
Abstract
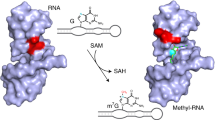
Methylation is a prevalent post-transcriptional modification encountered in coding and non-coding RNA. For RNA methylation, cells use methyltransferases and small organic substances as methyl-group donors, such as S-adenosylmethionine (SAM). SAM and other nucleotide-derived cofactors are viewed as evolutionary leftovers from an RNA world, in which riboswitches have regulated, and ribozymes have catalyzed essential metabolic reactions. Here, we disclose the thus far unrecognized direct link between a present-day riboswitch and its inherent reactivity for site-specific methylation. The key is O6-methyl pre-queuosine (m6preQ1), a potentially prebiotic nucleobase which is recognized by the native aptamer of a preQ1 class I riboswitch. Upon binding, the transfer of the ligand’s methyl group to a specific cytidine occurs, installing 3-methylcytidine (m3C) in the RNA pocket under release of pre-queuosine (preQ1). Our finding suggests that nucleic acid-mediated methylation is an ancient mechanism that has offered an early path for RNA epigenetics prior to the evolution of protein methyltransferases. Furthermore, our findings may pave the way for the development of riboswitch-descending methylation tools based on rational design as a powerful alternative to in vitro selection approaches.
Similar content being viewed by others

Introduction
In recent years, numerous discoveries of metabolite-sensing riboswitches hint at RNA World ribozymes that could have promoted chemical transformations1,2,3. One hypothesis suggests that present-day riboswitches for common enzyme cofactors may have evolved from ancient ribozymes that depended on these cofactors4,5,6,7. What has stood the test of time are RNA sensors of cofactors for the regulation of gene expression8 while the reactivity part has been taken over by more efficient protein-based modification machineries9,10,11,12. Riboswitches typically employ a highly conserved aptamer domain to sense the small molecule ligand with high specificity and selectivity, and an adjoining expression platform to regulate the expression of genes associated with ligand biosynthesis and transport8,13,14. Thereby, the partially overlapping RNA sequences of the two domains assist in converting the ligand-binding event into a change in gene expression by employing ligand-mediated RNA folding changes. It seems reasonable to assume that ligand-induced structural adaptions may also be exploited to trigger a chemical reaction. A single example exists for a known naturally occurring RNA, namely the glms riboswitch-ribozyme which binds glucosamine-6-phosphate whose protonated amino group then directly participates in reaction catalysis of RNA backbone cleavage15. Moreover, support for an advanced reactivity scope of cofactor-RNA systems has been provided by ribozyme engineering. The first methyltransferase ribozyme that catalyzes the site-specific installation of 1-methyladenosine in a substrate RNA, using a small-molecule cofactor has been created by in vitro evolution very recently16.
Numerous natural riboswitches are known to selectively sense nucleotide-derived metabolites connected to methyl-group transfer or one-carbon metabolism, comprising SAM, methylene tetrahydrofolate (THF) and adenosylcobalamin (vitamin B12)17. The aptamers of these riboswitches bind their dedicated ligands in the nano- to micromolar range. For all of them, different subclasses are found, meaning that the ligand is recognized by distinct RNA architectures with both specific binding pockets as well as overall folds18. For SAM-binding riboswitches, a total of six subclasses is known to date; they accommodate the ligand with its reactive methyl group in conformations that strictly avoid self-methylation19,20.
Therefore, it has remained an open question whether riboswitches can catalyze site-specific methylation reactions to produce defined methylated RNA products9,11. Besides the above-mentioned ubiquitous enzyme cofactors, other small organic compounds could serve for RNA-catalyzed RNA methylation in modern riboswitches, as implicated by the recently in vitro selected RNA methyltransferase that utilizes O6-methyl guanine16. An obvious link between guanine riboswitches and the near-cognate ligand O6-methyl guanine was tested even prior to the in vitro selected methyltransferase, however, methylation was not observed21. Nevertheless, we speculated that other riboswitches sensing guanine-type ligands may possess the inherent reactivity for methyl group transfer that we were searching for. Here, we report on the identification of a riboswitch motif that uses O6-methyl pre-queuosine (m6preQ1) as a small-molecule methyl-group donor, catalyzing site-specific methylation of a cytosine at the N3-atom, resulting in RNA sequence-specific installation of 3-methylcytidine (m3C) (Fig. 1).

RNA sequence-specific installation of N3-methyl cytidine (m3C) using the small molecule m6preQ1 as cofactor has been discovered and is described in this study. The direct link between a present-day riboswitch scaffold and RNA-catalyzed methylation is reported.
Results
Reactivity predictions for the preQ1 riboswitch and ligand requirements
We started our ambitious scheme to uncover RNA methylation activity with the analysis of ligand binding sites of various riboswitches and soon focused on preQ1 class I riboswitches (Fig. 2a) for which high-resolution crystal structures of both ligand-free and ligand-bound states were available (Fig. 2b, c)22,23,24. This choice was also guided by our hypothesis that the methylated version of the cognate ligand preQ1, namely m6preQ1, might be an excellent candidate for methyl group donation since m6preQ1 should retain the majority of interactions with the RNA as encountered in the genuine ligand-riboswitch system. Moreover, the principal reactivity of m6preQ1 is expected to be comparable to m6G that serves as cofactor in the recently in vitro selected RNA methyltransferase16.

a Sequence and secondary structure of the Tt preQ1-I riboswitch (minimal aptamer motif in black). b Cartoon representation of the three-dimensional fold of the preQ1-bound aptamer (pdb code 3Q50; DOI: 10.2210/pdb3q50/pdb). c Stick representation of the ligand-free pocket revealing that A14 slides into the pocket and fills the space of preQ1 while C15 flips out and becomes exposed (pdb code 3Q51; DOI: 10.2210/pdb3q51/pdb). d Stick representation of the preQ1-bound riboswitch pocket with crucial atom numbers annotated in black. e Chemical structures illustrating the hydrogen bond network of preQ1 in the riboswitch pocket and the 2’-OH group of G11 that comes close the Hoogsteen face of preQ1. f Structure of a putative base pair between C15 (imino tautomer) and m6preQ1 in the preQ1-I riboswitch pocket.
