Pd nanoparticles are efficient to activate hydrogen at low temperatures, but their strong binding with ethylene favors sequential hydrogenation of ethylene to ethane10,11. Isolated Pd site catalysts including Pd single-atom catalysts12,13,14,15 and Pd-based intermetallic compounds16,17,18,19,20,21,22 feature weak π-bonding with ethylene and thus good selectivity in the acetylene hydrogenation reaction, but their concomitant weakened hydrogen activation requires a relatively high reaction temperature (>100 °C) to achieve high conversion of acetylene, which potentially leads to a safety concern in the reactor beds21. Decreasing the size of oxide supports to nanocluster scale would remarkably change their coordination number (CN)23, surface termination24, and d-band character25, which allow a strong chemical and electronic interaction with Pd to continuously regulate the size and electronic structure of Pd. Among them, ligand-free Pd clusters stabilized by a nanometer metal/oxide interface are expected to bridge the size and performance gaps between Pd nanoparticles and single atoms with maximized interfacial effects.
Herein we propose a facile stepwise photochemical strategy to fabricate hybrid Pd/Bi2O3 clusters where the Pd clusters are stabilized by the ~1 nm ordered Bi2O3 cluster through the nanometer interfaces. These hybrid clusters are well and stably dispersed on TiO2 substrate with a high Pd loading up to 2.3 wt.%. They exhibit a low Pd–Pd CN of 4.7 and more importantly a Pd–Bi spatial correlation ascribed to the heterografting between Pd and Bi-terminated Bi2O3 clusters. Interestingly, these hybrid clusters feature an intra-cluster electron transfer toward Pd and result in a deeper d-band center compared with other Pd metals, which enables much weaker ethylene binding without compromising the hydrogen activation activity. As a result, a 90% conversion of acetylene together with a 91% selectivity to ethylene is achieved in excess of ethylene and at a temperature as low as 44 °C.
Results and discussion
Synthesis and structural characterizations
Pd1.0/Bi2O3/TiO2 catalysts were prepared by a stepwise photo-deposition of Bi and Pd on TiO2 with a molar ratio of Bi/Pd kept at 1.0 (please see details in “Methods”). In brief, Bi3+ was reduced by the photogenerated electrons to produce Bi0 clusters on TiO2. Subsequently, Pd was deposited onto Bi/TiO2 to form Pd/Bi/TiO2. When Bi/TiO2 and Pd/Bi/TiO2 were exposed to the air, Bi was spontaneously oxidized into Bi2O3 because the Gibbs free energy of the reaction is minus26. The synthetic procedure is schematically illustrated in Fig. 1a. Inductively coupled plasma–atomic emission spectroscopy (ICP-AES) suggests that the mass loading of Bi and Pd are 4.9 wt.% and 2.3 wt.%, respectively, close to the feeding ratio. Our previous study has proved that TiO2 has a strong interaction with Bi3+, characterized by an unprecedented 1.5 eV upshift of Ti 2p27. Such interaction ensures the formation of highly dispersed Bi on TiO2 during the following reduction of Bi3+ 28. As shown in Supplementary Fig. 1a, b, Bi species are uniformly dispersed on TiO2 as ~1 nm clusters with a Bi loading up to 5 wt.%. Interestingly, close inspection of individual Bi cluster on the TiO2 support by using aberration-corrected annular dark-field scanning transmission electron microscopy (ADF-STEM) indicates that the Bi cluster has an ordered α-Bi2O3 structure with a highly distorted lattice, which exhibits relatively weak diffuse peaks in the fast Fourier transform (FFT; Fig. 1b). This is consistent with the literature result26, which indicates that the mild oxidation of Bi0 in the air is thermodynamically spontaneous and usually forms monoclinic α-Bi2O3 (monoclinic, P21/c(14)). In addition to the intrinsic lattice distortion that blurs the image contrast, the beam-induced structural dynamics of the highly beam-sensitive Bi2O3 clusters may further introduce image blurring, mainly arising from the remarkable knock-on displacements and radiolysis. Despite these beam-induced effects, careful inspection on these ADF-STEM images allows the identification of local contrasts that closely resemble the projected structures and simulated ADF-STEM image of Bi2O3 along [100] (Fig. 1b). Specifically, the structural projection and simulated ADF-STEM image of ordered Bi2O3 feature a wave-shaped arrangement of Bi atomic columns from the [100] projection, while the experimental ADF-STEM image exhibits a similar contrast but with more lattice distortions (Fig. 1b). Accordingly, the FFT pattern of the experimental image exhibits rather diffuse spots compared with those in the FFT pattern of simulated image.

a Schematic illustration of the synthetic procedures. b–d STEM images of Bi2O3/TiO2 (b) and Pd1.0/Bi2O3/TiO2 (c, d). The insets from upper to lower and left to right: HRSTEM images, projected structural models, simulated ADF-STEM images, FFTs from dashed circular regions in HRSTEM images, and FFTs from simulated ADF-STEM images, respectively. e Elemental mapping of Pd1.0/Bi2O3/TiO2.
Followed by the secondary deposition of Pd, the clusters of Bi2O3 intergrown with Pd particles can be found according to the low-magnification ADF-STEM images (Supplementary Fig. 1). Besides, Pd1.0/Bi2O3/TiO2 exhibits no signals for Pd in the X-ray diffraction (XRD) patterns (Fig. 2a) while photo-deposited Pd/TiO2 with an identical Pd loading (2.5 wt.%) shows a characteristic Pd(111) diffraction peak29 at 40.1o, suggesting that pre-deposited Bi2O3 clusters assist in the high dispersion of Pd species in Pd1.0/Bi2O3/TiO2. According to the literatures30,31, pre-deposited component with high work function can serve as a sink of photo-induced electrons, which preferentially reduce the second metal on the surface of the pre-deposited metal. By fixing the loading of Bi at 5.0 wt.%, we can also fabricate Pd0.5/Bi2O3/TiO2 (Supplementary Fig. 2), Pd0.2/Bi2O3/TiO2, and Pd0.1/Bi2O3/TiO2 with high dispersion of Pd (Pdx/Bi2O3/TiO2, x represents Pd-to-Bi molar ratio). In contrast, when we increase Pd/Bi ratio to 3.0, the excess Pd also deposits directly on TiO2. As a result, Pd nanoparticles are obtained (~6.7 nm, Supplementary Fig. 3). Taking all above results together, Pd species are most likely deposited on the existing Bi2O3 clusters although the discrimination between Pd and Bi2O3 components is not straightforward by STEM imaging due to their small size and irradiation vulnerability. The nanoscopic elemental distribution can be mapped by a Super-X energy dispersive X-ray spectroscope (EDS) system with superior sensitivity. As shown in Fig. 1e and Supplementary Fig. 4, elemental mappings of Pd and Bi components suggest that their spatial distribution in most cases are correlated. In other words, these clusters are bicomponent with segregated Pd- and Bi-containing hemi-clusters, which unambiguously confirm the Pd-grafted Bi2O3 hybrid cluster structure. Careful inspection on several representative ADF-STEM images of Pd1.0/Bi2O3/TiO2 (Fig. 1c, d and Supplementary Fig. 5) allows the discrimination of Pd- and Bi-containing hemi-clusters from their distinct contrasts for adjacent clusters. The hemi-clusters featuring less bright contrast (marked by circle) are directly attached to brighter ones assigned to Bi2O3 hemi-clusters, further confirming that Pd is grafted onto the surface of Bi2O3 clusters. Notably, most such Pd clusters are identified to bond and hybridize with Bi2O3 clusters without any fixed orientation relationship or facet preference, probably due to the ultra-small size and large strain of the clusters. Two representative atomic-resolution ADF-STEM images of Pd-Bi2O3 nanoclusters with well-resolved Pd- and Bi-containing hemi-clusters are shown in Fig. 1c, d, of which the image contrasts and FFT patterns closely resemble the simulated ones of artificially constructed hybrid cluster models with different orientation relationships between two types of hemi-clusters made of Pd and α-Bi2O3 structures, respectively. The above observations unambiguously validate the proposed Pd–Bi2O3 hybrid structural model. Quite rarely, such Pd clusters are observed to directly nucleate onto the TiO2 substrate.

