
Abstract
Determining the nature and extent of covalency of early actinide chemical bonding is a fundamentally important challenge. Recently, X-ray absorption, electron paramagnetic, and nuclear magnetic resonance spectroscopic studies have probed actinide-ligand covalency, largely confirming the paradigm of early actinide bonding varying from ionic to polarised-covalent, with this range sitting on the continuum between ionic lanthanide and more covalent d transition metal analogues. Here, we report measurement of the covalency of a terminal uranium(VI)-nitride by 15N nuclear magnetic resonance spectroscopy, and find an exceptional nitride chemical shift and chemical shift anisotropy. This redefines the 15N nuclear magnetic resonance spectroscopy parameter space, and experimentally confirms a prior computational prediction that the uranium(VI)-nitride triple bond is not only highly covalent, but, more so than d transition metal analogues. These results enable construction of general, predictive metal-ligand 15N chemical shift-bond order correlations, and reframe our understanding of actinide chemical bonding to guide future studies.
Similar content being viewed by others
Introduction
Determining the nature and extent of covalency, that is the extent of electron sharing between two elements, in actinide-ligand (An–L) bonding is a central and enduring fundamental goal of actinide science1,2,3. For this reason, the study of An–L multiple bonding is of burgeoning interest since such bonds tend to inherently exhibit levels of covalency that are practical to investigate4,5,6,7,8,9. The generally accepted chemical bonding picture is that for early actinides the bonding varies from ionic to polarised-covalent as a function of An-oxidation state and ligands, with this range sitting intermediate to the ionic lanthanides and usually much more covalent d transition metal complexes5,7,8,10,11. Probing actinide covalency is challenging, but in recent years progress has been made using experimental approaches, underpinned by quantum chemical calculations, including K-edge X-ray absorption near edge spectroscopy12,13,14,15,16,17,18, pulsed electron paramagnetic resonance spectroscopy19, and nuclear magnetic resonance (NMR) spectroscopy20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36. These investigations have begun to place the bonding descriptions of An–L bonding on a rigorous, quantitative footing, and have been consistent with the status quo bonding description of early actinides.
Regarding the use of NMR spectroscopy to probe An–L covalency, solution, and solid-state studies encompassing 1H20, 13C21,22,23,24, 15N25,26, 17O27,28,29,30, 19F31,32,33,34, 35/37Cl35, 77Se36, and 125Te36 nuclei have revealed that their chemical shift properties are highly sensitive to interactions with actinide ions, thus constituting a direct, powerful experimental probe of chemical bonding and covalency when the individual bonding contributions to shielding tensors are analysed in detail20,21,22,24,25,26,36,37. A potential benchmark at the interface of NMR spectroscopic and An–L multiple bond investigations is the terminal uranium(VI)-nitride triple bond, due to its formally closed-shell diamagnetic formulation rendering it amenable to study by NMR spectroscopy. However, despite the sustained nature of An-L multiple bond chemistry, terminal actinide-nitrides remain rare, with only two classes of isolable terminal uranium-nitride reported. In 2012, the Liddle group reported [UV(N)(TrenTIPS)][Na(12C4)2] (TrenTIPS = N(CH2CH2NSiPri3)3, 12C4 = 12-crown-4 ether)38, and in 2013 disclosed its oxidation to give [UVI(N)(TrenTIPS)] (1)39. In 2020 the Mazzanti group40 reported [UVI(N){OSi(OBut)3}4][NBun4].
Prior quantum chemical modelling of 1 suggested a highly covalent U ≡ N triple bond rivalling, if not exceeding the covalency in group 6 nitride triple bonds39,41, which would be a significant result if experimentally confirmed given the dominance of 5f- over 6d-orbital participation in this bond, and exceptional chemical shielding tensors have thus been predicted for uranium-nitride linkages25. While some comparative uranium(V/VI)-nitride reactivity studies have begun to build a picture consistent with the view that the uranium(VI)-nitride linkage is highly covalent42,43,44, definitive experimental spectroscopic confirmation of the computational description of high covalency that exceeds d transition metal analogues has remained lacking. However, it has previously been shown to be possible to prepare the 15Nnitride enriched [UVI(N*)(TrenTIPS)] (1*, N* = 50:50 14N:15N)39, presenting an opportunity for benchmarking NMR studies to experimentally test the above predictions. Indeed, to the best of our knowledge only one other actinide-nitride complex has been studied by 15N NMR spectroscopy, namely the bridging dithorium-nitride complex [(N″)3Th(μ-N)Th(N″)3][K(18C6)(THF)2] (N″ = N(SiMe3)2) described by the Hayton, Autschbach, and Cho groups26.
Here, we report a solution- and solid-state 15N NMR spectroscopic study of the terminal actinide-nitride complex 1*. We find exceptional 15N chemical shift and chemical shift anisotropy that to the best of our knowledge redefine the range for N-containing compounds. We find that these 15N properties are dominated by the paramagnetic shielding term, with spin–orbit contributions being minor. The 15N NMR data experimentally confirm that the U ≡ N triple bond is highly covalent, and indeed more so than group 4–6 transition metal nitrides. This reframes our understanding of the nature and range of the covalency of An–L chemical bonding, and permits construction of metal-nitride 15N chemical shift correlated to several quantum chemical measures of bond order as general predictive models.
Results
Synthesis of 1*
We prepared a sample of 50% 15Nnitride enriched 1* according to Fig. 1. This is a modified method to the previously reported preparation of 1* (refs. 39,41,42), where use of [K(B15C5)2]+ (B15C5 = benzo-15-crown-5 ether) instead of [Na(12C4)2]+ as the cation component of the [U(14/15N)(TrenTIPS)][M(L)2] (M = Na, L = 12C4; M = K, L = B15C5) precursor to 1* is found to be more reliable and thus practicable. The purity and stability of 1* was checked and confirmed by 1H and 29Si NMR spectroscopies in D8-THF, 50:50 D8-THF:C6D6, and C6D6 (Supplementary Figs. 1–4).

The initial chloride complex is converted to the corresponding azide by salt elimination. Reduction releases N2 to produce a bridging nitride dimer, which can be cleaved into a separated ion pair by treatment with the appropriate crown ether. Oxidation of the separated ion pair gives neutral, 50% 15N-labelled 1* for the NMR investigations described in this study. B15C5 = benzo-15-crown-5 ether.
Solution and solid-state 15N NMR spectroscopic investigation of 1*
Previously, we were unable to detect the 15Nnitride resonance for 1*39, attributed to the low molar receptivity (~0.1% of 1H) and high longitudinal relaxation time constant of the 15N nucleus. However, using a low flip angle acquisition (10° r.f. pulse, 1 s acquisition, 1.7 s recycle delay), we were able to clearly observe the 15Nnitride resonance of 1*. The 15N{1H} NMR spectrum of 1* in D8-THF (Fig. 2a) exhibits a single isotropic chemical shift (δiso) resonance at 968.9 ppm relative to the IUPAC standard of MeNO2 at 0 ppm45. The analogous spectrum of 1* in D6-benzene exhibits a resonance at δiso 972.6 ppm, demonstrating little polar/non-polar solvent medium dependence of the 15Nnitride δiso of 1*.

a Solution spectrum in D8-THF. b Solid-state spectrum with MAS = 0 kHz. c Solid-state spectrum with MAS = 2.5 kHz. d Solid-state spectrum with MAS = 9.0 kHz. In each case the black line is the experimental spectrum, and the red line is the simulated spectrum.