Figure 2a shows the sequence and secondary structure of the 33 nt long aptamer of the Thermoanaerobacter tengcongensis (Tt) preQ1 riboswitch which adopts an H-type pseudoknot structure exhibiting low nanomolar affinity for preQ1 (Fig. 2b). The ligand is recognized by forming a Watson-Crick base pair with C15 (Fig. 2d,e) while the ligand’s N3 hydrogen acceptor and the C2-NH2 group are recognized in bidentate fashion via the Watson-Crick face of A29, complemented by a single H-bond between N9-H and the carbonyl oxygen of U6. Furthermore, a critical interaction responsible for the high affinity of preQ1 is attributed to the 7-aminomethyl group and the carbonyl oxygen of G5. The tight binding pocket is further defined by preQ1 stacking between the G5-C16 base pair and the nucleobase of G11 whose ribose 2′-OH is directed toward the preQ1 O6 atom and the 7-aminomethyl group (Fig. 2e). Importantly, the crystal structure of the ligand-free Tt RNA (Fig. 2c) points at the high flexibility and structural dynamics of parts of the binding pocket: C15 is now directed outwards while the nucleobase of its next neighbor (A14) slides into the stacked position filling the space of the former C15-preQ1 base pair.
Because of the above-highlighted characteristics in structure and structural dynamics of the preQ1 class I binding site we anticipated a high probability that the ligand congener m6preQ1 transfers its methyl group to the RNA: First of all, recognition of m6preQ1 by wild-type C15 should be easily possible if the tautomeric equilibrium is shifted towards the imino tautomer imC15 (Fig. 2f). Second, the wild-type RNA pocket provides sufficient space to accommodate the methyl group (Fig. 2b,d). Third, there are three potential nucleophilic groups for methylation available in the vicinity of the methyl group if m6preQ1 becomes recognized by the pocket: these are the 2′-OH of G11, the C4-NH2 of C15, and the N3 of C15 (Fig. 2 d,e).
Identification of methylated RNA product and reaction conditions
A methylation product of the preQ1 riboswitch would be difficult to distinguish from the unmodified RNA by gel shift or HPLC assays because its expected retention time will hardly differ. Therefore, we set out for an advanced mass spectrometric technology, Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometry (MS), which can determine the mass-to-charge ratio (m/z) of ions with very high precision by measuring the frequency of their cyclotron motion in a static magnetic field25,26. A major strength of such a set-up is that an RNA can be directly sequenced along with the identification of modifications and their localization (top-down RNA characterization) as we have demonstrated earlier27.
We prepared the 33 nt long Tt preQ1 riboswitch by RNA solid-phase synthesis, synthesized the m6preQ1 ligand, and for initial trials, incubated both in a riboswitch-typical aqueous buffer system (2.5 µM RNA, 75 µM m6preQ1, 50 mM MOPS, 100 mM KCl, 2 mM MgCl2, pH 7.0). The reaction mixture was left overnight at room temperature, after which ultrafiltration with centrifugal concentrator devices was applied to remove the ligand and salts from the RNA for direct characterization by electrospray ionization (ESI) FT-ICR MS. Encouragingly, the high-resolution FT-ICR mass spectrum indeed showed the anticipated signal with a 14.0157 Da mass increase, consistent with a methyl group attached to the RNA (Fig. 3a). The power of FT-ICR mass spectrometry is further manifested in the fact that backbone cleavage by collisionally activated dissociation (CAD) produced a complete set of c and y fragments (Fig. 3b,c) that allowed for sequence determination (Fig. 3c). The fragment mass values unequivocally revealed the site of methylation at the C15 nucleoside. Furthermore, the loss of methyl-C from RNA ions (Fig. 3d) is direct evidence that the methyl group is located at the nucleobase. With respect to the precise position, the observed ratio for C to methyl-C of 3:1, which is higher than the statistical ratio of 8:1 according to the number of Cs in the sequence, suggests the N3 atom because methylation at the imino group generates a positive charge that weakens the glycosidic bond and in consequence facilitates base loss.

a ESI mass spectrum of the 33 nt m3C Tt preQ1-I RNA; the inset shows the isotopically resolved signal of the (M–10H)10- ions; asterisks indicate piperidine RNA adducts. b Collisionally activated dissociation (CAD) of (M–nH)n– ions of RNA in the collision cell produces c and y fragment ions from RNA backbone cleavage. c Fragment-ion map illustrating sequence coverage from CAD of the methylated Tt preQ1-I RNA. The numbering of c and y fragments starts from the 5’ and 3’ terminus, respectively (top). MS signals of unmodified and methylated c13, c14, c15, c16, and complementary y20, y19, y18, y17 fragments from CAD of (M−7H)7− and (M–10H)10− ions reveal the site of methylation (C15); the calculated isotopic profiles for unmodified and singly methylated RNA are indicated by red open circles. d Loss of methylated cytosine (red) in spectra from collisionally activated dissociation (CAD) of (M–10H)10- ions of RNA is direct evidence for C nucleobase methylation; the 1:3 ratio of m3C/C nucleobase losses is consistent with destabilization of the glycosidic bond as a result of methylation at the N3 position.