a XRD patterns of TiO2, Pd/TiO2, Bi2O3/TiO2, and Pd1.0/Bi2O3/TiO2; b Fourier transform spectra of Pd K-edge EXAFS for Pd/TiO2, Pd1.0/Bi2O3/TiO2, and oxidized Pd1.0/Bi2O3/TiO2 (Pd1.0/Bi2O3/TiO2-ox); c Fourier transform spectra of Bi L3-edge EXAFS; d Bi L3-edge XANES spectra for Bi2O3/TiO2, Pd1.0/Bi2O3/TiO2, and Pd1.0/Bi2O3/TiO2-ox. Bi and Bi2O3 powder were used as references. e Pd K-edge XANES spectra for Pd/TiO2, Pd1.0/Bi2O3/TiO2, and Pd1.0/Bi2O3/TiO2-ox. Pd foil was used as a reference. f CO-adsorbed FT-IR spectra for various samples. Source data are provided in a Source data file.
To confirm the coordination environment of Pd clusters, X-ray absorption fine structure (XAFS) of Pd K-edge and Bi L3-edge were performed, as shown in Fig. 2b–e. In the Fourier transformed extended X-ray absorption fine structure (FT-EXAFS) data of Bi L3-edge, it is important to note that a new coordination peak in R-space at about 2.6 Å is observed on Pd1.0/Bi2O3/TiO2 (Fig. 2c), but it is totally absent on other samples containing Bi2O3 (i.e., Bi2O3 and Bi2O3/TiO2). Considering that the only difference between Pd1.0/Bi2O3/TiO2 and Bi2O3/TiO2 is the secondary deposition of 2.3 wt.% Pd on Bi2O3/TiO2, the peak at 2.6 Å should be contributed by the Bi–Pd interaction. Interestingly, the peak can be well fitted by a single Bi–Pd shell (Supplementary Fig. 6 and Supplementary Table 1), suggesting it is a characteristic peak of Bi–Pd coordination. To further prove this idea, the Pd1.0/Bi2O3/TiO2 is mildly treated under air at 150 °C (denoted as Pd1.0/Bi2O3/TiO2-ox) to oxidize Pd species while maintaining the structural integrity of Bi2O3 clusters32. In Fig. 2e, Pd1.0/Bi2O3/TiO2-ox shows clearly blue shift of the absorption edge position comparing with Pd1.0/Bi2O3/TiO2, indicating the oxidation of Pd after the treatment. Meanwhile, the peak at ~2.6 Å of Bi L3-edge EXAFS diminishes significantly upon oxidation of Pd1.0/Bi2O3/TiO2 (Fig. 2c). The fitting results reveal that the Bi–Pd CN is decreased from 2.9 for Pd1.0/Bi2O3/TiO2 to 1.1 for Pd1.0/Bi2O3/TiO2-ox (Supplementary Table 1). Accordingly, this experiment identifies that the peak at 2.6 Å is from Bi–Pd bond and in turn proves the Pd–Bi pairs across the interfaces of Pd/Bi2O3 hybrid clusters observed by ADF-STEM in Fig. 1c, d. The Bi–Pd interaction eventually changes the coordination environment of Pd in Pd1.0/Bi2O3/TiO2. As compared to Pd foil and Pd/TiO2, the significantly weaker and slightly broader coordination peak in the Pd K-edge EXAFS of Pd1.0/Bi2O3/TiO2 (Fig. 2b) implies a decreased CN of Pd–Pd pairs and distorted structure for the Pd clusters similarly as observed by STEM imaging33. The fitting of Pd K-edge EAXFS (Supplementary Fig. 7 and Supplementary Table 2) further confirms that the presence of Pd–Bi coordination (CN = 4.6) decreases the Pd–Pd CN from 10.0 for Pd/TiO2 to 4.7 for Pd1.0/Bi2O3/TiO2, which is consistent with Pd cluster structure observed by ADF-STEM. The Pd–Bi bond length obtained from the fitting of Pd K and Bi L3 edge is 2.79 ± 0.03 Å, suggesting the observed spatial correlation of Pd–Bi pairs arises from the direct bonding between Pd- and Bi-terminated clusters instead of the Pd–O–Bi moieties that attain a larger interatomic distance (~3.5 Å). Photo-deposition procedure is critical to ensure the formation of direct Pd–Bi bonding in Pd/Bi2O3 clusters as depicted in Fig. 1a. Theoretically, Bi2O3 favors an oxygen termination. Pd supported on Bi2O3 would be in direct contact with O rather than Bi. We prepared a Pd/Bi2O3 sample by directly depositing Pd onto commercial Bi2O3 support. Interestingly, no characteristic peak at ~2.6 Å was observed in the Bi L3 EXAFS of Pd/Bi2O3, excluding the presence of Pd–Bi bond (Fig. 2c). In this study, photo-deposition procedure ensures the Pd cluster deposited on reduced Bi0 clusters and allows the formation of Pd–Bi bonding during the synthesis as depicted in Fig. 1a. The Pd–Bi bond preserves during the mild oxidation of Bi to Bi2O3 at room temperature (RT) that occurred after the deposition of Pd onto Bi/TiO2, as evidenced by the characteristic Bi–Pd peak in Bi L3 EXAFS (Fig. 2c). More interestingly, 36% of this peak preserves even after oxidation in air at a higher temperature of 150 °C (Pd1.0/Bi2O3/TiO2-ox). These results clearly indicate the good stability of Pd–Bi bond in Pd1.0/Bi2O3/TiO2 and suggest that Pd supported on Bi-terminated Bi2O3 as the structural model for Pd1.0/Bi2O3/TiO2 in the hydrogenation reaction conditions. Taken together, the above-mentioned results unambiguously confirmed that the Pd species are predominantly in the form of nanometer-sized Pd clusters embedded in the Pd/Bi2O3 hybrid clusters through chemical adhesion.