Few molecular metal nitrides have been characterised by 15N NMR spectroscopy in solution, and while some have been referenced to MeNO2 = 0 ppm, others are referenced to NH3(l) = 0 that would make MeNO2 = 380.2 ppm under the measurement conditions used; in the latter reference frame the 15Nnitride δiso of 1* is then 1349.1/1352.8 ppm, respectively. Accordingly, we list pertinent nitrides with both associated δiso values (MeNO2/NH3(l) = 0 ppm): 1* (δiso = av. 971/1351 ppm); [V(N)(LMeDipp)(ODipp)] (δiso = 679/1059 ppm, LMeDipp = HC(CMeNDipp)2, Dipp = 2,6-Pri2C6H3)46; [V(N)(LMeDipp)(NTol2)] (δiso = 669/1049 ppm, Tol = 4-Me-C6H4)47; [V(N)(LMeDipp){N(Tol)(Mes)}] (δiso = 655/1035 ppm, Mes = 2,4,6-Me3C6H2)47; [Ti(N)(NP)2][K(2,2,2-crypt)] (δiso = 578/958 ppm, NP = MesNC6H3-3-Me-2-PPri2)48,49; [Ti(μ-N)(NP)2K(18C6)] (δiso = 542/922 ppm)48,49; [Ti(μ-N)(NP)2K(OEt2)]2 (δiso = 512/892 ppm)48,49; [Mo(N){N(But)(C6H3-3,5-Me2}3] (δiso = 460/840 ppm)50; [(N″)3Th(μ-N)Th(N″)3][K(18C6)(THF)2] (δiso = 299/679 ppm)25. Thus, the 15Nnitride resonance for 1* is the most downfield (highest frequency) deshielded δiso value to date, extending the known 15N NMR δiso range by ~300 ppm.
Given the downfield 15Nnitride δiso value of 1*, we recorded its solid-state NMR spectrum, Fig. 2b–d. The spectrum of the static sample is broad, but the general shape of the spectrum can be discerned. Under magic angle spinning (MAS) conditions, an improved signal-to-noise ratio is obtained and rotational side-bands are well-defined at 2.5 kHz spinning frequency that highlight a chemical shift anisotropy of ~2000 ppm. The signal-to-noise ratio is further improved at a MAS frequency of 9.0 kHz, but now the frequency-dependent rotational side-bands extend beyond the available pulse bandwidth. Nevertheless, taken together these data enable reliable spectral simulation, yielding a δiso value of 950 ppm, which is in good agreement with the solution δiso value (Δsol-ss = 21 ppm), and chemical shift tensor δxx, δyy, δzz, Ω (tensor span, δxx − δzz), and κ (skew, [3(δyy − δiso)]/δxx − δzz) values of 1617, 1603, −370, 1987 ppm, and 0.99, respectively.
The Ω value for 1* is notably large, and can be compared to those of [Ti(N)(NP)2][K(2,2,2-crypt)] (Ω = 1470 ppm)48,49, [Mo(N){N(But)(C6H3-3,5-Me2}3] (Ω = 1187 ppm)50, [Ti(μ-N)(LButDipp)(NTol2)K]2 (Ω = 1155 ppm)51, and [(N″)3Th(μ-N)Th(N″)3][K(18C6)(THF)2] (Ω = 847 ppm)26. Thus, though there are comparatively few data for comparison, 1* exhibits the largest Ω of any nitrogen-containing molecule, enlarging the known tensor span range by ~500 ppm. Like most other molecular metal nitrides, the κ value for 1* is close to one, and the line shape of the spectra are characteristic of an axially symmetric shift tensor. These findings are consistent with the structure of the uranium-nitride linkage in 1*, which is highly axial41, residing along a three-fold symmetry axis defined by TrenTIPS.
Computational benchmarking of the 15N NMR spectroscopic properties of 1*
The 15Nnitride NMR spectroscopic data for 1* prompted us to computationally model its NMR properties in detail. We used coordinates from geometry optimisation (see Supplementary Table 1) at the BP86 level39, noting the computed U ≡ N bond length of 1.7795 Å compares well to the crystallographically determined distance of 1.799(7) Å39. To give context to the U ≡ N bond length distance in 1/1*, the corresponding distances in [UV(N)(TrenTIPS)][Na(12C4)2] and [UVI(N){OSi(OBut)3}4][NBun4] are 1.825(15) and 1.769(2) Å, respectively38,40. We then calculated 15Nnitride δiso values from single point energy calculations with a range of functionals, corrected for the solvent (THF, since that is the most accurately determined δiso for 1*) and reference (MeNO2 = 0 at the same functional level). We found (Supplementary Table 2), irrespective of using scalar relativistic (SR) or two-component spin–orbit relativistic (SOR) effects, that BP86 and SAOP functionals underestimate the δiso value (Δcalc-exp = −166 to −359 ppm) whereas PBE0 and PBE0-HF40% over-estimate the δiso value (Δcalc-exp = +150 to +401 ppm). However, the B3LYP functional gave better agreement, with δiso at 1040 ppm (scalar) or 1044 ppm (spin–orbit) (Δcalc-exp = ~+70 ppm). Inclusion of dispersion did not improve the level of agreement, because optimised geometries returned unrealistically short U ≡ N bond lengths suggesting over-compensated dispersion, so we adopted an established empirical correction approach (Supplementary Table 3 and Supplementary Fig. 5, R2 = 0.9951)52,53, giving a corrected computed δiso value of 966 ppm (Δcalc-exp = −5 ppm). To provide support for the computed 15Nnitride δiso of 1*, we calculated the 29Si NMR δiso for the SiPri3 groups in 1* at the B3LYP level, and find a value of −0.61 ppm, in good agreement with the experimental δiso of 3.8 ppm in THF54.
Focussing on the B3LYP two-component SOR model, we calculated Namide and Namine δiso values of −123 and −311 ppm, respectively. for the TrenTIPS ligand in 1*. The computed Namide δiso of 1* agrees well with the reported 15N NMR δiso of −198 ppm for the Th-NH2 unit in [Th(NH2)(N″)3]25, and we note that the computed Nnitride, Namide, and Namine δiso values for 1* are positively correlated (Supplementary Figs. 6 and 7, R2 = 0.9983 or 0.9988) when plotted against their U–N Mayer bond orders (2.94, 0.91, 0.44) or U–N delocalisation indices (DI, 2.649, 0.790, 0.377), respectively. As expected, there is little variation in the computed diamagnetic isotropic shielding tensors (σd) for the Nnitride (320 ppm), Namide (311 ppm), and Namine (309 ppm) centres, since σd derives principally from tightly-bound core electron densities55. In contrast, the paramagnetic isotropic shielding tensors (σp) show substantial variation, being −1433 (Nnitride), −363 (Namide), and −186 (Namine) ppm, respectively, reflecting that σp derives from polarisable valence electron densities that directly report different chemical bonding environments arising from magnetically induced-mixing of the ground state with excited states55. Interestingly, the SO shielding tensors (σso) are relatively small, being −29 (Nnitride), −40 (Namide), and −20 (Namine) ppm.
With the 15Nnitride solution δiso value of 1* satisfactorily reproduced computationally, we analogously modelled (Supplementary Table 4 and Supplementary Fig. 8, R2 = 0.9977) the solid-state data, computing a solid-state 15Nnitride δiso of 935 ppm (Δcalc-exp = −15 ppm). The computed σp, σd, and σso values (−1492, 318, and −27.0 ppm, respectively) are similar to the computed solution parameters; also, the computed δxx, δyy, δzz, Ω, and κ values of 1590, 1584, −378, 1969 ppm, and 0.99 are in excellent agreement with the simulation of the experimental solid-state spectra. These data suggest that the modest difference in solution and solid-state δiso values are due to solvent effects, and that fast dynamic averaging effects do not operate for 1* in the solid-state at ambient temperature.
Computational electronic structure analysis of 1*
Having established that the B3LYP functional appropriately models the experimental 15N NMR data for 1*, we re-analysed the electronic structure of 1* in order to rationalise its downfield 15Nnitride δiso. Previously reported calculations on 1* using the BP86 functional39 revealed the the highest occupied molecular orbital (HOMO) to be the U ≡ N σ-bond, the HOMO-1 and -2 are U ≡ N π-bonds, then resides a manifold of U–Namide bonding combinations. B3LYP calculations on 1* reveal essentially the same gross U ≡ N σ > π energy bonding description, but now the U ≡ N combinations are more greatly delocalised with the U–Namide manifold, resulting in three groups of three MOs each, with each triad exhibiting varying orbital coefficient weightings of the U ≡ N σ→π-combinations mixed in with U–Namide combinations. However, the principal U ≡ N σ- and π-bonds can be identified as HOMO−3 to −5.