To be able to rapidly analyze the methylation reaction, we optimized anion-exchange HPLC conditions, resulting in clear baseline separation of the methylation product from the unmodified RNA (Fig. 4a). We then tested different reaction conditions. Most importantly, when we decreased the pH from 7.0 to 6.0, the reaction yields increased to a significant extent, approaching 50% after 24 h which is the maximum yield possible, considering that the cognate preQ1 ligand (with low nanomolar affinity)21,28 released during the methylation reaction will be bound by the riboswitch/ribozyme, thereby blocking the binding of m6preQ1 (Fig. 4b and Supplementary Fig. 1a). We note that upon removal of the low molecular weight compounds by ultrafiltration, followed by adding a new batch of m6preQ1 the reaction yields were increased further (Supplementary Fig. 1a). Importantly, the reaction rate was dependent on m6preQ1 concentration, with an apparent Michaelis constant Km of about 230 μM (Fig. 4c and Supplementary Fig. 1b). In this context, we mention that when a one-fold excess of m6preQ1 over RNA was applied only, methylation yields still amounted to 25% (Supplementary Fig. 1b).

a Anion-exchange HPLC analysis of the preQ1-I RNA-catalyzed reaction with m6preQ1 at 37 °C; 2.5 μM RNA substrate, 225 μM m6preQ1, 2.0 mM MgCl2, pH 6.0. HPLC traces of unmodified RNA, the reacted mixture, and the corresponding m3C-modified synthetic RNA reference is shown for comparison (for reaction time course see Supplementary Fig. 1b). The m3C-modified RNA elutes earlier. Incubation of m3C preQ1-I RNA (isolated from a typical methylation reaction with m6preQ1) under harsh basic conditions produced m3U from m3C under concomitant RNA hydrolysis (for FT-ICR MS characterization of m3U-RNA see Supplementary Fig. 1e); chemical structures of m3C-to-m3U hydrolysis. b preQ1-I RNA reactivity analysis using diverse buffer conditions, nucleobase mutagenesis, atomic mutagenesis, and near-cognate cofactors; bars (gray) show mean ± s.e.m. (n = 3 independent experiments); HPLC traces are depicted in Supplementary Fig. 2, 3; source data are provided in the Source Data file. c The reaction rate is dependent on m6preQ1 concentration. The observed rate constants kobs were determined based on HPLC trace analysis at five concentrations of m6preQ1, ranging from 25 to 300 μM. The red line represents a curve fit to kobs = kmax[m6preQ1]/(Km,app + [m6preQ1]). Individual data points (open circles) (n = 3 independent experiments), mean ± s.e.m. (black circles); source data are provided in the Source Data file. d Chemical structures for c3C and dG used in atomic mutagenesis reactivity assay. e Proposed reaction mechanism of preQ1-I RNA–catalyzed methylation using m6preQ1 as cofactor; key features are the syn-conformation of the methyl iminoester moiety and N1 protonation of m6preQ1; furthermore, coordination of the 2’-OH G11 to O6 of m6preQ1 assists in cofactor alignment and contributes to improve leaving group quality. f UV-spectroscopic determination of the pKa value of m6preQ1 (n = 3 independent experiments); source data are provided in the Source Data file. g Representative isotherm and ‘One set of sites’ bindingmodel fit for the preQ1 class I riboswitch titrated with preQ1 (left) and m6preQ1 (right); the indicated Kd values were determined at pH 6 (n = 3 independent experiments); source data are provided in the Source Data file; one experiment for preQ1 is depicted (left): N = 0.97, KA = 2.37 × 107 M−1, ΔH = −27.57 × 103 cal mol1, and ΔS = −58.7 cal (mol K)1; one experiment for m6preQ1 is depicted (right): N = 0.99, KA = 1.36 × 104 M−1, ΔH = −12.85 × 103 cal mol1, and ΔS = −24.2 cal (mol K)1. h Chemical structures of other potential cofactors tested.
To unequivocally confirm the site of methylation, we synthesized m3C phosphoramidite for RNA solid-phase synthesis and prepared the corresponding m3C15 containing RNA reference (Fig. 4a). This RNA had the same retention time as the product obtained by methylation using m6preQ1. We further point out that the synthetic m4C15 and m5C15 RNA references had retention times that were undistinguishable from the unmodified RNA in our HPLC assay (Supplementary Fig. 1c). Therefore, we separated the m3C RNA product from a methylation reaction mixture and analyzed the remaining RNA by FT-ICR mass spectrometry, but no further methylation products were detected (Supplementary Fig. 1d). Furthermore, we exposed the isolated m3C15 RNA to harsh basic conditions (pH 10) and observed the expected m3C-to-m3U conversion product (beside emerging fragments from RNA degradation) which additionally confirms that m3C15 RNA was formed selectively (Fig. 4a and Supplementary Fig. 1e).
RNA sequence requirements and reaction mechanism
Next, we set out to investigate the Tt preQ1 class I riboswitch sequence requirements for methyl group transfer. We observed no methylation products when C15 was mutated either to 3-deazacytidine (c3C), U, A, or G (Fig. 4b,d and Supplementary Fig. 2). In contrast, when C15 was mutated to 5-methylcytidine (C15m5C), methylation occurred almost as efficiently as for wildtype RNA (Fig. 4b and Supplementary Fig. 2). Furthermore, when the bulged-out nucleotide U12 close to the binding site, was deleted (mutant ΔU12), no methylation occurred (Fig. 4b). U12, therefore, seems essential for retaining the structural dynamics within the U12-A13-A14-C15 segment. Moreover, when U6 and A29 which are crucial for recognition of the N9–N3 face of preQ1 and m6preQ1 were mutated individually to C and G, methylation was not observed anymore (Fig. 4b and Supplementary Fig. 2). Importantly, compensatory mutation of the conserved base pair G5-C16 into C5-G16 gave a sevenfold decrease in yields for the m3C-modified RNA (Fig. 4b), consistent with the crucial interaction of the O6-G5 atom and the protonated 7-aminomethyl group of preQ1 or m6preQ1. Strikingly, we found evidence for a potential role of the 2′-OH group of G11 in the methyl transfer reaction. When we deleted this functional group (dG11 mutant), reaction yields decreased to one-fourth compared to the wild-type RNA (Fig. 4b,d). This finding together with the above-described pH dependence of the reaction leads to the proposed reaction mechanism depicted in Fig. 4e. The favorable, slightly acidic pH value of 5–6 is consistent with saturating protonation of the 7-aminomethyl group and its recognition by G5. Importantly, it is also consistent with protonation of N1 of m6preQ1, thereby facilitating stabilization of the preQ1 leaving group (Fig. 4f). Our observation that methylation activity becomes impaired at pH values lower than 5 suggests protonation of N3 of C15, and hence the loss of nucleophilicity needed for an attack at m6preQ1. Furthermore, the model is compatible with stabilization of the syn conformation of the methyl iminoester by a hydrogen bond between m6preQ1 O6 and the 2′-OH of G11.
