
Abstract
Single-atom catalysts (SACs) have emerged as a frontier in heterogeneous catalysis due to the well-defined active site structure and the maximized metal atom utilization. Nevertheless, the robustness of SACs remains a critical concern for practical applications. Herein, we report a highly active, selective and robust Ru SAC which was synthesized by pyrolysis of ruthenium acetylacetonate and N/C precursors at 900 °C in N2 followed by treatment at 800 °C in NH3. The resultant Ru1-N3 structure exhibits moderate capability for hydrogen activation even in excess NH3, which enables the effective modulation between transimination and hydrogenation activity in the reductive amination of aldehydes/ketones towards primary amines. As a consequence, it shows superior amine productivity, unrivalled resistance against CO and sulfur, and unexpectedly high stability under harsh hydrotreating conditions compared to most SACs and nanocatalysts. This SAC strategy will open an avenue towards the rational design of highly selective and robust catalysts for other demanding transformations.
Similar content being viewed by others

Introduction
Single-atom catalysts (SACs) have emerged as a class of heterogeneous catalysts for they offer the potential to maximize the metal utilization efficiency and the opportunity to probe into the reaction mechanism at the atomic scale in a manner of resembling molecular catalysts1,2,3,4,5. In most SAC systems, the metal single atoms are bonded to the support via strong electronic interaction, which renders them to be positively charged and thereby weakens their capability for activating hydrogen molecule6,7. Such a weak to moderate ability to activate hydrogen is just what is required for a certain of chemoselective hydrogenation reactions8, and this has been sufficiently demonstrated in the chemoselective hydrogenation of substituted nitroarenes since the birth of SACs9,10,11,12,13. Nevertheless, SACs have not been explored for more challenging reactions which involve both hydrogen and other strongly adsorbed molecules.
The reductive amination of aldehydes/ketones to produce primary amines is such a reaction that involves the activation of both hydrogen and NH3 molecules14. In the past few years, this reaction has attracted intensive attention attributed to the broad applications of primary amines as key building blocks for pharmaceuticals, agrochemicals, polyamides, and other fine chemicals15,16. In particular, the aldehydes/ketones are readily available from lignocellulosic biomass, which provides the opportunity for the sustainable production of primary amines from renewables and thus contributes to reducing the carbon footprint17,18. Nevertheless, the selectivity maneuvering toward primary amines remains a great challenge owing to the occurrence of many side reactions. Taking the reductive amination of furfural (FAL) to furfural amine (FAM) as an example19, it involves a complex reaction network as shown in Fig. 1. While the target product FAM (2a) can be facilely produced through the condensation of FAL (1a) with NH3 followed by the subsequent hydrogenation of primary imine (7a), there are at least three side reactions to compete with the main reaction due to the high reactivity of aldehyde (1a) as well as the high instability of the intermediate imine (7a): the direct hydrogenation of FAL to furfural alcohol (6a), the trimerization of 7a and the subsequent cyclization to form 3a, and the condensation of 2a with 1a or 7a to form a stable intermediate, Schiff base (secondary imine, 4a), which can be further hydrogenated to secondary amine (5a). This network involving many possible reaction pathways imposes a great challenge for the selective synthesis of primary amines. Moreover, different from simple hydrogenation, reductive amination requires the catalyst to be able to survive in the presence of excess ammonia and amine, which are usually strongly adsorbed on metal catalysts and become a possible poison for hydrogenation reactions20.

The transformation of furfural (1a) to the target product furfural amine (2a) is accompanied by a number of side reactions (3a– 8a).