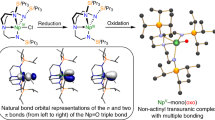
In order to provide a chemically more intuitive model, we examined the NLMOs of 1* at the B3LYP level (Fig. 3) using NBO6. The U ≡ N σ-bond is 39.9% U (7s:7p:6d:5f = 3:1:10:86%) and 58.9% N (2s:2p = 8:92%) character, whereas the U ≡ N π-bonds are 31% U (7s:7p:6d:5f = 0:0:24:76%) and 68% N (2s:2p = 0:99%). The B3LYP NLMO (NBO6) data are in good agreement with previously reported BP86 NBO (NBO5) analysis of 1*39, and emphasise the highly covalent nature, and dominance of 5f U and 2p N orbitals, in the bonding of the U ≡ N linkage in 1*.

These three NLMOs represent the U ≡ N σ- and π-bonds. Hydrogen atoms are omitted for clarity.
Computational chemical shift analysis of 1*
Ramsey’s formula, Eq. (1), examines NMR interactions from a quantum–mechanical perspective, decomposing magnetic shielding contributions into σd and σp components that are dependent on electron orbital angular momenta56,57,58. While this does not directly translate to the MO approach of hybrid DFT, it provides a framework with which to rationalise NMR magnetic shieldings using DFT B3LYP calculations when SO contributions are included, Eq. (2)44.
Since for 1* σd varies little for any of the N-environments and σso is fairly small, we focus on σp. This term can be presented in reduced form as:
Here, r is the radial expansion of the shielding electrons from the nucleus being examined, N denotes the NMR nucleus, ΣM QNM is the sum of the charge density and bond order matrix elements over the relevant atoms (M), and ΔE is the energy separation between the ground and excited states in question36,37,45,56,57,58. It follows that σp is proportional to the magnetic field-induced mixing of magnetically coupled orbitals and inversely proportional to r3. For the latter this arises because as a nucleus (M) withdraws charge from the NMR nucleus (N) the valence orbitals of N will contract so the 1/r3 term will become larger (more deshielded NMR nucleus) resulting in a larger σp term. Put another way, the larger the bond order of, so more covalent, the bond involving the NMR nucleus the larger σp will become36. The σp term is also inversely proportional to the energy gap between the occupied and virtual orbitals that become magnetically coupled, so smaller ΔE gaps produce larger σp values. We note in passing that the HOMO-LUMO gap of 1* is unremarkable (see Supplementary Table 5), and so would be expected to be subordinate to the 1/r3 term in terms of its contribution to σp. The mixing by magnetic coupling of occupied and virtual orbitals must be symmetry allowed, since the angular momentum operators belong to the same irreducible representations as the rotational operators. Canonical MOs are often delocalised, and so contributions to deshielding can be distributed over many components and so become difficult to fully identify. However, noting the above and using the ADF NMR analysis package enables identification of the principal components that contribute to the σp term of 1* (Fig. 4) as being magnetic field-induced coupling between U ≡ N σ ↔ π* and π ↔ σ* MOs; these MO combinations conform to the requirement for rotational orthogonality and also being spatially and energetically proximate.

a Mixing of the occupied U ≡ N σ-bond with unoccupied U ≡ N π*-bonds. b Mixing of the occupied U ≡ N π-bonds with unoccupied U ≡ N σ*-bond. Hydrogen atoms are omitted for clarity.
Field-induced magnetic mixing of the ground state with low-lying, thermally inaccessible, paramagnetic states in 1*—i.e. temperature-independent paramagnetism, TIP—is confirmed experimentally by SQUID magnetometry measurements on powdered 1* (Fig. 5 and Supplementary Figs. 9–11), which reveal a small (χMT < 0.04 cm3 mol−1 K) magnetic moment, but a shallow positive slope for the temperature-dependent χMT data even though 1S0 uranium(VI) is formally a 5f 06d0 ion. Linear regression analysis reveals a χTIP value of 0.9527 × 10−4 cm3 mol−1 K for 1*, which compares to analogous values of 3.43 × 10−4 and 6.26 × 10−4 cm3 mol−1 K determined for two uranium(VI)-acetylides24. This suggests that TIP effects on the σp term will be modest.

Experimental data points are represented by black circles and the line is a guide to the eye only. For comparison, the hypothetical magnetic moments of 5f1 and 5f2 ions are represented by the blue and red dotted lines, respectively.
We note that the δxx, δyy, δzz, and Ω values for N2 are 136, 136, −539, and 675 ppm59, which when compared to the data for 1* reflects the presence of the TrenTIPS ligand in 1* and, since the Δδppm between 1* and N2 changes much less for δzz compared to δxx and δyy, the expectation of strong mixing of occupied and virtual orbitals when a magnetic field is applied perpendicular to the U ≡ N bond60,61,62. Since the σp term for 1* is notably large, it follows that the U ≡ N bond is highly covalent, as has been proposed before on the basis of QTAIM calculations39.
While the MO analysis provides a useful qualitatively instructive framework to probe the field-induced magnetic coupling of occupied and virtual orbitals, the delocalised nature of Kohn Sham MOs renders such analysis incomplete. In order to derive a more representative picture we analysed the shielding data in terms of NLMOs63,64 (Table 1) pertaining to the NLMOs in Fig. 3. These data confirm that the δiso and Ω values of the nitride in 1* is due primarily to the U ≡ N σ- and π-bonds, which induce large negative σxx and σyy nuclear shielding component contributions to the σp parameter.
Focussing on the SOR data, the σ-bonding NLMO contributes −1234 and −1233 ppm to the Lewis and non-Lewis (L + NL) σxx and σyy, of which −1262 and −1261 ppm are σp + σso, and 28 ppm of each are σd, respectively. Since the σzz value is close to zero this then produces a σiso of −824 ppm for the U ≡ N σ-bond NLMO. The two U ≡ N π-bonds present a slightly more complex picture, with L + NL σxx and σyy values of −870/−90 and −91/−866 for the σp + σso component and σd values of 28/15 and 14/28 ppm; these values reflect the strong response of a given π-orbital for a perpendicular magnetic field, but much weaker response for a parallel magnetic field, as shown most clearly by the SR data, but the SOR calculation resolves contributions resulting from the quasi-degeneracy, and thus symmetry breaking components, of the orthogonal σxx and σyy tensors that result from residing within the C3-symmetric ligand field environment of the TrenTIPS ligand. Again, σzz components are quite small, resulting in σiso values of −303 and −302 ppm for the two U ≡ N π-bonds, showing that overall although the two U ≡ N π-bonds are not equivalent, their slight non-degeneracy averages out at the σiso level.
The large U ≡ N σiso NLMO values are counter-balanced largely only by the σd value from the nitride 1s orbital of 240 ppm, since various contributions from uranium outer core orbitals and other minor contributions from N lone pairs tend to be mainly cancelled out by competing Lewis and Non-Lewis components, leaving the U ≡ N σ- and π-bonding components as the dominant nuclear shielding components that primarily determine the NMR characteristics of the nitride.
A notable feature of the NLMO and MO shielding component data is the consistently small size of σso, whereas σso of up to hundreds of ppm have been reported in NMR studies of uranium-carbon, -nitride, and chalcogenido bonding20,21,24,25,26,36. As shown consistently by NBO and NLMO analysis the N-component of the bonding of the nitride is dominated by 2p-orbital character, with only 8% 2s-character in the U ≡ N σ-bond. Transfer of SO-induced spin-polarisation to the NMR nucleus is mediated by the N s-orbital, and so with little 2s character a small Fermi contact will result, despite the large overall contributions of both U- and N-orbitals in the U ≡ N bond. Thus, despite the significant SOC that would be anticipated from a covalent U ≡ N linkage with significant 5f-character, there is little σso in 1*. Though the Δso values for the σ- and π-U ≡ N bonds are small (cf +22 and −12 (av.)) we note that their respective signs are in-line with SO-heavy-atom-light-atom effects37, and again highlight the dominant contribution of σ ↔ π* over π ↔ σ* mixing to the 15Nnitride δiso of 1*, as consistently evidenced by the MO and NLMO analyses.