Finally, we set out to estimate the affinity of m6preQ1 to the riboswitch. Using isothermal titration calorimetry (ITC), we first determined the dissociation constant of the cognate preQ1 ligand to wildtype preQ1 RNA at pH 6 as reference (Fig. 4g). The KdITC value amounted to 43.0 ± 1.9 nM. We then determined the affinity of m6preQ1 to the unreactive C15U RNA mutant and obtained a KdITC value of 72.2 ± 2.1 µM at pH 6.0. This micromolar Kd likely reflects the portion of pairing strength that arises from the identical recognition of the C7-CH2NH2 moieties and the N9–N3–C2-NH2 faces of preQ1 and m6preQ1, respectively, by the pocket through A29 and U6. We note that A29G and U6C mutants were both inactive (Supplementary Fig. 2). Further, we believe that the actual affinity of m6preQ1 to wild-type (C15) RNA is higher than to the unreactive C15U mutant because the imino tautomer imC15 as proposed in Fig. 2f could in principle provide a better match with respect to hydrogen donor-acceptor interactions compared to U15.
Near-cognate cofactors and other preQ1 riboswitch scaffolds
Encouraged by our finding that a simple, methylated nucleobase can act as cofactor for riboswitch methylation, we set out to synthesize and test structural congeners of m6preQ1, namely O6-ethyl-preQ1 (e6preQ1), O6-benzyl-preQ1 (bn6preQ1), and O6-methyl-preQ0 (m6preQo) (Fig. 4h and Supplementary Fig. 3). These derivatives, however, exhibited only very minor—or no—alkylation activities under optimized conditions (Fig. 4b and Supplementary Fig. 1f,g and Supplementary Fig. 3). Of further note, O6-methylguanine (m6G) and O6-(4-aminomethyl)benzylguanine (abn6G) did not result in detectable amounts of m3C15 RNA product (Fig. 4b). The observation that the potential methyl group donors m6G and m6preQ0 did not transfer their methyl groups can be rationalized by considering the significantly lower affinities of their non-methylated precursors, preQ0 (17-fold lower)22 and G (at least 25-fold lower)29 compared to the cognate ligand preQ1. Therefore, it is reasonable to assume that m6preQ1 is also significantly stronger bound compared to m6G and m6preQ0. It is the 7-aminomethyl substituent of preQ1 and m6preQ1 that has to be present together with its interaction partner, the G5-C16 base pair to reach high affinities22 and high methylation yields (see G5C-C16G mutant, Fig. 4b). We further note that e6preQ1 and bn6preQ1 which provide the 7-aminomethyl substituent exhibit lower yields likely because of steric hindrance of their larger O6 substituents with the binding pocket.
Next, we were wondering if representatives of the other known preQ1 riboswitch classes also possess self-methylation properties29. Both, preQ1 class II and III riboswitches have an identical architecture of their preQ1-bound pockets, however, this pocket architecture is different from class I30,31. Characteristically, class II and III apply a preQ1–C trans base pair instead of a cis Watson–Crick pair (class I), and significantly, the O6 atom of preQ1 is rather solvent-exposed with no obvious RNA functionalities for nucleophilic attack nearby. Nevertheless, we tested the well-studied preQ1 II riboswitch from Lactobacillus rhamnosus for methyl transfer of m6preQ1; as expected, methylation was not observed (Fig. 4b).
Noteworthily, class I preQ1 riboswitches are categorized into three subtypes, with the Tt riboswitch belonging to type 129. We, therefore, complemented our analysis of RNA sequence requirements with a type 2 (Bacillus subtilis) and a type 3 preQ1 riboswitch (Shigella dysenteriae). Both revealed no methyltransferase activity (Fig. 4b).
Discussion
Our study answers the long-standing question of whether contemporary riboswitches can act as cofactor-dependent ribozymes by a clear yes. Direct evidence became possible after careful analysis of three-dimensional riboswitch architectures in combination with rational ligand design and chemical intuition that finally led to the discovery of methyltransferase activity of the Tt preQ1 class I riboswitch. PreQ1 itself is a guanine-derived precursor of the nucleobase queuosine (Q) that is found in the wobble position of tRNAs containing the GUN anticodon32. The presence of Q in tRNAs improves their ability to read degenerate codons, while the absence of enzymes participating in Q biosynthesis or salvage results in deleterious phenotypes in many organisms33,34. Noteworthily, O6-methylated versions of the pre-queuosine heterocycle have been found in nature, in form of 2-amino-5-cyano-4-methoxypyrrolo[2,3-d]pyrimidine produced by Streptomyces35, and as the corresponding N2 -glyosylated natural products huimycin and dapiramicin36. Their occurrence hints at potential cellular availability of the near-cognate ligand m6preQ1 for preQ1 riboswitches. In this context it is also interesting that preQ1 and SAM aptamers have been found in a putative tandem riboswitch architecture which might hint at the regulatory utility of linking m6preQ1 biosynthesis to the concentrations of SAM and preQ129,35,36. At this time, one can only speculate about the biological relevance of a riboswitch with potential self-methylation activity. Such a scenario implies that m3C methylation in the riboswitch pocket impairs recognition of the cognate ligand preQ1 and thereby impacts the gene regulatory function of the riboswitch. Thus far, the m3C modification itself is known for tRNAs in position 32 in diverse organisms37,38, ranging from yeast39, S. Pombe40, parasites such as Trypanosoma brucei41, to mice, rat, and human42. The modification has attracted additional interest when m3C has been identified as dynamic modification also in mRNA, and corresponding writer (METTL2, METTL6, METTL8) and eraser enzymes (ALKBH1, ALKBH3) have been characterized43. The emerging importance of the modification stimulated the development of novel RNA sequencing approaches, AlkAniline-Seq44 and HAC-seq45, respectively, to reveal m3C sites transcriptome-wide.