In order to identify the electronic structure of the as-synthesized Pd/Bi2O3 hybrid clusters in reaction conditions, in situ X-ray photoelectron spectra (XPS) was collected at 100 °C under H2 atmosphere (Supplementary Fig. 8). As shown in Supplementary Fig. 8b, there are symmetric Bi 4f peaks at 158.8/164.1 eV, confirming that the majority of Bi are in the form of Bi2O334. Both Bi2O3/TiO2 and Pd1.0/Bi2O3/TiO2 exhibit similar Bi L3-edge structures with Bi2O3 by inspecting the X-ray absorption near edge structure (XANES), indicating similar valence states among them (Fig. 2d). It is frequently reported that intermetallic compounds (IMC) could be possibly formed by a so-called reactive metal–support interaction35. However, according to the density functional theory (DFT) calculations (Supplementary Table 3), the Pd–Bi distance in PdBi IMC ranges from 2.85 to 3.03 Å, which is obviously larger than that of Pd1.0Bi/TiO2 (2.79 Å, Supplementary Table 2). This result excludes PdBi IMC as the main phase of Pd1.0/Bi2O3/TiO2. To better illustrate the difference between Pd–Bi2O3 nanoclusters and PdBi intermetallic compounds, we synthesized a PdBi IMC (denoted as PdBi/TiO2) for comparison (Supplementary Fig. 9). XRD patterns (Supplementary Fig. 9a) of PdBi/TiO2 exhibit characteristic peaks at 39.9 and 42.8°, corresponding to (102) and (110) of hexagonal PdBi IMC (sobolevskite, P63/mmc(194), a = b = 4.22 Å, c = 5.709 Å, α = β = 90°, and γ = 120°). Pd–M (M = Pd or Bi) coordination peaks in R space of PdBi/TiO2 obviously shift (~0.06 Å) from that of Pd1.0/Bi2O3/TiO2, confirming that PdBi IMC have longer Pd–Bi distances than Pd–Bi2O3 nanoclusters (Supplementary Fig. 9b). More importantly, a massive Bi0 peak at 156.6/162.0 eV (Supplementary Fig. 9d) is observed in the Bi 4f of PdBi/TiO2 but is absent in that of Pd1.0/Bi2O3/TiO2 (Supplementary Fig. 8b). These results clearly indicate that Pd1.0/Bi2O3/TiO2 is composed of Pd–Bi2O3 hybrid clusters rather than PdBi IMC. It is reasonable because the reaction temperature in this study is too low to reduce Bi2O3 to Bi, which is a prerequisite for the formation of PdBi IMC. On the other side, the Pd 3d patterns of Pd/TiO2 and Pd1.0/Bi2O3/TiO2 exhibit a slightly asymmetric lineshape (Supplementary Fig. 8a), which is most likely due to the many-body screening response of conduction electrons to the photoemission of a core electron36. The predominant Pd 3d signals of Pd/TiO2 and Pd1.0/Bi2O3/TiO2 locate at ~334.9/340.0 eV, which are characteristic of Pd0. It is important to note that, due to the differences in the extra-atomic relaxation of metal particles of different sizes, decreasing the particle size of Pd generally upshifts Pd 3d to higher binding energy (BE)37. In this study, the Pd 3d and 3p of Pd1.0/Bi2O3/TiO2 downshift slightly to lower BE although the particle size of Pd1.0/Bi2O3/TiO2 is significantly smaller than that of Pd/TiO2 (Supplementary Figs. 8a, c). These results, opposite to the particle-size-induced BE shift, indicate a charge transfer from Bi to Pd. Similar result was also reported in Au/TiO2 system38.
The more important feature associated with the electronic structure of Pd clusters is observed in the Pd K-edge and L3-edge structures as shown in Fig. 2e and Supplementary Fig. 10. Specifically, the Pd K-edge XANES profile of Pd/TiO2 is very similar to that of Pd foil. In contrast, Pd1.0/Bi2O3/TiO2 shows a marked red-shift of the absorption edge energy and decrease in the white line intensity. This indicates the electron-richness of Pd atoms in Pd1.0Bi/TiO2 compared to those in Pd foil and Pd/TiO2, which most likely arises from the Bi2O3-to-Pd intra-cluster electron transfer. Moreover, Pd1.0/Bi2O3/TiO2 shows a much weaker Pd L3 white line intensity at ~3173 eV than Pd/TiO2 (Supplementary Fig. 10), which indicates an enhanced d-band filling. Such phenomenon is similarly predicted by the Bader charge analysis over Pd8 clusters supported by the Bi2O3 cluster as shown in Supplementary Fig. 11. It is quite surprising to observe such an enhanced d-band filling and associated negative shift of d-band center away from the Fermi level for nanometer-sized Pd clusters, because the strong “size effect” of most supported metals usually leads to a decreased d-band filling and thus positive shift of d-band center toward Fermi level for smaller nanoparticles39. Actually, this has become a limiting factor for applying supported metal nanoparticles in the selective acetylene hydrogenation reaction due to the strong adsorption of ethylene molecules on electron-deficient metals and results in over-hydrogenation. The metal termination of Bi2O3 observed here at the Pd–Bi interface of the hybrid cluster could lead to a strong downshift of its surface conduction band. Similar results were also reported in metal–metal interface of Pd/MgO, Pd/ZnO, Ru/MgO, and Au/TiO238,40,41,42. Electrons transferred from terminated magnesium or zinc to adsorbed Pd can also result in negatively charged Pd. This charge transfer was ascribed to the band filling modification and the orbital hybridization between substrate and metal atoms40. Specifically, for Pd deposited on magnesium termination, the surface Mg conduction band is shifted toward higher energy and is emptied, while the Pd d band is shifted in the opposite direction and becomes filled. Similar band filling modification and orbital hybridization might also apply to Pd–Bi2O3 system. This allows the Bi2O3-to-Pd intra-cluster electron transfer through direct Pd–Bi bonding across the interface, which leads to a greater filling of high-lying d bands in Pd clusters and largely circumvented over-hydrogenation problem. It is important to note that the charge transfer between Pd and Bi is localized in the Pd–Bi interface. Considering the relatively higher concentration of Bi2O3 comparing with Pd, the signal of electron-deficient Bi is likely overwhelmed by the signal of unaffected Bi2O3 and is therefore not observed by XPS and XANES.
The unique atomic and electronic structures of heterografted Pd cluster further result in its largely modulated gas adsorption behaviors compared with other supported Pd metals, which can be investigated by the in situ diffuse reflectance infrared Fourier transform spectroscopy using CO as the probe molecule. As shown in Fig. 2f, signals at 2089 and 2059 cm−1 for linear-bonded CO43 over Pd/TiO2 redshift to 2076 and 2055 cm−1 for those over Pd1.0/Bi2O3/TiO2, suggesting a strengthened π-back donation of metal d-electrons to π* orbitals of CO over Pd1.0/Bi2O3/TiO2 and a downshift of d-band center44. This result is well consistent with the XPS and XANES results. It is noteworthy that signals for bridge-bonded CO (1996 and 1948 cm−1) are observed over Pd/TiO2 but invisible over Pd1.0/Bi2O3/TiO2, likely arising from combined size and electronic effects. Specifically, the downshifted d-band center weakens the adsorption strength of CO on Pd44. More importantly, the small size and large structural distortion of Pd clusters disfavor the bridge adsorption mode of CO molecules due to the low average CN and broad distance distribution of Pd–Pd pairs.
Catalytic performance in acetylene hydrogenation
With the unique hybrid cluster structure and Bi2O3-to-Pd intra-cluster electron transfer, Pd1.0/Bi2O3/TiO2 readily serves as a model catalyst to demonstrate the essential catalytic role of a nanometer metal/oxide interface. The catalytic properties were evaluated in selective hydrogenation of acetylene with excess ethylene, mimicking the front-end condition. In this condition, the thermodynamically favored over-hydrogenation of ethylene with the large excess hydrogen generally leads to an unsatisfied C2H4 selectivity at high C2H2 conversion, accompanied by a thermal runaway. To overcome this problem, a small amount of CO is usually added in the feed stream to reduce the reaction rate so as to improve the C2H4 selectivity10. At very low CO levels, high C2H4 selectivity and C2H2 conversion are difficult to achieve simultaneously. In this study, no CO is added in the feed stream. Both Pd/TiO2 and well-established Pd1Ag3/Al2O3 catalysts30,45 were evaluated for comparison with Pd1.0/Bi2O3/TiO2. The composition and synthesis procedure of the PdAg3/Al2O3 catalyst is the same as OleMax@251, a widely used industrial catalyst for acetylene hydrogenation45. The carbon balances are all >99%. Negligible oligomers were formed during the hydrogenation process, likely due to the short contact time and high concentration of hydrogen. According to the literatures, the large excess of hydrogen would change the adsorption modes of C2H2 and C2H4 from C-CH2 vinylidene and C-CH3 ethylidyne to weak π-bonded C2H2 and C2H446,47. As a result, the possible reaction between vinylidene and acetylene to form C4 species as well as the hydrocarbon isomerization and decomposition are suppressed.