It is instructive to consider the DFT σso/2s-character NLMO data for the nitride (−29 ppm/8%), amide (−40 ppm/37% av.), and amine (−20 ppm/17%). Recalling the Mayer and DI data above, the nitride is bonded to uranium very covalently with high 5f- but low 2s-character; therefore, σso translates to being intermediate of the three. In contrast, the amide has lower, but significant enough, covalent bonding to uranium but much greater 2s-character to facilitate larger σso than the nitride, whereas the amine has essentially double the 2s-character of the nitride, but it engages in much more electrostatic bonding to uranium giving a lower σso value than the nitride.
The observation of little σso for 1* may at first seem contradictory to previous reports of large σso contributions to uranium(VI) chemical bonding. However, we propose that 1* simply reflects an extreme case of highly covalent bonding combined with little s-character in the bonding of the NMR nucleus. Where other ionic linkages are concerned, there is either more s-character in the bonding of the NMR nucleus, giving greater Fermi contact that would increase σso, or there is likely heavy-atom heavy-atom shielding effects37. Thus, even though 1* has major 5f-bonding contributions and very high covalency, dominant 2p-character of the N bonding results in small σso—this may be a hallmark of such covalent bonding with low s-character bonded ligands, and conversely it may be a characteristic feature of more ionic bonds, but ones which have enough covalency and s-character to have large σso that is increased by 5f-bonding character21,22,23,24,25,26,36.
Correlating 15N NMR spectroscopic chemical shift to metal-nitride bond order
The MO and NLMO data consistently suggest essentially constant σd and small σso contributions but large σp contributions to the 15Nnitride δiso resonance of 1*, which when taken together with the unremarkable HOMO-LUMO gap and small χTIP value of 1* are consistent with the σp parameter being the decisive factor for the 15Nnitride δiso. Since σp is a direct reporter of covalency, then plots of the experimental 15N δiso vs computed bond order metrics should produce linear correlations across all formal M–N bond orders. For this condition to be valid, the HOMO-LUMO gaps, which constitute a proportionate representation of the extent of magnetic field-induced mixing of occupied and virtual orbitals that contribute to σiso, for complexes under consideration should all be similar. This is indeed the case, spanning a range of ≤2.8 eV and we note that the HOMO-LUMO gap of 1* is consistently close to the mean and median computed HOMO-LUMO gap over BP86, PBE0, and B3LYP functionals (Supplementary Table 5). Indeed, separately plotting 15N δiso values for the three unique, correlated U–N bonds in 1* (experimental nitride δiso and extrapolated computed amide and amine δiso values) and available experimental data for 16 literature complexes25,26,46,47,48,49,50,51,65—various M-NR2, M = NR, and M ≡ N (terminal and capped) bonds involving Th, Ti, Zr, V, and Mo—against Mayer, Nalewajski-Mrozek (NM), Delocalisation Index (DI), and interatomic exchange-correlation energy (VXC)66 bond order metrics produces satisfactory linear correlated relationships with R2 values ranging from 0.7240 (δiso vs VXC) to 0.8621 (δiso vs Mayer) (Fig. 6 and Supplementary Figs. 12–14 and Supplementary Table 6).

Linear regression: Mayer bond order = (0.0019 × δiso(exp)) + 1.3461, R2 = 0.8621.
Considering the wide range of metals, metal oxidation states, supporting ligands, bridging and terminal nitrides, formal complex charges, solvent variation (THF and benzene), and focus on δiso that makes no distinction between dependency on σp or (σp + σso), the agreement of the four correlations is remarkably good. Furthermore, focussing on δiso vs Mayer (Fig. 6) if [(N″)3Th(μ-N)Th(N″)3][K(18C6)(THF)2]25,26 is omitted from the comparison, which is reasonable since that complex alone in the series exhibits large σso effects (79 ppm) that compromise the linear σp response, R2 becomes 0.905. These plots with associated δiso-bond order equations thus present reasonable models for making 15N NMR spectroscopic δiso and bond order predictions. These correlation plots all consistently suggest that the U ≡ N bond in 1* is highly covalent, and indeed more covalent than transition metal analogues, thus experimentally confirming the previous computational prediction39.
Discussion
We have reported solution and solid-state 15N NMR spectroscopic characterisation of 1*, revealing exceptional 15N chemical shift and chemical shift anisotropy for N-containing compounds. These properties provide a benchmark for 15N NMR spectroscopy, and result from large σp values (and almost negligible σso values) that originate from the U ≡ N triple bond σ- and π-components. Thus, the U ≡ N triple bond in 1* is experimentally confirmed to be highly covalent, and one that by computational and now experimental measures is more covalent than group 4–6 transition metal nitrides. The small σso contributions reflect the low 2s-character of the nitride bonding orbitals, which reduces the expected large SOC from a highly covalent U ≡ N triple bond with significant 5f-bonding character in 1*. The data presented here have been combined with literature data to confirm correlated linear relationships between 15N δiso data for M ← NR3, M–NR2, M = NR, and M ≡ N linkages and four measures of bond order in good agreement, which can be used for 15N NMR spectroscopic δiso and bond order predictions. By providing experimental confirmation of a previous theoretical prediction, these results demonstrate the powerful nature of using NMR spectroscopy to directly address the central, fundamental goal in actinide science of quantifying the nature and extent of covalency in An–L chemical bonding. These experimental data also serve to highlight the predictive power of modern 5f quantum chemistry.
Methods
Safety
Depleted uranium (0.2% 235U, 99.8% 238U) is a weak α-emitter (4.197 MeV) with a half-life of 4.47 × 109 years. While most manipulations can be carried out safely using Schlenk line or glove boxes in an appropriate personal protective equipment, logging, and monitoring regime, particular care should be exercised when embarking on solid-state magic angle spinning (SS-MAS) NMR experiments. A full risk assessment and protocols for loading, unloading, and actions in the event of a rotor crash and contamination should be completed before undertaking SS-MAS-NMR experiments.
General experimental details
All manipulations were carried out under an inert atmosphere of dry nitrogen using Schlenk techniques, or an MBraun UniLab glovebox operating under an atmosphere of dry nitrogen. THF, toluene, and pentane solvents were dried by passage through activated alumina towers and degassed before use. Hexanes and benzene were distilled from potassium. All solvents were stored over potassium mirrors except for ethers which were stored over activated 4Å sieves. Deuterated solvents were distilled from potassium, degassed by three freeze–pump–thaw cycles and stored under nitrogen prior to use. Iodine was used as purchased. Sodium-1-15N azide (98%) and benzo-15-crown-5 (B15C5) were dried under vacuum for 8 h before use. KC867 and [U(TrenTIPS)(Cl)]38 were prepared using literature methods.