We furthermore anticipate that the discovered methyltransferase activity of the preQ1 class I riboswitch can serve as starting point for engineering in vitro and in vivo methylation tools. Applications for m3C installation are conceivable not only in cis, but also in trans. First experimental attempts towards this aim by splitting the aptamer into two halves have been successful and led to remarkable 38% of methylation for the preQ1 stem-loop by simply adding the 3′-terminal preQ1 RNA fragment in the presence of cofactor (Fig. 5). Beyond methyltransferase activity and the particular m3C modification investigated here, our work may stimulate the search for other ribozyme-catalyzed reactions that are lying dormant in present-day riboswitches. Given the large number and great diversity of riboswitches that have been identified to date, a broad repertoire of chemical reactivities should be directly retrievable without the need for nucleic acids evolution that builds on constructed RNA libraries of naturally occurring riboswitch scaffolds46 or on de novo in vitro RNA selection approaches47.

a Sequences of the two Tt preQ1-I fragments used and anion-exchange HPLC traces of the reaction mixture at time points 0 and 48 h (25 μM each RNA strand, 1.5 mM m6preQ1, 2.0 mM MgCl2, 100 mM KCl, MES buffer pH 6.0, 37 °C); as expected, the m3C-modified RNA product elutes earlier. b Secondary structure of the m3C-modified preQ1-stem-loop RNA and CAD-MS of co-isolated (M − 5H)5- ions of unreacted 21 nt RNA (61.1%, mexp. 6683.926 Da, mcalc. 6683.929 Da) and methylated RNA (38.9%, mexp. 6697.942, mcalc. 6697.945 Da) produced at 65 eV laboratory frame energy c and y fragments with and without methylation. c Thereby, the fraction of methylated c fragments (cm) increased from 0% to 36.8 ± 2.6% at site 15 (mean of values for sites 15 to 20 ± standard deviation). d The fraction of methylated y fragments (ym) decreased from 38.4 ± 1.4% to 0% at site 15 (mean of values for sites 1 to 14 ± standard deviation), which both are consistent with C15 as methylation site and with the yields independently obtained by HPLC analysis. Source data are provided in the Source Data file.
Finally, we stress the importance of our findings for the RNA world hypothesis which postulates that possible early life on Earth relied on catalytic RNA properties essential for evolving sophisticated RNA-based life5,6,7,8,9,10,48. The idea is strongly supported by the original discovery of bacterial riboswitches that regulate—without the aid of proteins—the expression of genes that are involved in the biosynthesis of metabolites6. Some of these riboswitches may have evolved from ancient cofactor-dependent ribozymes preserving their regulatory capacity while the reactivity part has been taken over by more efficient protein enzyme machineries. Our findings may also hint at a prebiotic version of RNA epigenetics with methylated nucleobases taking the role as cofactors for ancient methyltransferase ribozymes49,50,51,52.
Methods
RNA synthesis
RNA oligonucleotides were prepared by solid-phase synthesis using phosphoramidite chemistry (2′-O-TOM-protected) on controlled-pore glass solid supports53. RNA sequences are given in Supplementary Table 1. Modified phosphoramidites for atomic mutagenesis and reference oligonucleotides were purchased (ChemGenes, Glen Research, Berry Asscociates) or prepared in-house, following published procedures54,55. RNA oligonucleotides were deprotected with ammonia and butylamine instead of ammonia and methylamine to avoid any possible methylated RNA from deprotection, followed by 1 M tetrabutylammonium fluoride in THF, desalted (Sephadex G25), and purified by denaturing anion exchange chromatography (Dionex DNAPac PA100, 9 × 250 mm, at 80 °C; solvent A was 25 mM Tris-HCl (pH 8.0) and 20 mM NaClO4 in 20% aqueous acetonitrile; solvent B was 25 mM Tris-HCl (pH 8.0) and 0.6 M NaClO4 in 20% aqueous acetonitrile; the gradient was: linear, 25–40% (25–45 for longer sequences) with slope of 5 % solvent B per column volume). Customized deprotection conditions were used for m3C RNA (3:1 (v/v) 28–30% aqueous NH3 and EtOH, at 40 °C for 5 h), m4C RNA (O4-chlorophenyl U convertible nucleoside approach, 7 M NH3 in MeOH at 42 °C for 18 h). The quality of RNAs (purity and identity) was analyzed by anion-exchange HPLC (Dionex DNAPac PA100, 2 × 250 mm, eluents as above, the gradient was: linear, 22–35% solvent B, with slope of 0.87% solvent B per column volume), and HR–ESI–MS (Thermo Fisher Orbitrap, negative-mode) or FT ICR MS (see below). Measured and calculated masses are listed in Supplementary Table 1.
Characterization of m6preQ1, e6preQ1, and bn6preQ1
Analytical data (NMR spectra) of m6preQ1, e6preQ1, and bn6preQ1 are shown in Supplementary Figs. 4–11. m6G and abn6G were purchased from commercial sources (Carbosynth).