Figure 3a plots the C2H4 selectivity as a function of C2H2 conversion on various catalysts. Consistent with the literature results30, the C2H4 selectivity drops rapidly on well-established Pd1Ag3/Al2O3 catalyst once the C2H2 conversion exceeds 40%. On the contrary, Pd1.0/Bi2O3/TiO2 and Pd0.2/Bi2O3/TiO2 catalysts preserve very high selectivity toward C2H4 at much higher C2H2 conversions. Figure 3b compares the C2H4 selectivity at 95% C2H2 conversion. Interestingly, all catalysts except Pd1.0/Bi2O3/TiO2 and Pd0.2/Bi2O3/TiO2 exhibit negative selectivity toward C2H4 due to the over-hydrogenation of ethylene to ethane. Specifically, Pd1.0/Bi2O3/TiO2 exhibits much higher selectivity than 2.3 wt.% Pd/TiO2 regardless of the same Pd loading. In addition, we also synthesized Bix/Pd/TiO2 (x = 0.5, 1, x is the molar ratio of Bi to Pd) by photo-depositing Bi onto 2.3 wt% Pd/TiO2. Interestingly, both Bi0.5/Pd/TiO2 and Bi1.0/Pd/TiO2 exhibit 100% conversion of C2H2 and negative selectivity toward ethylene (−123% for Bi0.5/Pd/TiO2 and −143% for Bi0.5/Pd/TiO2) at RT. These results suggest that a simple site blocking mechanism cannot explain the beneficial effect of Bi in this study. PdBi/TiO2 composed of PdBi IMC (Supplementary Fig. 9) shows 100% conversion of C2H2 and negative selectivity toward ethylene (−1319%) at RT. The strong exothermic effect of unselective acetylene hydrogenation eventually leads to a runaway temperature up to 58.5 °C. These results exclude PdBi IMC as the active site for Pd1.0/Bi2O3/TiO2 and further indicates the critical role of the nanometer Pd/Bi2O3 interface in the catalytic selectivity of Pd. It is important to highlight that 91% selectivity of C2H4 with 90% conversion of C2H2 is achieved by Pd1.0/Bi2O3/TiO2 at a temperature as low as 44 °C (Fig. 3c). Such an excellent low-temperature performance of acetylene hydrogenation has never been reported previously, suggesting the unique structure and catalytic properties of Pd1.0/Bi2O3/TiO2. Moreover, the C2H2 conversion and C2H4 selectivity remain almost constant over 24 h operating at 40 °C (Fig. 3d). XRD and XAFS also confirm that Pd/Bi2O3 hybrid cluster structure is still maintained (Fig. 2), demonstrating a good long-term stability of Pd1.0/Bi2O3/TiO2.

a Selectivity as a function of acetylene conversion over Pd1.0/Bi2O3/TiO2, Pd0.2/Bi2O3/TiO2, and PdAg3/Al2O3. b The selectivity to C2H4 for 95% acetylene conversion over different catalysts. c Reaction temperature (T90) and C2H4 selectivity for 90% acetylene conversion. *For Pd3.0/Bi2O3/TiO2, hydrogen dissociation easily takes place at room temperature. The strong exothermic effect of unselective acetylene hydrogenation eventually leads to a runaway temperature up to 63.5 °C. d C2H2 conversion and C2H4 selectivity with time on stream over Pd1.0/Bi2O3/TiO2 at 40 °C. e H2-TPR profiles for Pd/TiO2, Bi2O3/TiO2, and Pd1.0/Bi2O3/TiO2. f Microcalorimetric studies of C2H4 pulse adsorption over Pd/TiO2 and Pd1.0/Bi2O3/TiO2. Source data are provided in a Source data file.
The variation of Pd-to-Bi ratio leads to an evolution of Pd/Bi2O3 hybrid clusters and major alteration of their catalytic performances in selective acetylene hydrogenation. Usually, the high Pd-to-Bi ratio results in the formation of Pd nanoparticles along with decreased Pd/Bi2O3 clusters, while low Pd-to-Bi ratio results in Pd single atoms. Here the reaction temperature (T90) and C2H4 selectivity at 90% conversion of acetylene are utilized to evaluate the catalytic performance of the samples. As shown in Fig. 3c, T90 increases along with the decrease of Pd-to-Bi ratio, implying that small Pd size is unfavorable to hydrogenation. Under an extreme condition when Pd is atomically dispersed (Pd-to-Bi ratio ≤0.1), a very high T90 (>90 °C) will be obtained, likely due to its poor ability of hydrogen activation21. In contrast, the moderate size of Pd in the hybrid clusters allows the efficient activation of hydrogen at much lower temperatures. All these catalysts exhibit quite high C2H4 selectivity. The Pd3.0/Bi2O3/TiO2 with an increased Pd-to-Bi ratio generates Pd nanoparticles and can even convert >90% of acetylene at RT. However, it suffers from the negative C2H4 selectivity at 90% conversion of acetylene. The above results experimentally confirm the trade-off between the conversion of acetylene and the selectivity of C2H4 upon the size effect of Pd, while an optimal catalytic performance is achieved over the Pd clusters stabilized by a nanometer metal–oxide interface.
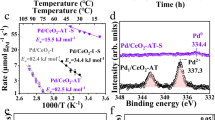
The origin of such inherent trade-off in the catalytic performance of Pd lies in two aspects: the hydrogen activation and ethylene adsorption. It is generally accepted that the facile dissociative activation of hydrogen on Pd nanoparticles produces too much active H species, which migrate into the Pd lattice and generate β-hydride phase that leads to over-hydrogenation of ethylene43. Accordingly, a negative peak (55–75 °C, Fig. 3e) characteristic for the decomposition of β-hydride phase is observed from the temperature programmed reduction (TPR) profile of Pd/TiO2, which is, however, not observed from that of Pd1.0/Bi2O3/TiO2. These results are consistent with literatures that report high-coordinated Pd ensembles (the case of Pd/TiO2) as active sites for the formation of β-hydride43. As for Pd1.0/Bi2O3/TiO2, the Pd/Bi2O3 hybrid clusters feature a small Pd–Pd CN and nanometer Pd–Bi interface, which prevent the formation of β-hydride. The lack of β-hydride suppresses the over-hydrogenation of ethylene to ethane and therefore contribute to the high C2H4 selectivity48. It is important to highlight that the reduced size of Pd cluster does not compromise the hydrogen activation and thus the catalytic activity. As a result, a low T90 could be achieved on Pd1.0/Bi2O3/TiO2 (Fig. 3c).