Based on the above understanding of the reaction mechanism, we propose that a highly selective catalyst for reductive amination should possess weak to moderate adsorption to hydrogen in the presence of NH3. If a catalyst has strong hydrogenation activity such as Pd and Pt, the secondary amine and/or alcohol would be favorably obtained21. On the other hand, if a catalyst cannot activate hydrogen, imine trimerization would occur to a great extent22. Therefore, to rationally design catalysts for the reductive amination of aldehydes/ketones towards the primary amines, one needs to be able to effectively tune the hydrogenation activity and to establish a structure–performance relationship at an atomic/molecular scale. Unfortunately, for conventional nanocatalysts, the fine-tuning of their hydrogenation capability is hindered by the heterogeneity in shape, size, and composition of the metal nanoparticles23,24. Taking Ru nanocatalysts as an example, the oxidation state, size, and shape of Ru nanoparticles were all reported to govern the catalytic performances in reductive amination reaction19,25,26,27. For instance, we previously found that a small proportion of positively charged RuOx in Ru/ZrO2 catalyst played a critical role in the activation of carbonyl group25. Hara’s group stated the weak electron donation of Ru in Ru/Nb2O5 catalyst suppressed the over-hydrogenation of furan ring in the reductive amination of furfuraldehyde19. Khodakov et al. proposed the dehydrogenation of primary amines was structure-sensitive such that the selectivity of secondary amines increased with the size of Ru nanoparticle26. Chandra et al. found the flat-shaped fcc-Ru nanoparticles exposing mainly {111} facets exhibited much higher catalytic activity27. While these catalysts exhibited moderate to high selectivity toward the primary amines, no unambiguous structure–performance relationship has been established, which hinders the rational design of efficient and selective catalysts. In addition to Ru nanocatalysts, it was recently reported that nanoparticles (NPs) of Ni28,29 or Co30, when they were encapsulated with an N-doped carbon layer via pyrolysis of organic ligands, exhibited greatly enhanced selectivity to the primary amines. Again, due to the heterogeneity in metal size and the complex structure of the N-doped carbon layer, the underlying mechanism is not clear yet.
Thanks to the uniform and tunable structure at the atomic scale, SACs provide a unique platform to probe into the structure–performance relationship31,32,33,34. We and other research groups have reported the fine modulation of the hydrogenation activities of Pt1/FeOx33, Co–N–C35, and Ni–N–C36 SACs through engineering the coordination environment of SACs. Herein, we apply SACs to a more demanding reaction, the reductive amination of biomass-derived aldehydes/ketones for the selective synthesis of primary amines. Ru1/NC SACs with finely tuned active sites including RuN5, RuN4, and RuN3 are successfully fabricated by changing the synthesis temperatures. It is found that the oxidation state of Ru single atoms is +2, yet the electron density increases with a decrease in the Ru–N coordination number, which results in a gradual enhancement in the hydrogen activation even in the case of strong pre-adsorption of NH3. As a consequence, the selectivity to primary amines is greatly enhanced up to 99%. More interesting, compared with conventional Ru/AC as well as other state-of-the-art Ru nanocatalysts, the Ru1/NC SAC with RuN3 structure not only possesses the highest primary amine productivity per atom Ru per hour but also exhibits far superior resistance against being poisoned by sulfur and CO, as well as much higher tolerance to harsh treatment including hydrogen reduction at 600 °C. To our knowledge, the as-obtained RuN3 moiety is the most robust yet highly active and selective single-atom structure reported thus far.