Reaction of preQ1 RNA with m6preQ1 or analogs
A typical methylation reaction was carried out in a volume of 200 μl containing 0.5 nmol RNA (2.5 µM) and 75 μM ligand (30 equiv) in buffer solution (2 mM MgCl2, 100 mM KCl, 50 mM 2-(N-morpholino) ethanesulfonic acid (MES), pH 6.0). After heating the mixture to 90 °C for 2 min, it was incubated for 48 h at 37 °C. After desalting using a Sep-Pak C18 cartridge, the methylated RNA product was directly analyzed in the mixture or isolated by AE HPLC, desalted again (Sep-Pak C18), and subjected to FT ICR-ESI-MS (which included a further desalting step using centrifugal concentrators; see below). For experiments at pH 4 and 5, sodium acetate buffer was used instead of MES; for pH 7, 7.5, and 8, MOPS was used. Several of the experiments were additionally analyzed using 60 and 120 equiv of the cofactor.
The hydrolysis of m3C to m3U RNA was examined in a volume of 280 μl with 7 nmol RNA (25 µM) in 25 mM Na2CO3 buffer (pH 10) with 1 mM EDTA at 65 °C for 1.5 h. After desalting (Sep-Pak C18) the m3U RNA was isolated by AE HPLC and analyzed by FT ICR–ESI–MS (see below).
FT-ICR mass spectrometric analysis of RNA methylation/alkylation products
Methanol was HPLC grade (Acros), ammonium acetate (≥99.0%, Na ≤5 mg/kg, K ≤ 5 mg/kg), piperidine (≥99.5%), and imidazole (≥99.5%, Na ≤ 50 mg/kg, K ≤ 50 mg/kg) were from Sigma-Aldrich, and H2O was purified to 18 MΩ·cm at room temperature using a Milli-Q system (Millipore). Experiments were performed on a 7T Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (Bruker APEX ultra) equipped with an ESI source for (M − nH)n− ion generation and a collision cell through which a flow of Ar gas was maintained for CAD. The mass resolving power of this instrument is routinely (broadband detection, 2 M data points for a ∼2 s transient) ∼220,000, ∼120,000, and ∼80 000 at m/z 500, 1000, and 1500, respectively, and the mass accuracy is ∼1 ppm with internal calibration and ∼20 ppm with external calibration (Supplementary Table 1). RNA was electrosprayed (flow rate 1.5 µl/min) from 1–2 µM solutions in 1:1 or 9:1 H2O/CH3OH vol/vol with piperidine (2–10 mM) and imidazole (0–10 mM) as additives. Prior to dissociation by CAD, the (M − nH)n− ions under study were isolated in a linear quadrupole; for a more detailed description of the experimental setup for CAD see56. For statistical reasons, between 25 and 500 scans were added for each spectrum (20–50 for ESI, 100–500 for CAD), and data reduction utilized the SNAP2 algorithm (Bruker). For desalting, 400 µl of an ammonium salt solution (100 mM ammonium acetate in H2O) was added to 100 µl RNA solution (about 1 nmol in H2O) and concentrated to 100 µl using Vivaspin 500 centrifugal concentrators (Sartorius, MWCO 3000). The process was repeated five to seven times, followed by six to seven cycles of concentration and dilution with H2O. RNA concentration was determined by UV absorption at 260 nm using a NanoPhotometer (Implen).
Kinetic assays of RNA-catalyzed methylation reactions
The methylation reactions were carried out as described above (on a 1.5 nmol scale; 600 µl reaction volume) and 50-μl aliquots were taken at desired time points and quenched immediately by adding 50 μl of stop solution (12 M urea). The samples were analyzed by AE HPLC and UV-detection. The peak areas were quantified. The yield versus time data were fit to (fraction unreacted) = Y(1 − e–kt), in which k = kobs and Y = final yield, using OriginPro (2020). All kinetic assays were carried out as three independent replicates.
Isothermal titration calorimetry (ITC)
ITC measurements were performed on a MicroCal iTC200 instrument at 20 °C in 50 mM MES, 100 mM KCl and 2 mM MgCl2 (pH 6.0). For preQ1, the concentration of Tte preQ1 class I RNA in the measuring cell was typically 0.012 mM; preQ1 ligand was in the syringe at a concentration that was 10-fold higher than the RNA. The c-value was typically around 300. For m6preQ1, the concentration of the Tte preQ1 class I RNA in the measuring cell was typically 0.19 mM; m6preQ1 cofactor was in the syringe at a concentration that was 15-fold higher than the RNA. The c-value was typically around 3. Both Kd determinations were carried out as three independent replicates.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
Custom software that would need code availability was not used for this study. Software used for data collection: Bruker Daltronics Compass Apex Control 3.0.0, Bruker Topspin 3.6, UNICORN 5.0, Chromeleon 6.8. Software used for data analysis: Microsoft Excel 16.16.22, OriginPro 2020.
References
Breaker, R. R. Imaginary ribozymes. ACS Chem. Biol. 15, 2020–2030 (2020).
Cochrane, J. C. & Strobel, S. A. Riboswitch effectors as protein enzyme cofactors. RNA 14, 993–1002 (2008).
Micura, R. & Höbartner, C. Fundamental studies of functional nucleic acids: aptamers, riboswitches, ribozymes and DNAzymes. Chem. Soc. Rev. 49, 7331–7353 (2020).
Joyce, G. F. & Szostak, J. W. Protocells and RNA self-replication. Cold Spring Harb. Perspect. Biol. 10, a034801 (2018).
Nelson, J. W. & Breaker, R. R. The lost language of the RNA World. Sci. Signal. 10, eaam8812 (2017).
Jadhav, V. R. & Yarus, M. Coenzymes as coribozymes. Biochimie 84, 877–888 (2002).
Kirschning, A. Coenzymes and their role in the evolution of life. Angew. Chem. Int. Ed. 60, 6242–6269 (2021).
Garst, A. D., Edwards, A. L. & Batey, R. T. Riboswitches: structures and mechanisms. Cold Spring Harb. Perspect. Biol. 3, a003533 (2011).
Motorin, Y. & Helm, M. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA 2, 611–631 (2011).