Solution 1H and 29Si{1H} spectra were recorded on a Bruker AVIII 400 spectrometer operating at 400 and 79 MHz, respectively; chemical shifts are quoted in ppm and are relative to TMS (1H, 29Si), respectively. Solution 15N NMR spectra were acquired using a Bruker AVIII HD 400 spectrometer with 5 mm Prodigy probe. (1H TMS frequency 400 MHz, Me15NO2 frequency 40.5541637 MHz). Spectra were referenced to an external MeNO2 (neat) standard and acquired both unlocked without adjusting the field correction relative to the standard (to ensure accurate chemical shift measurement), and locked. Acquisition was performed by averaging 24576 transients of 16384 complex points acquired using a 10° r.f. pulse with an acquisition time of 1 s and a recycle delay of 1.7 s. For processing, a matched filter (3.2 Hz Lorentzian broadening) was applied to provide an optimum signal-to-noise ratio. Solid-state {1H-}15N cross-polarisation (CP) NMR spectra were recorded using a Bruker 9.4 T (400 MHz 1H Larmor frequency) AVANCE III spectrometer equipped with a 4 mm HFX MAS probe. Experiments were acquired at ambient temperature using both static and MAS conditions. Samples were packed into 4 mm o.d. zirconia rotors in a glovebox, and sealed with a Kel-F rotor cap. The 1H π-pulse duration was 5 μs, and the 15N π-pulse duration was 40 μs for static experiments and 22 μs for MAS experiments. 15N spin-locking was applied for 5 ms at ~12.5 kHz, with corresponding 1H spin-locking at ~12.5 kHz, for static CP experiments and at ~23 kHz, with corresponding ramped (70–100%) 1H spin-locking, for CPMAS experiments. One hundred kilohertz of SPINAL-64 (ref. 68) heteronuclear 1H decoupling was used throughout ~6 ms of signal acquisition (with 6.1 and 2.1 μs dwell-time between complex data points for static and MAS experiments, respectively). A Hahn-echo τr–π–τr sequence of two rotor periods total duration was applied to 15N after CP to circumvent receiver dead-time. For static and fast MAS experiments (νr = 9 kHz), the transmitter frequency offset was 1420 ppm and 912,576 and 204,800 transients were co-added for static and fast MAS experiments, respectively, with repetition delays of 0.6 s. For slow MAS experiments (νr = 2.5 kHz), transmitter frequency offsets of 1820, 1420, 1020, and −380 ppm were used and the resulting magnitude spectra were added. Spectral simulations were performed in the solid line-shape analysis (SOLA) module v2.2.4 in Bruker TopSpin v4.0.9. The 15N chemical shifts were referenced to MeNO2 externally using glycine (−347.2 ppm)69. Static variable-temperature magnetic moment data were recorded in an applied dc field of 1 T on a Quantum Design MPMS 3 superconducting quantum interference device (SQUID) magnetometer using recrystallised powdered samples. Care was taken to ensure complete thermalisation of the sample before each data point was measured and samples were immobilised in an eicosane matrix to prevent sample reorientation during measurements. Diamagnetic corrections were applied using tabulated Pascal constants and measurements were corrected for the effect of the blank sample holders (flame sealed Wilmad NMR tube and straw) and eicosane matrix.
Preparation of [U(TrenTIPS)(15/14N14N14/15N)]
This synthesis was performed in an analogous manner to the preparation of the previously reported [U(TrenTIPS)(N3)]41, using 98% isotopically enriched sodium-1-15N azide in place of isotopically normal sodium azide. THF (40 ml) was added slowly to a pre-cooled stirring mixture of [U(TrenTIPS)(Cl)] (1.77 g, 2.00 mmol) and sodium-1-15N azide (0.20 g, 3.00 mmol) at −78 °C. After addition, the brown solution was allowed to warm to room temperature and stirred for 3 days to ensure all the uranium chloride starting material was fully converted to the uranium azide product, which was confirmed by 1H NMR of the reaction before following work-up. Volatiles were removed in vacuo and the product was extracted into toluene (30 ml). The mixture was filtered, and volatiles were removed in vacuo to give a brown crude product, which was washed with cold pentane (2 × 10 ml) to yield [U(TrenTIPS)(15/14N14N14/15N)] as a green solid. Yield: 1.34 g, 75%. The purity of [U(TrenTIPS)(15/14N14N14/15N)] was confirmed by 1H NMR spectroscopy, evidenced by an identical spectrum to that of the non-labelled analogue39.
Preparation of [U(TrenTIPS)(N*)][K(B15C5)2] (N* =50:50 14/15N)
This synthesis was carried out in an analogous manner to the preparation of the reported [U(TrenTIPS)(N)][K(B15C5)2]41,42, using [U(TrenTIPS)(15/14N14N14/15N)] instead of non-15N-labelled [U(TrenTIPS)(N3)]39. Toluene (30 ml) was added to a pre-cooled stirring mixture of [U(TrenTIPS)(15/14N14N14/15N)] (1.34 g, 1.50 mmol) and KC8 (0.20 g, 1.50 mmol) at −78 °C, and then the resulting mixture was allowed to warm to ambient temperature and stirred for further 6 days to afford a dark brown suspension. After this time, volatiles were removed in vacuo and the product was washed with pentane (3 × 10 ml). The dark brown solid residue was then extracted into hot benzene (80 °C) and quickly filtered through a frit to remove the graphite precipitate. The filtrate was stored at 5 °C for 24 h to yield dark red crystals of [{U(TrenTIPS)}2(µ-N*K)2] that were isolated by filtration and dried in vacuo. Yield: 0.57 g, 42% (crystalline). It should be noted that the isolated [{U(TrenTIPS)}2(µ-N*K)2] and the following complexes [U(TrenTIPS)(N*)][(B15C5)2] and 1* are only 50% 15N-labelled due to the loss of 50% 15N-labelled nitrogen as dinitrogen in this reduction step. The bridging complex [{U(TrenTIPS)}2(µ-N*K)2] was converted to the terminal species [U(TrenTIPS)(N*)][(B15C5)2] by further reacting with 2.0 equivalents of benzo-15-crown-5 ether; therefore, toluene (20 ml) was added to the obtained [{U(TrenTIPS)}2(µ-N*K)2] (0.57 g, 0.63 mmol) with benzo-15-crown-5 (B15C5, 0.34 g, 1.30 mmol) at −78 °C. The red brown mixture was allowed to warm to room temperature and stirred for 16 h. The solvent was removed in vacuo to yield a brown residue which was washed with pentane (2 × 10 ml) and dried in vacuo to yield [U(TrenTIPS)(N*)][(B15C5)2] as a brown powder. Yield: 0.58 g, 64%. The purity of [U(TrenTIPS)(N*)][(B15C5)2]was confirmed by 1H NMR spectroscopy, evidenced by an identical spectrum to that of the non-labelled analogue42.
Preparation of [U(TrenTIPS)(N*)] (1*, N* =50:50 14/15N)
The synthesis of 1* was carried out in an analogous manner to the reported preparation of 1 (ref. 39), using [U(TrenTIPS)(N*)][(B15C5)2] as the reacting precursor in place of non-15N-labelled normal uranium(V) nitride complex [U(TrenTIPS)(N)][(B15C5)2]42. A solution of I2 (0.05 g, 0.20 mmol) in toluene (10 ml) was added dropwise to a stirring solution of [U(TrenTIPS)(N*)][(B15C5)2] (0.58 g, 0.40 mmol) in toluene (10 ml) at −78 °C. The brown solution was allowed to warm to room temperature with stirring over 16 h under dark as 1* is light sensitive. After this time, volatiles were removed in vacuo and the product was washed with pentane (2 × 5 ml). The brown residue was extracted into toluene (10 ml) and filtered to remove the [K(B15C5)2]I salt. The resulting red filtrate was concentrated to 3 ml and stored at −35 °C for 2 days, yielding 1* as red crystals. Yield: 0.18 g, 51%. The purity of the product was confirmed by 1H NMR in C6D6, evidenced by an identical spectrum to that of the non-labelled analgoue39. Complex 1* is poorly soluble in pentane, and only partially soluble in benzene/toluene, but more soluble in THF, so the solution 15N NMR spectrum was obtained in D8-THF, the 1H NMR of 1* was also recorded in D8-THF, indicating good stability of 1* in THF solvent in the absence of ambient light. 1H NMR (400 MHz, D8-THF, 298 K): δ (ppm) 1.56 (d, 54H, CH(CH3)2), 2.20 (septet, 9H, CH(CH3)2), 3.04 (t, 6H, CH2CH2), 5.42 (t, 6H, CH2CH2). 1H NMR (400 MHz, D8-THF and C6D6 = 50:50, 298 K): δ (ppm) 1.54 (d, 54H, CH(CH3)2), 2.16 (septet, 9H, CH(CH3)2), 2.68 (t, 6H, CH2CH2), 5.20 (t, 6H, CH2CH2). 1H NMR (400 MHz, C6D6, 298 K): δ (ppm) 1.69 (d, 54H, CH(CH3)2), 2.31 (septet, 9H, CH(CH3)2), 2.48 (t, 6H, CH2CH2), 5.17 (t, 6H, CH2CH2). 29Si{1H} NMR (79 MHz, D8-THF, 298 K): δ (ppm) 3.78. 29Si{1H} NMR (79 MHz, D8-THF and C6D6 = 50:50, 298 K): δ (ppm) 5.60. 29Si{1H} NMR (79 MHz, C6D6, 298 K): δ (ppm) 5.87. 15N{1H} NMR (41 MHz, D8-THF, 298 K): δ (ppm) 968.9. 15N{1H} NMR (41 MHz, D8-THF and C6D6 = 50:50, 298 K): δ (ppm) 970.4. 15N{1H} NMR (41 MHz, C6D6, 298 K): δ (ppm) 972.6.