Results and discussion
Preparation and textual properties of Ru1/NC SACs
Ru1/NC SACs were prepared by ball milling of a mixture of L-cysteine, dicyandiamide, and ruthenium acetylacetonate (Ru(acac)3), followed by pyrolysis under N2 atmosphere (Fig. 2a), similar to that earlier reported by Liu et al. for the synthesis of Ni/NC37. During the pyrolysis process, the C, N-containing l-cysteine, and dicyandiamide underwent polymerization and condensation reactions to form an N-doped carbon matrix, which then captured the Ru atoms to form RuNx moieties. With the attempt to tailor the Ru–N coordination environment, the samples were pyrolyzed at different temperatures in the range of 700–1000 °C, and denoted as Ru1/NC-T (T refers to the pyrolysis temperature). In addition, the Ru1/NC-900 sample was subjected to further treatment by NH3 at 800 °C (denoted as Ru1/NC-900–800NH3) in order to increase the accessibility of RuNx active sites through etching of carbonaceous materials by NH3. The as-synthesized Ru1/NC-T samples show two-dimensional (2D) few-layer graphene morphology (Fig. 2b) with the specific surface area of 216–286 m2/g and mesoporous structure (Supplementary Fig. 1 and Supplementary Table 1). After NH3 treatment, the specific surface area is significantly increased from 216 to 471 m2/g due to the creation of plenty of micropores. Inductively coupled plasma optical emission spectrometry (ICP-OES) analysis (Supplementary Table 1) reveals that the mass loading of Ru increases steadily from 0.8 wt% to 1.6 wt% with the pyrolysis temperature, and rises further to 2.3 wt% upon NH3 treatment, suggesting the gradual decomposition of the N-doped carbon matrix at elevated temperatures, especially in the presence of NH3.

a Schematic illustration for preparation of Ru1/NC-T catalysts. b Transmission electron microscopy image of Ru1/NC-900. c, d High-angle annular dark-field scanning transmission electron microscopy images of Ru1/NC-900 and Ru1/NC-900–800NH3. e Electron energy loss spectrometer spectra of Ru1/NC-900–800NH3.
The X-ray diffraction (XRD) patterns of all the above samples do not show any reflections of metallic or oxidic Ru species (Supplementary Fig. 2), which, in combination with the absence of NPs in low-magnification scanning transmission electron microscopy (STEM) images (Supplementary Fig. 3), indicates that Ru species are highly dispersed in the N-doped carbon matrix. We then employed sub-Ångström-resolution high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) technique to probe the highly dispersed Ru-containing species. Representative images are shown in Fig. 2c, d, and more images are shown in Supplementary Fig. 4. It can be clearly seen that a high density of Ru single atoms (bright dots) are uniformly dispersed on the N-doped carbon materials. The surface density of single atoms based on Ru loading and the specific surface area of the Ru1/NC-900 is calculated to be 0.5 Ru atom/nm2, which is lower than that estimated from HAADF-STEM images (1.2 Ru atom/nm2), possibly due to that some Ru single atoms were imbedded into the NC material. Notably, NH3 treatment of Ru1/NC-900 at 800 °C does not change the atomic dispersion of Ru species, manifesting the effectiveness of our method to prepare Ru SACs. It should be emphasized that the acid-leaching step widely employed to remove the metal nanoparticles in the preparation of Co(Fe, Ni)–N–C single-atom catalysts35,36,38 is absolutely unnecessary in our method because all of the Ru species are atomically dispersed, which is highly desirable from the viewpoint of saving precious metals and environmentally benign process. When the sample was pyrolyzed at 1000 °C, however, serious aggregation began to take place leading to the formation of a large number of Ru NPs with the average size of 5.2 nm (Supplementary Fig. 5). Obviously, in order to achieve stable Ru single atoms on the NC surface, the pyrolysis temperature should not exceed 1000 °C. The microenvironment of Ru single atoms is further probed with high-resolution energy-dispersive spectrometer (EDS) and electron energy loss spectrometer (EELS). As shown in Fig. 2e, both Ru (note, Ru and C EELS spectra are overlapped and therefore cannot be distinguished) and N spectra are detected when the electron beam is focused on an individual Ru single atom, which suggests that Ru is bonded to N in accordance with that reported in M–N–C catalysts37,39. The elemental mapping by EDS (Supplementary Fig. 6) shows that Ru and N elements are uniformly dispersed throughout the whole sample, again suggesting that they are bonding together.