In addition to the formation of β-hydride, the adsorption behavior of ethylene is also strongly influenced by the Pd structures, which leads to different microcalorimetric profiles49. Figure 3f shows the heat flow during the C2H4 pulse adsorption process as a function of time for Pd1.0/Bi2O3/TiO2, Pd/TiO2, and Bi2O3/TiO2 at 40 °C. Obvious heat flow signals are observed on Pd/TiO2 and Pd1.0/Bi2O3/TiO2 but are absent on Bi2O3/TiO2 and blank test, demonstrating that C2H4 is adsorbed on Pd instead of on Bi2O3. It is interesting to note that the amplitude of the heat flow signal of Pd1.0/Bi2O3/TiO2 is much smaller than that of Pd/TiO2.The calculated adsorption heat of Pd1.0/Bi2O3/TiO2 (~5.9 kJ mol-1) is significantly lower than that of Pd/TiO2 (~234.5 kJ mol−1), clearly indicating a much weaker ethylene adsorption on Pd1.0/Bi2O3/TiO2. These observations can be attributed to the unique geometric and electronic structure of hybrid cluster, similar to the results reported in the alloying of Pd50,51. The low Pd–Pd coordination and the Bi2O3-to-Pd intra-cluster electron transfer likely change the adsorption configuration of C2H4 from stable ethylidyne to weak π-bonded C2H4 and promote the desorption of ethylene as the desired product. To confirm this hypothesis, we further performed the temperature programmed desorption of ethylene by monitoring the mass signal of m/e = 27 (Supplementary Fig. 12). According to the literature, the peak at ~65 °C could be assigned to weak π-bonded ethylene species, which readily desorb without decomposition52. The peak centered at ~115 °C originates from di-σ-bonded ethylene, which undergoes decomposition followed by the recombination of surface hydrocarbon species and hydrogen to produce ethylene and ethane52. Compared with Pd/TiO2, Pd1.0/Bi2O3/TiO2 exhibits a much weaker peak at ~115 °C but a significantly larger peak at 65 °C. These results confirm that the adsorption configuration of C2H4 is changed from the strong σ-bonding for Pd/TiO2 to weak π-bonding for Pd1.0/Bi2O3/TiO2.
Reaction mechanism investigated by DFT calculations
DFT calculations were performed to further provide insights into the molecular-level mechanisms of acetylene hydrogenation on experimental Pd1.0/Bi2O3/TiO2 catalyst. Model of Bi2O3-supported Pd8 cluster catalyst was built according to the experimental characterization results and the structure of which is shown in Fig. 4a (please see the details of model development in Supplementary Information). In this model, the size of Pd cluster on Bi2O3(100) is around 1.6 nm × 1.5 nm. In addition, this Pd cluster shows an average Pd–Pd CN of 4.0, which is close to the experimental values measured, i.e., 4.7 ± 0.5 (Supplementary Table 2). The Pd–Bi pair distribution function of the Pd8 cluster structure is shown in Supplementary Fig. 13. In this figure, the dominant peak appears at ~2.75 Å, which is smaller than that in the PdBi intermetallic model (~2.91 Å, Supplementary Table 3). In addition, Bader charge analysis suggests that Pd atoms in PdBi IMC (average charge of −0.36 e) model are more electron-rich than those in Pd-Bi2O3 hybrid clusters model (−0.21 e). These results are well consistent with Pd K-edge XANES and Pd 3d XPS shown in Supplementary Fig. 9, which therefore validate the reliability of the Pd8 cluster model. The possibility of hydride formation over the Pd cluster was studied, and it was found that hydrogen atoms prefer to adsorb at surface Pd sites after optimization (Supplementary Fig. 14), suggesting that formation of Pd hydride from this cluster is difficult, which again agrees well with the results shown in Fig. 3e.

a Optimized Pd cluster structure for DFT calculation (Pd: cyan, Bi: purple, O: red) and b energy profile of acetylene hydrogenation to ethane on Pd(111) and Pd8 cluster supported on Bi2O3(100). c DOS projected onto d electrons over Pd atom of Pd(111) and Pd8 cluster structures. A surface Pd atom of Pd(111), and the most active Pd atom of Pd8 cluster structure (on which C2H4 adsorbs most strongly) are chosen to plot the DOS. The position of d-band center (εd) is highlighted with a red bar. d Eads of C2H4 as a function of εd over different Pd atom on Pd cluster surface (black squares). The most stable adsorption configuration is shown as solid square, while the other less stable adsorption structures are denoted by hollow squares. A surface Pd atom of Pd(111) is also shown as red solid square for comparison. The blue fitted line is a guide for the eyes. It shows that a more negative εd corresponds to a more positive Eads of C2H4. Source data are provided in a Source data file.
It was reported that C-CH2 vinylidene and C-CH3 ethylidyne are energetic stable and important spectator species in acetylene hydrogenation53,54,55. However, in the front-end condition where H2 is in large excess, vinylidene and ethylidyne are readily hydrogenated and are insignificant at steady-state conditions10. Besides, the adsorption configurations of C2H2 and C2H4 also depend on the catalyst structure. For the Pd cluster structure studied in this work, it is found that the hydrogenation of CH≡CH to CHCH2 has the lowest activation energy (Ea) of 0.74 eV compared with the dehydrogenation or hydrogen shift of CH≡CH (Supplementary Table 4). This strongly suggests that formation of CCH2 would be unfavorable on the Pd cluster structure. Similarly, the hydrogenation of CHCH2 to CH2CH2 has the lowest Ea among all reactions starting from CHCH2, again indicating that the spectator species CCH3 is hard to form on Pd cluster model (please see Supplementary Information for details). To this end, the effects of these species are not discussed here. The energy profile of acetylene hydrogenation to ethane over this Pd cluster is shown in Fig. 4b, and the corresponding optimized configurations of surface intermediates and transition states are shown in Supplementary Fig. 15. Meanwhile, the energy profile on Pd(111) representing Pd foil is also shown in Fig. 4b for comparison. The calculated barriers are generally consistent with the reported values (Supplementary Table 5), demonstrating that our calculated results are reliable. In addition, we further calculated the vibrational frequency based on the transition state structures on Pd(111). All the transition states were characterized to possess only one imaginary frequency along the bond formation of hydrogenation reactions. These results further demonstrate the reliability of the constrained minimization method that we used in this work. As can be seen from Fig. 4b, the adsorption of C2H2 over Pd(111) and Pd8 cluster is exothermic, and the transition state energies of C2H2 hydrogenation to C2H4 on both surfaces are below the energy of gaseous C2H2, suggesting that the hydrogenation processes should be facile. The semi-hydrogenation product C2H4 would either desorb from the surface or undergo further hydrogenation to ethane (C2H6). In Fig. 4b, the transition state energy of C2H4 hydrogenation on Pd8 cluster is above the gaseous C2H4 energy, suggesting that desorption of C2H4 from the Pd8 cluster may be favored compared with its further hydrogenation to C2H5. Herein the difference between the barriers for further hydrogenation and desorption of ethylene can be used to estimate the possibility of selective C2H4 formation17,56,57,58,59,60. Within this framework, a more positive value of Ea,hydro − |Eads,C2H4|, where Ea,hydro is the effective hydrogenation barrier of C2H4 to C2H6 and |Eads,C2H4| is the absolute value of C2H4 adsorption energy, corresponds to higher C2H4 selectivity. The calculated values of Ea,hydro − |Eads,C2H4| over Pd8 cluster and Pd(111) are 0.53 and 0.31 eV, respectively, demonstrating higher C2H4 selectivity over the Pd8 cluster than Pd(111), which is consistent with the experimental results.
We further carried out electronic structure analysis to understand the weaker adsorption of reaction intermediates over the Pd cluster than Pd(111). In Fig. 4c, we plotted the density of states projected onto d-electrons of the surface Pd atom where C2H4 adsorbs over Pd(111) and Pd cluster, and the d-band center (εd) were calculated to be −1.53 and −1.79 eV, respectively. According to the d-band center theory61, the more negative εd indicates weaker binding to adsorbates, which is in agreement with the trend of adsorption energies calculated for the reaction intermediates over Pd(111) and Pd cluster. In addition, the εd of each Pd atom on Pd cluster was found to be correlated with Eads of C2H4 as shown in Fig. 4d, and a more negative εd generally corresponds to weaker adsorption of C2H4. The most stable adsorption of ethylene, corresponding to the black solid square in Fig. 4d and *C2H4 in Fig. 4b, gives rise to the Eads of −0.54 eV, which is slightly weaker than that over Pd(111), i.e., −0.57 eV. However, one can also see that C2H4 adsorbs much weaker at other Pd sites over the Pd8 cluster (shown as black hollow squares), therefore Pd8 cluster shows weaker adsorption on average compared with those sites over Pd(111) where all the surface sites are identical. C2H4 would be more likely to desorb from the Pd cluster, resulting in the high C2H4 selectivity observed.