General computational details
Geometry optimisations were performed using coordinates derived from the respective crystal structures as the starting points. No constraints were imposed on the structures during the geometry optimisations. The calculations were performed using the Amsterdam Density Functional (ADF) suite version 2017 with standard convergence criteria70,71. The DFT geometry optimisations employed Slater type orbital (STO) TZP polarisation all-electron basis sets (from the Dirac and ZORA/TZP database of the ADF suite). Scalar relativistic approaches (spin–orbit neglected) were used within the ZORA Hamiltonian72,73,74 for the inclusion of relativistic effects and the local density approximation with the correlation potential due to Vosko et al. was used in all of the calculations75. Generalised gradient approximation corrections were performed using the functionals of Becke and Perdew76,77.
Scalar and two-component spin–orbit relativistic (ZORA) single point energy calculations were then run on the geometry optimised coordinates. For 1* the functionals screened included SAOP-TZ2P, BP86-TZ2P, PBE0-TZP, PBE0-TZ2P, PBE0-HF40-TZ2P, and B3LYP-TZ2P, the latter of which gave the closest agreement of computed NMR properties compared to experiment. Once B3LYP was identified as the functional of choice, scalar relativistic (ZORA) single point energy calculations on the literature compounds used for the standardisation of empirical corrections were run with the B3LYP functional and STO TZ2P polarisation all-electron basis sets. The conductor-like screening model (COSMO) was used to simulate solvent effects, with the appropriate solvent matched to the solvent medium used in the literature report for the specific compound being modelled. MOs were visualised with ADFView.
NLMO analysis of 1* was carried out using NBO6 (ref. 78) and the B3LYP STO TZ2P scalar relativistic ZORA COSMO single point energy data. These calculations used the Hartree–Fock RI scheme to suspend the dependency key and avoid numerical issues. The NLMOs were visualised using ADFView.
Mayer and Nalewajski-Mrozek values were computed using ADF and the PBE0 functional. DI and VXC bond metrics were computed using AIMAll79, from WFX files generated from single point energy calculations performed with Gaussian 16 (ref. 80). In these calculations, the PBE0 (refs. 81,82) density functional approximation was employed. Dunning’s correlation consistent basis sets of polarised triple-ζ quality83,84,85,86 was used for all non-actinide atoms, except for K which was treated with Pople’s 6-311G* basis set87. U and Th atoms were treated with the all-electron SARC basis sets88,89,90, including the second-order Douglas–Kroll–Hess (DKH2) Hamiltonian to account for scalar relativistic effects91,92,93. The PBE0 functional was used since NBO and NLMO outputs from the ADF and Gaussian 16 programmes could be checked to ensure consistency of outputs.
NMR shielding calculations were carried out using the NMR programme within ADF63,64,94,95,96,97,98. Calculated nuclear shieldings were converted to chemical shifts by subtraction from the calculated nuclear shielding of MeNO2 in neat MeNO2 calculated at the same level. Canonical MO contributions to the nuclear shieldings were calculated at the scalar and two-component spin–orbit levels, the former with the FAKESO key. Scalar and two-component spin–orbit NLMO calculations of the computed nuclear shieldings were carried out using NBO6 and ADF. These calculations used the Hartree–Fock RI scheme to suspend the dependency key and avoid numerical issues.
Data availability
All data are available within this article, the Supplementary Information (Supplementary Figs. 1–14 and Table 1–6), or from S.T.L. on reasonable request.
References
Glueckauf, E. & McKay, H. A. C. Possible f-shell covalency in the actinide elements. Nature 165, 594–595 (1950).
Neidig, M. L., Clark, D. L. & Martin, R. L. Covalency in f-element complexes. Coord. Chem. Rev. 257, 394–406 (2013).
Kaltsoyannis, N. Does covalency increase or decrease across the actinide series? Implications for minor actinide partitioning. Inorg. Chem. 52, 3407–3413 (2013).
Hayton, T. W. Metal-ligand multiple bonding in uranium: structure and reactivity. Dalton Trans. 39, 1145–1158 (2010).
gJones, M. B. & Gaunt, A. J. Recent developments in synthesis and structural chemistry of nonaqueous actinide complexes. Chem. Rev. 113, 1137–1198 (2013).
Hayton, T. W. Recent developments in actinide-ligand multiple bonding. Chem. Commun. 49, 2956–2973 (2013).
La Pierre, H. S. & Meyer, K. Activation of small molecules by molecular uranium complexes. Prog. Inorg. Chem. 58, 303–415 (2014).
Liddle, S. T. The renaissance of non-aqueous uranium chemistry. Angew. Chem. Int. Ed. 54, 8604–8641 (2015).
Schädle, D. & Anwander, R. Rare-earth metal and actinide organoimide chemistry. Chem. Soc. Rev. 48, 5752–5805 (2019).
Lukens, W. W. et al. Quantifying the σ and π interactions between U(V) f orbitals and halide, alkyl, alkoxide, amide, and ketimide ligands. J. Am. Chem. Soc. 135, 10742–10754 (2013).
Seaman, L. A. et al. Probing the 5f orbital contribution to the bonding in a U(V) ketimide complex. J. Am. Chem. Soc. 134, 4931–4940 (2012).
Smiles, D. E. et al. The duality of electron localization and covalency in lanthanide and actinide metallocenes. Chem. Sci. 11, 2796–2809 (2020).
Su, J. et al. Energy-degeneracy-driven covalency in actinide bonding. J. Am. Chem. Soc. 140, 17977–17984 (2018).
Cross, J. N. et al. Covalency in Americium(III) hexachloride. J. Am. Chem. Soc. 139, 8667–8677 (2017).
Minasian, S. G. et al. New evidence for 5f covalency in actinocenes determined from carbon K-edge XAS and electronic structure theory. Chem. Sci. 5, 351–359 (2014).
Spencer, L. P. et al. Tetrahalide complexes of the [U(NR)2]2+ ion: synthesis, theory, and chlorine K-edge X-ray absorption spectroscopy. J. Am. Chem. Soc. 135, 2279–2290 (2013).
Minasian, S. G. et al. Determining relative f and d orbital contributions to M-Cl covalency in MCl62− (M=Ti, Zr, Hf, U) and UOCl5− using Cl K-edge X-ray absorption spectroscopy and time-dependent density functional theory. J. Am. Chem. Soc. 134, 5586–5597 (2012).
Kozimor, S. A. et al. Trends in covalency for d- and f-element metallocene dichlorides identified using chlorine K-edge X-ray absorption spectroscopy and time-dependent density functional theory. J. Am. Chem. Soc. 131, 12125–12136 (2009).
Formanuik, A. et al. Actinide covalency measured by pulsed electron paramagnetic resonance spectroscopy. Nat. Chem. 9, 578–583 (2017).
Hrobárik, P., Hrobáriková, V., Greif, A. H. & Kaupp, M. Giant spin-orbit effects on NMR shifts in diamagnetic actinide complexes: guiding the search of uranium(VI) hydride complexes in the correct spectral range. Angew. Chem. Int. Ed. 51, 10884–10888 (2012).