Zhao, B. S., Roundtree, I. A. & He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42 (2017).
Wiener, D. & Schwartz, S. The epitranscriptome beyond m6A. Nat. Rev. Genet. 22, 119–131 (2021).
Andexer, J. N. & Rentmeister, A. Challenging nature’s preference for methylation. Nat. Chem. 12, 791–792 (2020).
Serganov, A. & Patel, D. J. Molecular recognition and function of riboswitches. Curr. Opin. Struct. Biol. 22, 279–286 (2012).
Schwalbe, H., Buck, J., Fürtig, B., Noeske, J. & Wöhnert, J. Structures of RNA switches: insight into molecular recognition and tertiary structure. Angew. Chem. Int. Ed. 46, 1212–1219 (2007).
Ferré-D’Amaré, A. R. The glmS ribozyme: use of a small molecule coenzyme by a gene-regulatory RNA. Q. Rev. Biophys. 43, 423–447 (2010).
Scheitl, C. P. M., Ghaem Maghami, M., Lenz, A. K. & Höbartner, C. Site-specific RNA methylation by a methyltransferase ribozyme. Nature 587, 663–667 (2020).
Bédard, A. V., Hien, E. D. M. & Lafontaine, D. A. Riboswitch regulation mechanisms: RNA, metabolites and regulatory proteins. Biochim. Biophys. Acta Gene Regul. Mech. 1863, 194501 (2020).
McCown, P. J., Corbino, K. A., Stav, S., Sherlock, M. E. & Breaker, R. R. Riboswitch diversity and distribution. RNA 23, 995–1011 (2017).
Price, I. R., Grigg, J. C. & Ke, A. Common themes and differences in SAM recognition among SAM riboswitches. Biochim. Biophys. Acta 1839, 931–938 (2014).
Sun, A. et al. SAM-VI riboswitch structure and signature for ligand discrimination. Nat. Commun. 10, 5728 (2019).
Gilbert, S. D., Reyes, F. E., Edwards, A. L. & Batey, R. T. Adaptive ligand binding by the purine riboswitch in the recognition of guanine and adenine analogs. Structure 17, 857–868 (2009).
Jenkins, J. L., Krucinska, J., McCarty, R. M., Bandarian, V. & Wedekind, J. E. Comparison of a preQ1 riboswitch aptamer in metabolite-bound and free states with implications for gene regulation. J. Biol. Chem. 286, 24626–24637 (2011).
Klein, D. J., Edwards, T. E. & Ferré-D’Amaré, A. R. Cocrystal structure of a class I preQ1 riboswitch reveals a pseudoknot recognizing an essential hypermodified nucleobase. Nat. Struct. Mol. Biol. 16, 343–344 (2009).
Schroeder, G. M. et al. Analysis of a preQ1-I riboswitch in effector-free and bound states reveals a metabolite-programmed nucleobase-stacking spine that controls gene regulation. Nucleic Acids Res. 48, 8146–8164 (2020).
Han, X., Jin, M., Breuker, K. & McLafferty, F. W. Extending top-down mass spectrometry to proteins with masses greater than 200 kilodaltons. Science 314, 109–112 (2006).
Liko, I., Allison, T. M., Hopper, J. T. & Robinson, C. V. Mass spectrometry guided structural biology. Curr. Opin. Struct. Biol. 40, 136–144 (2016).
Glasner, H., Riml, C., Micura, R. & Breuker, K. Label-free, direct localization and relative quantitation of the RNA nucleobase methylations m6A, m5C, m3U, and m5U by top-down mass spectrometry. Nucleic Acids Res. 45, 8014–8025 (2017).
Frener, M. & Micura, R. Conformational rearrangements of individual nucleotides during RNA-ligand binding are rate-differentiated. J. Am. Chem. Soc. 138, 3627–3630 (2016).
McCown, P. J., Liang, J. J., Weinberg, Z. & Breaker, R. R. Structural, functional, and taxonomic diversity of three preQ1 riboswitch classes. Chem. Biol. 21, 880–889 (2014).
Liberman, J. A., Salim, M., Krucinska, J. & Wedekind, J. E. Structure of a class II preQ1 riboswitch reveals ligand recognition by a new fold. Nat. Chem. Biol. 9, 353–355 (2013).
Liberman, J. A. et al. Structural analysis of a class III preQ1 riboswitch reveals an aptamer distant from a ribosome-binding site regulated by fast dynamics. Proc. Natl Acad. Sci. USA 112, E3485–E3494 (2015).
Iwata-Reuyl, D. An embarrassment of riches: the enzymology of RNA modification. Curr. Opin. Chem. Biol. 12, 126–133 (2008).
Durand, J. M. B., Dagberg, B., Uhlin, B. E. & Björk, G. R. Transfer RNA modification, temperature and DNA superhelicity have a common target in the regulatory network of the virulence of Shigella flexneri: the expression of the virF gene. Mol. Microbiol. 35, 924–935 (2000).
Rakovich, T. et al. Queuosine deficiency in eukaryotes compromises tyrosine production through increased tetrahydrobiopterin oxidation. J. Biol. Chem. 286, 19354–19363 (2011).
Iijima, M. et al. A guanine derivative as a new MEK inhibitor produced by Streptomyces sp. MK63-43F2. J. Antibiotics 71, 135–138 (2018).
Shuai, H., Myronovskyi, M., Nadmid, S. & Luzhetskyy, A. Identification of a biosynthetic gene cluster responsible for the production of a new pyrrolopyrimidine natural product-huimycin. Biomolecules 10, 1074 (2020).
Ignatova, V. V. et al. METTL6 is a tRNA m3C methyltransferase that regulates pluripotency and tumor cell growth. Sci. Adv. 6, eaaz4551 (2020).