Our findings demonstrate the essential catalytic roles of a nanometer metal/oxide interface in selective hydrogenation of acetylene. Pd–Bi2O3 hybrid clusters feature a small Pd–Pd CN and intra-cluster electron transfer, which enables a weak C2H4 adsorption without compromising the H2 activation activity. The superior low-temperature catalytic performance of Pd–Bi2O3 nanocluster ensembles over Pd single atom and nanoparticles might open new opportunities for fundamental research of hybrid nanoclusters. Besides, the demonstrated stepwise photochemical strategy also provides a new path for fabricating hybrid nanoclusters and nanometer metal/oxide interface.
Methods
Synthesis of Pdx/Bi2O3/TiO2
Pdx/Bi2O3/TiO2 (x is the nominal molar ratio of Pd to Bi) catalysts were prepared by a two-step photo-deposition method using a high-pressure Xe lamp (300 W) as the light source. Typically, 100 mg of TiO2 and 11.6 mg of Bi(NO3)3·5H2O (Sinopharm chemicals, 99%) were dispersed in 4 mL of ethylene glycol in a Pyrex glass reactor. Prior to ultraviolet irradiation for 1 h, the suspension was bubbled with Ar for 30 min to eliminate dissolved O2. Subsequently, 8 mL of PdCl2 aqueous solution (the concentration is determined by x) was added into the suspension. After irradiation in Ar for another 1 h, the precipitates were collected by centrifugation, washed twice by water and ethanol, and then dried in an oven at 40 °C. When the sample was exposed in the air, Bi was oxidized to Bi2O3 spontaneously. The catalyst was activated by H2 at 100 °C for 1 h and then cooled to RT in N2 prior to the catalytic reaction and characterizations. The synthetic procedures of Bi2O3/TiO2, Pd/TiO2, Pd1.0/Bi2O3/TiO2-ox, PdBi/TiO2, and Pd/Bi2O3 are presented in Supplementary Information.
Characterization
Powder XRD patterns were recorded on a Rigaku Ultimate IV diffractometer using Cu Kα radiation operated at 40 mA and 40 kV (scan rate: 5o min−1). XPS measurements were performed in a VG Scientific ESCALAB Mark II spectrometer. All BEs were referenced to the C 1s peak at 284.8 eV of the surface adventitious carbon to correct the shift caused by charge effect. In situ near ambient pressure XPS (NAP-XPS) was conducted using a Specs NAP-XPS system with a PHOIBOS NAP hemispherical energy analyzer in Vacuum Interconnected Nanotech Workstation, Suzhou Institute of Nano-Tech and Nano-Bionics. The NAP operation was conducted under 1 mbar H2 atmosphere. The actual loading of Pd and Bi were analyzed by ICP-AES using a Profile Spec ICP-AES spectrometer (Leeman, USA). Structural characterizations and chemical composition distribution were determined by a probe Cs-corrected electron microscope (FEI Titan) equipped with a Super-X EDS. Microcalorimetric measurements of C2H4 adsorption were carried out on a home-designed adsorption microcalorimetry system consisting of a chemisorption apparatus (Micromeritics Autochem II 2920) and a microcalorimeter (Setaram Sensys EVO 600).
The XAFS spectra at Pd K-edge and Bi L3-edge of the samples were measured at beamline 14W of Shanghai Synchrotron Radiation Facility in China62. The output beam was filtered by Si(311) monochromator. Pd foil was used to calibrate the energy.
In situ Fourier transform infrared (FT-IR) adsorption spectroscopy of CO experiments were recorded on a Nicolet iS50 instrument. Prior to CO chemisorption, the catalysts were activated by H2 at 100 °C for 1 h and then cooled down to RT in Ar. The FT-IR spectrum of Ar at RT was taken as the background spectrum and subtracted automatically from subsequent spectra. Then the catalysts were exposed to a CO flow for 10 min and degassed by Ar for 10 min to desorb the physical adsorbed CO, and IR spectra were recorded.
TPR measurements were conducted on Micromeritics ChemiSorb 2920, equipped with a thermal conductivity detector. Prior to TPR measurements, 100 mg of the catalyst was activated by H2 at 100 °C for 1 h and then cooled down to RT in Ar. Afterwards, the catalyst was subjected to 10 vol% H2/Ar at a flow rate of 30 mL min−1 and heated to 150 °C at 10 °C min−1.
Catalytic tests
Selective hydrogenation of acetylene in excess ethylene was carried out in a fixed bed vertical quartz reactor, with a space velocity of 120,000 mL h–1 g–1. The reaction gas consisting of 1.0 vol% C2H2, 20.0 vol% C2H4, 20.0 vol.% H2, and 59 vol.% N2 was fed at a flow rate of 60 mL min−1, simulating the front-end hydrogenation conditions. Typically, 30 mg of the catalyst (diluted by 400 mg of quartz sand) was activated by H2 (20 mL min–1) at 100 °C for 1 h and then cooled to RT in N2 prior to the catalytic reaction. The gas components from the microreactor outlet were analyzed by online gas chromatography (GC; Shimadzu GC-2010) equipped with a flame ionization detector. It is important to note that the pretreatment in H2 is important as the long-term exposure of Pd1.0/Bi2O3/TiO2 in air might oxidize Pd and demolish Pd–Bi bonding. Control experiment also suggests that untreated Pd1.0/Bi2O3/TiO2 has poor selectivity toward ethylene. To this end, all catalysts were pretreated in H2 before catalytic measurements.
C2H4 and C2H6 were the only products detected by GC. Negligible oligomers were formed during the hydrogenation process, likely due to the short contact time and high concentration of hydrogen10. C2H2 conversion and C2H4 selectivity were calculated as:
Computational details
In this work, Vienna Ab initio Simulation Package63,64,65,66 was used to conduct density functional calculations within the generalized gradient approximation of RPBE functional developed by the Nørskov group67. Ionic cores and electrons were described by the projector augmented wave method68,69. The energy cutoff was set to be 500 eV and the force threshold was 0.05 eV Å−1. We used constrained minimization method59,70,71,72 to locate the transition state structures. The adsorption energies of C2Hx (x = 2, 4, and 6) were calculated as: Eads = EC2Hx+slab − (EC2Hx + Eslab), where EC2Hx+slab is the energy of the system after adsorption of C2Hx species, EC2Hx is the energy of the gas-phase C2Hx adsorbate, and Eslab is the energy of the slab.
The optimized lattice parameters of Bi2O3 were a = 5.981 Å, b = 8.340 Å, and c = 7.591 Å, which are close to experimental values (monoclinic, P21/c(14), a = 5.849 Å, b = 8.166 Å, and c = 7.510 Å). A Bi2O3(100) slab was built with a 2 × 2 supercell with nine atomic layers, and the Pd cluster structure was built by adding 8 Pd atoms onto the Bi2O3(100) surface, followed by structure optimization. To build a valid catalyst model of Pd cluster, we followed these rules: (i) Pd would disperse on the Bi2O3 surface with a size <2 nm; (ii) the structure should give rise to similar CNs listed in Supplementary Table 2; (iii) the model should be stable and would not deform under reaction condition, which has been widely discussed. During the optimizations involving Pd cluster structure, the bottom two layers of atoms of Bi2O3(100) component were fixed to simulate bulk structure of Bi2O3, while the other atoms were fully relaxed. The vacuum layer was set higher than 12 Å to avoid spurious interaction between adjacent slabs. The k-point grid used in the surface Brillouin zone was 1 × 1 × 1 for all the calculations. More details about the structure development are provided in Supplementary Information (Supplementary Figs. 16 and 17).