Seaman, L. A. et al. A rare uranyl(VI)-alkyl ate complex [Li(DME)1.5]2[UO2(CH2SiMe3)4] and its vomparison with a homoleptic uranium(VI)-hexaalkyl. Angew. Chem. Int. Ed. 52, 3259–3263 (2013).
Smiles, D. E., Wu, G., Hrobárik, P. & Hayton, T. W. Synthesis, thermochemistry, bonding, and 13C NMR chemical shift snalysis of a phosphorano-stabilized carbene of thorium. Organometallics 36, 4519–4524 (2017).
Rungthanaphatsophon, P., Huang, P. & Walensky, J. R. Phosphorano-stabilized carbene complexes with short Thorium(IV)- and Uranium(IV)-carbon bonds. Organometallics 37, 1884–1891 (2018).
Mullane, K. C. et al. 13C NMR shifts as an indicator of U-C bond covalency in uranium(VI) acetylide complexes: an experimental and computational study. Inorg. Chem. 58, 4152–4163 (2019).
Staun, S. L., Sergentu, D. C., Wu, G., Autschbach, J. & Hayton, T. W. Use of 15N NMR spectroscopy to probe covalency in a thorium nitride. Chem. Sci. 10, 6431–6436 (2019).
Sergentu, D. C. et al. Probing the electronic structure of a thorium nitride complex by solid-state 15N NMR spectroscopy. Inorg. Chem. 59, 10138–10145 (2020).
Cho, H., De Jong, W. A. & Soderquist, C. Z. Probing the oxygen environment in UO22+ by solid-state 17O nuclear magnetic resonance spectroscopy and relativistic density functional calculations. J. Chem. Phys. 132, 084501 (2010).
Martel, L. et al. High-resolution solid-state oxygen-17 NMR of actinide-bearing compounds: an insight into the 5f chemistry. Inorg. Chem. 53, 6928–6933 (2014).
Martel, L. et al. Structural investigation of uranium−neptunium mixed oxides using XRD, XANES, and 17O MAS NMR. J. Phys. Chem. C 118, 27640–27647 (2014).
Martel, L. et al. Magnetization, specific heat, 17O NMR, and 237Np Mössbauer study of U0.15Np0.85O2. Phys. Rev. B Condens. Matter Mater. Phys. 98, 014410 (2018).
Devore, M. A., Klug, C. A., Kriz, M. R., Roy, L. E. & Wellons, M. S. Investigations of uranyl fluoride sesquihydrate (UO2F2·1.57H2O): combining 19F solid-state MAS NMR spectroscopy and GIPAW chemical shift calculations. J. Phys. Chem. A 122, 6873–6878 (2018).
Gabuda, S. P., Falaleeva, L. G. & Gagarinskii, Y. V. Dipolar broadening of the NMR dpectrum of 19F in UF4. Phys. Status Solidi B 33, 435–438 (1969).
Capan, C., Dempsey, R. J., Sinkov, S., McNamara, B. K. & Cho, H. Probing the Pu4+ magnetic moment in PuF4 with 19F NMR spectroscopy. Phys. Rev. B Condens. Matter Mater. Phys. 93, 224409 (2016).
Martel, L. et al. Insight into the crystalline structure of ThF4 with the combined use of neutron diffraction, 19F magic-angle spinning-NMR, and density functional theory calculations. Inorg. Chem. 57, 15350–15360 (2018).
Mounce, A. M. et al. Nuclear magnetic resonance measurements and electronic structure of Pu(IV) in [Me4N]2PuCl6. Inorg. Chem. 55, 8371–8380 (2016).
Smiles, D. E., Wu, G., Hrobárik, P. & Hayton, T. W. Use of 77Se and 125Te NMR spectroscopy to probe covalency of the actinide-chalcogen bonding in [Th(En){N(SiMe3)2}3]- (E = Se, Te; N = 1, 2) and their oxo-uranium(VI) congeners. J. Am. Chem. Soc. 138, 814–825 (2016).
Vĺcha, J. et al. Relativistic heavy-neighbor-atom effects on nmr shifts: concepts and trends across the periodic table. Chem. Rev. 120, 7065–7103 (2020).
King, D. M. et al. Synthesis and structure of a terminal uranium nitride complex. Science 337, 717–720 (2012).
King, D. M. et al. Isolation and characterisation of a uranium(VI)-nitride triple bond. Nat. Chem. 5, 482–488 (2013).
Barluzzi, L., Scopelliti, R. & Mazzanti, M. Photochemical synthesis of a stable terminal uranium(VI) nitride. J. Am. Chem. Soc. 142, 19047–19051 (2020).
King, D. M. et al. Molecular and electronic structure of terminal and alkali metal-capped uranium(V)-nitride complexes. Nat. Commun. 7, 13773 (2016).
Cleaves, P. A. et al. Two-electron reductive carbonylation of terminal uranium(V) and uranium(VI) nitrides to cyanate by carbon monoxide. Angew. Chem. Int. Ed. 53, 10412–10415 (2014).
Chatelain, L. et al. Terminal uranium(V)-nitride hydrogenations involving direct addition or frustrated Lewis pair mechanisms. Nat. Commun. 11, 14221-y (2020).
Barluzzi, L. et al. Synthesis, structure, and reactivity of uranium(VI) nitrides. Chem. Sci. 12, 8096–8104 (2021).
Harris, R. K., Becker, E. D., Cabral de Menezes, S., Goodfellow, R. & Granger, P. NMR nomenclature. nuclear spin properties and conventions for chemical shifts. Pure Appl. Chem. 73, 1795–1818 (2001).
Tran, B. L. et al. A planar three-coordinate Vanadium(III) complex and the study of terminal vanadium nitrides from N2: a kinetic or thermodynamic impediment to N-N bond cleavage? J. Am. Chem. Soc. 134, 13055–13045 (2012).
Tran, B. L., Pink, M., Gao, X., Park, H. & Mindiola, D. J. Low-coordinate and neutral nitrido complexes of vanadium. J. Am. Chem. Soc. 132, 1458–1459 (2010).
Carroll, M. E., Pinter, B., Carroll, P. J. & Mindiola, D. J. Mononuclear and terminally bound titanium nitrides. J. Am. Chem. Soc. 137, 8884–8887 (2015).
Grant, L. N. et al. Molecular titanium nitrides: nucleophiles unleashed. Chem. Sci. 8, 1209–1224 (2017).
Sceats, E. L. et al. Complexes obtained by electrophilic attack on a dinitrogen-derived terminal molybdenum nitride: electronic structure analysis by solid state CP/MAS 15N NMR in combination with DFT calculations. Polyhedron 23, 2751–2768 (2004).
Thompson, R., Chen, C.-H., Pink, M., Wu, G. & Mindiola, D. J. A nitrido salt reagent of titanium. J. Am. Chem. Soc. 136, 8197–8200 (2014).
Lee, D. et al. Solid-state NMR and DFT combined for the surface study of functionalized silicon nanoparticles. Chem. Eur. J. 21, 16047–16058 (2015).
Xin, D. et al. Systematic investigation of DFT-GIAO 15N NMR chemical shift prediction using B3LYP/cc-pVDZ: application to studies of regioisomers, tautomers, protonation states and N-oxides. Org. Biomol. Chem. 15, 928–936 (2017).
Windorff, C. J. & Evans, W. J. 29Si NMR spectra of silicon-containing uranium complexes. Organometallics 33, 3786–3791 (2014).
Widdifield, C. M. & Schurko, R. W. Understanding chemical shielding tensors using group theory, MO analysis, and modern density-functional theory. Concepts Magn. Reson. Part A 34, 91–123 (2009).
Ramsey, N. F. Magnetic shielding of nuclei in molecules. Phys. Rev. 78, 699–703 (1950).
Ramsey, N. F. Dependence of magnetic shielding of nuclei upon molecular orientation. Phys. Rev. 83, 540–541 (1951).