Lentini, J. M., Alsaif, H. S., Faqeih, E., Alkuraya, F. S. & Fu, D. DALRD3 encodes a protein mutated in epileptic encephalopathy that targets arginine tRNAs for 3-methylcytosine modification. Nat. Commun. 11, 2510 (2020).
Han, L., Marcus, E., D’Silva, S. & Phizicky, E. M. S. cerevisiae Trm140 has two recognition modes for 3-methylcytidine modification of the anticodon loop of tRNA substrates. RNA 23, 406–419 (2017).
Arimbasseri, A. G. et al. Evolving specificity of tRNA 3-methyl-cytidine-32 (m3C32) modification: a subset of tRNAsSer requires N6-isopentenylation of A37. RNA 22, 1400–1410 (2016).
Rubio, M. A. T. et al. Editing and methylation at a single site by functionally interdependent activities. Nature 542, 494–497 (2017).
Xu, L. et al. Three distinct 3-methylcytidine (m3C) methyltransferases modify tRNA and mRNA in mice and humans. J. Biol. Chem. 292, 14695–14703 (2017).
Ma, C. J., Ding, J. H., Ye, T. T., Yuan, B. F. & Feng, Y. Q. AlkB homologue 1 demethylates N3-methylcytidine in mRNA of Mammals. ACS Chem. Biol. 14, 1418–1425 (2019).
Marchand, V. et al. AlkAniline-Seq: profiling of m7G and m3C RNA modifications at single nucleotide resolution. Angew. Chem. Int. Ed. 57, 16785–16790 (2018).
Cui, J., Liu, Q., Sendinc, E., Shi, Y. & Gregory, R. I. Nucleotide resolution profiling of m3C RNA modification by HAC-seq. Nucleic Acids Res. 49, e27 (2021).
Ishida, S., Terasaka, N., Katoh, T. & Suga, H. An aminoacylation ribozyme evolved from a natural tRNA-sensing T-box riboswitch. Nat. Chem. Biol. 16, 702–709 (2020).
Burke, D. H. & Gold, L. RNA aptamers to the adenosine moiety of S-adenosyl methionine: structural inferences from variations on a theme and the reproducibility of SELEX. Nucleic Acids Res. 25, 2020–2024 (1997).
Xu, J. et al. Selective prebiotic formation of RNA pyrimidine and DNA purine nucleosides. Nature 582, 60–66 (2020).
Höfer, K. & Jäschke, A. Epitranscriptomics: RNA modifications in bacteria and archaea. Microbiol. Spectr. 6, 3 (2018).
Waddell, T. G., Eilders, L. L., Patel, B. P. & Sims, M. Prebiotic methylation and the evolution of methyl transfer reactions in living cells. Orig. Life Evol. Biosph. 30, 539–548 (2000).
Traube, F. R. & Carell, T. The chemistries and consequences of DNA and RNA methylation and demethylation. RNA Biol. 14, 1099–1107 (2017).
Schneider, C. et al. Noncanonical RNA nucleosides as molecular fossils of an early Earth-generation by prebiotic methylations and carbamoylations. Angew. Chem. Int. Ed. 57, 5943–5946 (2018).
Pitsch, S., Weiss, P. A., Jenny, L., Stutz, A. & Wu, X. Reliable chemical synthesis of oligoribonucleotides (RNA) with 2′-O-[(triisopropylsilyl)oxy]methyl(2′-O-tom)-protected phosphoramidites. Helv. Chim. Acta 84, 3773–3795 (2001).
Höbartner, C. et al. The synthesis of 2′-O-[(triisopropylsilyl)oxy] methyl (TOM) phosphoramidites of methylated ribonucleosides (m1G, m2G, m22G, m1I, m3U, m4C, m6A, m62A) for use in automated RNA solid-phase synthesis. Monatshefte Chem. 134, 851–873 (2003).
Mao, S. et al. Base pairing and functional insights into N3-methylcytidine (m3C) in RNA. ACS Chem. Biol. 16, 76–85 (2020).
Taucher, M. & Breuker, K. Top-down mass spectrometry for sequencing of larger (up to 61 nt) RNA by CAD and EDD. J. Am. Soc. Mass Spectrom. 21, 918–929 (2010).
Acknowledgements
We thank Max Himmelstoß (Innsbruck) for mass spectrometric measurements, Christoph Kreutz (Innsbruck) for NMR spectroscopic support, and Daniel Fellner (Innsbruck) for technical support. We thank the following institutions for funding: Austrian Science Fund FWF [P31691, F8011-B to R.M., P30087 to K.B.]; Austrian Research Promotion Agency FFG [West Austrian BioNMR 858017]; Vienna Science and Technology Fund WWTF (LS17-003).
Author information
Authors and Affiliations
Contributions
L.F., K.B., and R.M. conceived the project and designed experiments. L.F. performed all experiments, except FT-ICR-MS experiments that were performed by S.H. and K.B. S.M. contributed to cofactor and nucleoside phosphoramidite synthesis. L.F. and R.M. analyzed and interpreted the data, with help from S.H., S.M., and K.B. R.M. supervised the project. R.M., K.B., and L.F. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Juan Alfonzo, Hashim Al-Hashimi and other, anonymous, reviewers for their contribtuions to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Flemmich, L., Heel, S., Moreno, S. et al. A natural riboswitch scaffold with self-methylation activity. Nat Commun 12, 3877 (2021). https://doi.org/10.1038/s41467-021-24193-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-24193-7
This article is cited by
-
A SAM analogue-utilizing ribozyme for site-specific RNA alkylation in living cells
Nature Chemistry (2023)
-
Ribozyme for stabilized SAM analogue modifies RNA in cells
Nature Chemistry (2023)
-
Structure and mechanism of the methyltransferase ribozyme MTR1
Nature Chemical Biology (2022)
-
A small RNA that cooperatively senses two stacked metabolites in one pocket for gene control
Nature Communications (2022)
-
Structure and mechanism of a methyltransferase ribozyme
Nature Chemical Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