Data availability
All the data supporting the findings of this study are available within the article and its Supplementary Information files or from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Cargnello, M. et al. Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts. Science 341, 771–773 (2013).
Matsubu, J. C. et al. Adsorbate-mediated strong metal–support interactions in oxide-supported Rh catalysts. Nat. Chem. 9, 120 (2016).
Kattel, S., Ramírez, P. J., Chen, J. G., Rodriguez, J. A. & Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 355, 1296–1299 (2017).
Suchorski, Y. et al. The role of metal/oxide interfaces for long-range metal particle activation during CO oxidation. Nat. Mater. 17, 519–522 (2018).
Ernst, F. Metal-oxide interfaces. Mater. Sci. Eng. R. 14, 97–156 (1995).
Picone, A. et al. Reactive metal–oxide interfaces: a microscopic view. Surf. Sci. Rep. 71, 32–76 (2016).
Campbell, C. T. Electronic perturbations. Nat. Chem. 4, 597 (2012).
Ro, I., Resasco, J. & Christopher, P. Approaches for understanding and controlling interfacial effects in oxide-supported metal catalysts. ACS Catal. 8, 7368–7387 (2018).
Kyriakou, G. et al. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science 335, 1209–1212 (2012).
Borodziński, A. & Bond, G. C. Selective hydrogenation of ethyne in ethene–rich streams on palladium catalysts. Part 1. Effect of changes to the catalyst during reaction. Catal. Rev. 48, 91–144 (2006).
Studt, F. et al. Identification of non-precious metal alloy catalysts for selective hydrogenation of acetylene. Science 320, 1320–1322 (2008).
Vilé, G. et al. A stable single‐site palladium catalyst for hydrogenations. Angew. Chem. Int. Ed. 54, 11265–11269 (2015).
Pei, G. X. et al. Ag alloyed Pd single-atom catalysts for efficient selective hydrogenation of acetylene to ethylene in excess ethylene. ACS Catal. 5, 3717–3725 (2015).
Huang, X. et al. Enhancing both selectivity and coking-resistance of a single-atom Pd1/C3N4 catalyst for acetylene hydrogenation. Nano Res. 10, 1302–1312 (2017).
Pei, G. X. et al. Performance of Cu-alloyed Pd single-atom catalyst for semihydrogenation of acetylene under simulated front-end conditions. ACS Catal. 7, 1491–1500 (2017).
Osswald, J. et al. Palladium–gallium intermetallic compounds for the selective hydrogenation of acetylene. Part II: Surface characterization and catalytic performance. J. Catal. 258, 219–227 (2008).
Armbrüster, M. et al. Pd−Ga intermetallic compounds as highly selective semihydrogenation catalysts. J. Am. Chem. Soc. 132, 14745–14747 (2010).
Prinz, J. et al. Adsorption of small hydrocarbons on the three-fold PdGa surfaces: the road to selective hydrogenation. J. Am. Chem. Soc. 136, 11792–11798 (2014).
Luo, Y. et al. Addressing electronic effects in the semi-hydrogenation of ethyne by InPd2 and intermetallic Ga–Pd compounds. J. Catal. 338, 265–272 (2016).
Krajčí, M. & Hafner, J. Selective semi-hydrogenation of acetylene: atomistic scenario for reactions on the polar threefold surfaces of GaPd. J. Catal. 312, 232–248 (2014).
Zhou, H. et al. PdZn intermetallic nanostructure with Pd–Zn–Pd ensembles for highly active and chemoselective semi-hydrogenation of acetylene. ACS Catal. 6, 1054–1061 (2016).
Feng, Q. et al. Isolated single-atom Pd sites in intermetallic nanostructures: high catalytic selectivity for semihydrogenation of alkynes. J. Am. Chem. Soc. 139, 7294–7301 (2017).
Zhou, X.-D. & Huebner, W. Size-induced lattice relaxation in CeO2 nanoparticles. Appl. Phys. Lett. 79, 3512–3514 (2001).
Henrich, V. E. & Cox, P. A. The Surface Science of Metal Oxides (Cambridge University Press, 1996).
Iablokov, V. et al. Catalytic CO oxidation over well-defined cobalt oxide nanoparticles: size-reactivity correlation. ACS Catal. 5, 5714–5718 (2015).
Xia, J.-Y., Tang, M.-T., Chen, C., Jin, S.-M. & Chen, Y.-M. Preparation of α-Bi2O3 from bismuth powders through low-temperature oxidation. Trans. Nonferrous Met. Soc. China 22, 2289–2294 (2012).
Zou, S. et al. Boosting hydrogen evolution activities by strong interfacial electronic interaction in ZnO@Bi(NO3)3 core–shell structures. J. Phys. Chem. C 121, 4343–4351 (2017).
Lou, B. et al. Selectively depositing Bi2O3 quantum dots on TiO2 nanotubes for efficient visible-light-driven photocatalysis. Mater. Lett. 288, 129413 (2021).
Zou, S. et al. Fabricating the AuPdPt ternary nanophase diagram at 800 °C to guide the exploration of optimal catalyst for n-hexane oxidation. J. Phys. Chem. C 121, 4074–4082 (2017).
Han, Y. et al. TiO2 supported Pd@Ag as highly selective catalysts for hydrogenation of acetylene in excess ethylene. Chem. Commun. 49, 8350–8352 (2013).
Yan, Y. et al. Site-specific deposition creates electron-rich Pd atoms for unprecedented C−H activation in aerobic alcohol oxidation. Chin. J. Catal. 41, 1240–1247 (2020).
Lichtenberger, J., Lee, D. & Iglesia, E. Catalytic oxidation of methanol on Pd metal and oxide clusters at near-ambient temperatures. Phys. Chem. Chem. Phys. 9, 4902–4906 (2007).
Yan, H. et al. Single-atom Pd1/graphene catalyst achieved by atomic layer deposition: remarkable performance in selective hydrogenation of 1,3-butadiene. J. Am. Chem. Soc. 137, 10484–10487 (2015).
Liu, J. et al. Synergistic effect between Pt0 and Bi2O3−x for efficient room-temperature alcohol oxidation under base-free aqueous conditions. Catal. Sci. Technol. 7, 1203–1210 (2017).
Penner, S. & Armbrüster, M. Formation of intermetallic compounds by reactive metal–support interaction: a frequently encountered phenomenon in catalysis. ChemCatChem 7, 374–392 (2015).
Cheung, T. T. P. Lineshape studies of the X-ray photoemission of small metal clusters. Surf. Sci. 127, L129–L134 (1983).
Kaden, W. E., Wu, T., Kunkel, W. A. & Anderson, S. L. Electronic structure controls reactivity of size-selected Pd clusters adsorbed on TiO2 surfaces. Science 326, 826–829 (2009).
Jiang, Z. et al. Direct XPS evidence for charge transfer from a reduced rutile TiO2(110) surface to Au clusters. J. Phys. Chem. C 111, 12434–12439 (2007).
Lopez, N. & Nørskov, J. K. Theoretical study of the Au/TiO2(110) interface. Surf. Sci. 515, 175–186 (2002).
Goniakowski, J. & Noguera, C. Characteristics of Pd deposition on the MgO(111) surface. Phys. Rev. B 60, 16120–16128 (1999).
Goniakowski, J. & Noguera, C. Microscopic mechanisms of stabilization of polar oxide surfaces: transition metals on the MgO(111) surface. Phys. Rev. B 66, 085417 (2002).