Ramsey, N. F. Chemical effects in nuclear magnetic resonance and in diamagnetic susceptibility. Phys. Rev. 86, 243–246 (1952).
Flygare, W. H. Magnetic interactions in molecules and an analysis of molecular electronic charge distribution from magnetic parameters. Chem. Rev. 74, 653–687 (1974).
Wu, G. et al. Unusual 31P chemical shielding tensors in terminal phosphido complexes containing a phosphorus-metal triple bond. J. Am. Chem. Soc. 118, 10654–10655 (1996).
Halbert, S., Copéret, C., Raynaud, C. & Eisenstein, O. Elucidating the link between NMR chemical shifts and electronic structure in d0 metathesis catalysts. J. Am. Chem. Soc. 138, 2261–2272 (2016).
Yamamoto, K. et al. Orbital analysis of carbon-13 chemical shift tensors reveals patterns to distinguish Fischer and Schrock carbenes. Angew. Chem. Int. Ed. 56, 10127–10131 (2017).
Autschbach, J. & Analyzing, N. M. R. Shielding tensors calculated with two-component relativistic methods using spin-free localized molecular orbitals. J. Chem. Phys. 128, 164112 (2008).
Autschbach, J. & Zheng, S. Analyzing Pt chemical shifts calculated from relativistic density functional theory using localized orbitals: the role of Pt 5d lone pairs. Magn. Reson. Chem. 46, S45–S55 (2008).
Grant, L. N., Pinter, B., Gu, J. & Mindiola, D. J. Molecular zirconium nitride super base from a mononuclear parent imide. J. Am. Chem. Soc. 140, 17399–17403 (2018).
Berryman, V. E. J. et al. Quantum chemical topology and natural bond orbital analysis of M-O covalency in M(OC6H5)4 (M = Ti, Zr, Hf, Ce, Th, Pa, U, Np). Phys. Chem. Chem. Phys. 22, 16804–16812 (2020).
Bergbreiter, D. E. & Killough, J. M. Reactions of potassium-graphite. J. Am. Chem. Soc. 100, 2126–2134 (1978).
Fung, B. M., Khitrin, A. K. & Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 142, 97–101 (2000).
Bertani, P., Raya, J. & Bechinger, B. 15N chemical shift referencing in solid state NMR. Solid State Nucl. Magn. Reson. 61-62, 15–18 (2014).
Fonseca Guerra, C., Snijders, J. G., Te Velde, G. & Baerends, E. J. Towards an order-N DFT method. Theor. Chem. Acc. 99, 391–403 (1998).
Te Velde, G. et al. Chemistry with ADF. J. Comput. Chem. 22, 931–967 (2001).
Van Lenthe, E., Baerends, E. J. & Snijders, J. G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 99, 4597–4610 (1993).
Van Lenthe, E., Baerends, E. J. & Snijders, J. G. Relativistic total energy using regular approximations. J. Chem. Phys. 101, 9783–9792 (1994).
Van Lenthe, E., Ehlers, A. E. & Baerends, E. J. Geometry optimization in the zero order regular approximation for relativistic effects. J. Chem. Phys. 110, 8943–8953 (1999).
Vosko, S. H., Wilk, L. & Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 58, 1200–1211 (1980).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. A 38, 3098 (1988).
Perdew, J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 33, 8822 (1986).
Glendening, E. D., Landis, C. R. & Weinhold, F. Erratum: NBO 6.0: natural bond orbital analysis program. J. Comput. Chem. 34, 2134–2134 (2013).
Keith, T. A. AIMA11, TK Gristmill Software http://aim.tkgristmill.com/ (2019).
Frisch, M. J. et al. Gaussian 16, Revision C.01 (Gaussian, Inc., 2016).
Ernzerhof, M. & Scuseria, G. E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 110, 5029–5036 (1999).
Adamo, C. & Barone, V. Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110, 6158–6170 (1999).
Dunning, T. H. J. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989).
Kendall, R. A., Dunning, T. H. & Harrison, R. J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 96, 6796–6806 (1992).
Woon, D. E. & Dunning, T. H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 98, 1358–1371 (1993).
Wilson, A. K., Van Mourik, T. & Dunning, T. H. Gaussian basis sets for use in correlated molecular calculations. VI. Sextuple zeta correlation consistent basis sets for boron through neon. J. Mol. Struct. THEOCHEM. 388, 339–349 (1996).
Blaudeau, J. P., McGrath, M. P., Curtiss, L. A. & Radom, L. Extension of Gaussian-2 (G2) theory to molecules containing third-row atoms K and Ca. J. Chem. Phys. 107, 5016–5021 (1997).
Pritchard, B. P., Altarawy, D., Didier, B., Gibson, T. D. & Windus, T. L. New basis set exchange: an open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model. 59, 4814–4820 (2019).
Pantazis, D. A. & Neese, F. All-electron scalar relativistic basis sets for the actinides. J. Chem. Theory Comput. 7, 677–684 (2011).
Pantazis, D. A. & Neese, F. All-electron basis sets for heavy elements. Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 363–374 (2014).
Douglas, M. & Kroll, N. M. Quantum electrodynamical corrections to the fine structure of helium. Ann. Phys. (NY) 82, 89–155 (1974).
Hess, B. A. Relativistic electronic-structure calculations employing a two-component no-pair formalism with external-field projection operators. Phys. Rev. A 33, 3742–3748 (1986).
Jansen, G. & Hess, B. A. Revision of the Douglas-Kroll transformation. Phys. Rev. A 39, 6016–6017 (1989).
Schreckenbach, G. & Ziegler, T. Calculation of NMR shielding tensors using gauge-including atomic orbitals and modern density functional theory. J. Phys. Chem. 99, 606–611 (1995).
Autschbach, J. & Zurek, E. Relativistic density-functional computations of the chemical shift of 129Xe in Xe@C60. J. Phys. Chem. A 107, 4967–4972 (2003).
Krykunov, M., Ziegler, T. & Van Lenthe, E. Hybrid density functional calculations of nuclear magnetic shieldings using Slater-type orbitals and the zeroth-order regular approximation. Int. J. Quantum Chem. 109, 1676–1683 (2009).
Wolff, S. K., Ziegler, T., Van Lenthe, E. & Baerends, E. J. Density functional calculations of nuclear magnetic shieldings using the zeroth-order regular approximation (ZORA) for relativistic effects: ZORA nuclear magnetic resonance. J. Chem. Phys. 110, 7689–7698 (1999).
Autschbach, J. The role of the exchange-correlation response kernel and scaling corrections in relativistic density functional nuclear magnetic shielding calculations with the zeroth-order regular approximation. Mol. Phys. 111, 2544–2554 (2013).
Acknowledgements
We gratefully acknowledge funding and support from the UK Engineering and Physical Sciences Research Council (grants EP/K024000/1, EP/M027015/1, and EP/S033181/1), European Research Council (grant CoG612724), and The University of Manchester including computational resources and associated support services from the Computational Shared Facility. The Alexander von Humboldt Foundation is thanked for a Friedrich Wilhelm Bessel Research Award to S.T.L. We thank Dr. Ashley J. Wooles (The University of Manchester) for MAS NMR sample radiological safety assessments and protocols.
Author information
Authors and Affiliations
Contributions
J.D. prepared and characterised the sample. J.A.S. performed and analysed the magnetic measurements. V.E.J.B. and N.K. conducted quantum chemical calculations to determine the bond metrics. R.W.A. and D.L. recorded and analysed the NMR spectroscopic data. S.T.L. conceived the central idea, conducted the DFT and NLMO NMR calculations, analysed the data, and wrote the manuscript with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, J., Seed, J.A., Berryman, V.E.J. et al. Exceptional uranium(VI)-nitride triple bond covalency from 15N nuclear magnetic resonance spectroscopy and quantum chemical analysis. Nat Commun 12, 5649 (2021). https://doi.org/10.1038/s41467-021-25863-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-25863-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