Saito, M., Wagner, T., Richter, G. & Rühle, M. High-resolution TEM investigation of structure and composition of polar Pd/ZnO interfaces. Phys. Rev. B 80, 134110 (2009).
Fan, Q. et al. Photodeposited Pd nanoparticles with disordered structure for phenylacetylene semihydrogenation. Sci. Rep. 7, 42172 (2017).
Bistoni, G. et al. How π back-donation quantitatively controls the CO stretching response in classical and non-classical metal carbonyl complexes. Chem. Sci. 7, 1174–1184 (2016).
Szesni, N. et al. Catalyst composition for selective hydrogenation with improved characteristics. U.S. patent 20200094226 A1 (2020).
Tysoe, W. T., Nyberg, G. L. & Lambert, R. M. Photoelectron spectroscopy and heterogeneous catalysis: benzene and ethylene from acetylene on palladium (111). Surf. Sci. 135, 128–146 (1983).
Pradier, C. M., Mazina, M., Berthier, Y. & Oudar, J. Hydrogenation of acetylene on palladium. J. Mol. Catal. 89, 211–220 (1994).
Huang, F. et al. Atomically dispersed Pd on nanodiamond/graphene hybrid for selective hydrogenation of acetylene. J. Am. Chem. Soc. 140, 13142–13146 (2018).
Lou, B. et al. Highly selective acetylene semihydrogenation catalyst with an operation window exceeding 150 °C. ACS Catal. 11, 6073–6080 (2021).
Hill, J. M., Shen, J., Watwe, R. M. & Dumesic, J. A. Microcalorimetric, infrared spectroscopic, and DFT studies of ethylene adsorption on Pd and Pd/Sn catalysts. Langmuir 16, 2213–2219 (2000).
Hamm, G. et al. The adsorption of ethene on Pd(111) and ordered Sn/Pd(111) surface alloys. Z. Phys. Chem. 223, 209 (2009).
Kim, E. et al. Pd catalyst promoted by two metal oxides with different reducibilities: properties and performance in the selective hydrogenation of acetylene. Appl. Catal. A Gen. 471, 80–83 (2014).
Podkolzin, S. G., Alcalá, R. & Dumesic, J. A. Density functional theory studies of acetylene hydrogenation on clean, vinylidene- and ethylidyne-covered Pt(111) surfaces. J. Mol. Catal. A 218, 217–227 (2004).
Gao, J., Zhao, H., Yang, X., Koel, B. E. & Podkolzin, S. G. Controlling acetylene adsorption and reactions on Pt–Sn catalytic surfaces. ACS Catal. 3, 1149–1153 (2013).
Gao, J., Zhao, H., Yang, X., Koel, B. E. & Podkolzin, S. G. Geometric requirements for hydrocarbon catalytic sites on platinum surfaces. Angew. Chem. Int. Ed. 53, 3641–3644 (2014).
Yang, B., Burch, R., Hardacre, C., Headdock, G. & Hu, P. Influence of surface structures, subsurface carbon and hydrogen, and surface alloying on the activity and selectivity of acetylene hydrogenation on Pd surfaces: a density functional theory study. J. Catal. 305, 264–276 (2013).
Yang, B., Burch, R., Hardacre, C., Headdock, G. & Hu, P. Understanding the optimal adsorption energies for catalyst screening in heterogeneous catalysis. ACS Catal. 4, 182–186 (2014).
Yang, B., Burch, R., Hardacre, C., Hu, P. & Hughes, P. Selective hydrogenation of acetylene over Cu(211), Ag(211) and Au(211): Horiuti-Polanyi mechanism vs. non-Horiuti-Polanyi mechanism. Catal. Sci. Technol. 7, 1508–1514 (2017).
Yang, B. et al. Evidence to challenge the universality of the Horiuti-Polanyi mechanism for hydrogenation in heterogeneous catalysis: origin and trend of the preference of a non-Horiuti-Polanyi mechanism. J. Am. Chem. Soc. 135, 15244–15250 (2013).
Yang, K. & Yang, B. Identification of the active and selective sites over a single Pt atom-alloyed Cu catalyst for the hydrogenation of 1,3-butadiene: a combined DFT and microkinetic modeling study. J. Phys. Chem. C 122, 10883–10891 (2018).
Nørskov, J. K., Bligaard, T., Rossmeisl, J. & Christensen, C. H. Towards the computational design of solid catalysts. Nat. Chem. 1, 37 (2009).
Yu, H.-S., Wei, X.-J., Li, J., Gu, S.-Q. & Zhang, S. The XAFS beamline of SSRF. Nucl. Sci. Tech. 26, 50102–050102 (2015).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised perdew-burke-ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Alavi, A., Hu, P., Deutsch, T., Silvestrelli, P. L. & Hutter, J. CO oxidation on Pt(111): an ab initio density functional theory study. Phys. Rev. Lett. 80, 3650–3653 (1998).
Michaelides, A. & Hu, P. Catalytic water formation on platinum: a first-principles study. J. Am. Chem. Soc. 123, 4235–4242 (2001).
Liu, Z.-P. & Hu, P. General rules for predicting where a catalytic reaction should occur on metal surfaces: a density functional theory study of C−H and C−O bond breaking/making on flat, stepped, and kinked metal surfaces. J. Am. Chem. Soc. 125, 1958–1967 (2003).
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (92045301, 91545113, 91845203, 21802122, 21703050), China Postdoctoral Science Foundation (2019M662020), and Shell Global Solutions International B. V. (PT71423, PT74557). Y.Z. acknowledges financial support from the Zhejiang Provincial Natural Science Foundation of China (LR18B030003), National Natural Science Foundation of China (51701181), and the Thousand Talents Program for Distinguished Young Scholars. We thank the HPC Platform of ShanghaiTech University for computing time. This research used resources of the 8-BM and 7-BM Beamline of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.
Author information
Authors and Affiliations
Contributions
J.F. and S.Z. designed the study. S.Z. and B.L. performed most of the experiments. W.Y., C.Z., Y.Z., and Y.W. performed the electron microscopy characterization. K.Y. and B.Y. finished the DFT calculations. L.M., Y.D., J.M., and Z.J. carried out the X-ray structure characterization and analysis. W.H. carried out the microcalorimetric measurements. Z.G. and Y.C. performed the in situ NAP-XPS studies. L.L., J.L., and L.X. performed the TPR and TPD measurements. S.Z., K.Y., J.F., B.Y., and Y.Z. wrote the paper. All authors interpreted the data and contributed to the preparation of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Ding Ma and the other anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zou, S., Lou, B., Yang, K. et al. Grafting nanometer metal/oxide interface towards enhanced low-temperature acetylene semi-hydrogenation. Nat Commun 12, 5770 (2021). https://doi.org/10.1038/s41467-021-25984-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-25984-8
This article is cited by
-
A MOF-supported Pd1–Au1 dimer catalyses the semihydrogenation reaction of acetylene in ethylene with a nearly barrierless activation energy
Nature Catalysis (2024)
-
Pt-doped Ru nanoparticles loaded on ‘black gold’ plasmonic nanoreactors as air stable reduction catalysts
Nature Communications (2024)
-
Synergistic combination of Pd nanosheets and porous Bi(OH)3 boosts activity and durability for ethanol oxidation reaction
Nano Research (2022)
-
Fundamental aspects of alkyne semi-hydrogenation over heterogeneous catalysts
Nano Research (2022)
-
Influence of different alumina phases on the catalytic properties of palladium-alumina catalysts for selective hydrogenation of acetylene to ethylene
Reaction Kinetics, Mechanisms and Catalysis (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