Probing into the electronic and coordinative structure
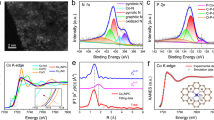
X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) characterizations were conducted to determine the chemical state and coordination environment of Ru1/NC-T catalysts. Due to the overlap in binding energies of Ru 3d and C 1s (284.6 eV), we detected Ru 3p XPS in spite of some loss in signal/noise ratio, as shown in Fig. 3. It is found that the Ru 3p3/2 binding energy is in the range of 462.0–462.6 eV, indicating that Ru single atoms have an oxidation state of +240. Moreover, a slight shift toward lower energy can be observed with an increase of the pyrolysis temperature, in particular for the Ru1/NC-900–800NH3 catalyst, indicating a slight increase in the electron density of Ru single atoms. Such a fine-tuning of the electronic property without altering the single-atom dispersion is highly important to SACs since it can allow for the de-coupling of activity and chemoselectivity on the condition that the latter is more dependent on the single-atom dispersion31,41,42. On the other hand, the N 1s XP spectra can be deconvoluted into five different N species: pyridinic N (397.9–398.4 eV), Ru–N (399.0–399.5 eV), pyrrolic N (400.5 eV), graphitic N (401.3–401.6 eV) and oxidized graphitic N (402.5–403.3 eV). It is interesting to note that the binding energies of the Ru–N species also slightly shift toward lower values with the increase of the pyrolysis temperature, suggesting the increased electron density on the N atoms, which are likely caused by electron donation of the neighboring C43. Moreover, with an increase of pyrolysis temperature, the contents of both pyridinic N and Ru–N decrease gradually while the other three types of N species increase (Supplementary Table 2), suggesting that Ru atoms are likely coordinated to pyridinic N and the resultant Ru–N coordination number decreases by elevating the pyrolysis temperature and further treatment with NH3.

a Ru 3p3/2. b N 1s.
The modulation of the electronic and coordinative structures is further supported by XAS technique. Figure 4a displays the X-ray absorption near-edge spectra (XANES) at Ru K-edge of Ru1/NC-T catalysts and references. The edge energies (E0) for all the Ru1/NC-T samples are lower than that of Ru(acac)3 yet higher than that of Ru foil, suggesting Ru atoms carry positive charges +δ (0 < δ < 3), consistent with the XPS characterization results. Meanwhile, a comparison of the E0 values suggests the oxidation state increases following the order of Ru1/NC-900–800NH3 < Ru1/NC-900 < Ru1/NC-800 ≈ Ru1/NC-700, again in line with the XPS result. The coordination environment of Ru single atoms is determined by the extended X-ray absorption fine structure spectra (EXAFS). As shown in the Fourier-transformed k2-weighted EXAFS spectra at the Ru K-edge (Fig. 4b), in contrast to the reference samples of Ru foil and RuO2, the Ru1/NC-T catalysts do not show any prominent peaks at the positions of either Ru–Ru shell (2.4 Å) or Ru–O–Ru (3.2) shell, excluding the existence of metallic Ru or RuOx nanoparticles. The absence of Ru–Ru scattering is further demonstrated by the wavelet transform (WT) technique. As shown in Fig. 4c, Ru foil affords a lobe at (2.4 Å, 9.2 Å−1), which is attributed to Ru–Ru coordination. The absence of lobe at high k value in Ru1/NC-900–800NH3 (Fig. 4d) indicates the central Ru does not bind to heavy atoms (e.g., Ru), on the contrary, there exists a lobe at low k values (1.5 Å, 3.5 Å−1), which can be ascribed to Ru–N coordination. Notably, in the series of Ru1/NC-T samples, the peak intensity of Ru–N contribution decreases with an increase of pyrolysis temperature (Fig. 4b, inset), suggesting the decrease of Ru–N coordination number. Exactly, the EXAFS data-fitting results (Table 1) show that the Ru–N coordination number gradually decreases from 4.9 in Ru1/NC-700 to 4.3 in Ru1/NC-800 and further to 3.1–3.3 in Ru1/NC-900 and Ru1/NC-900–800NH3, respectively, demonstrating RuN5, RuN4, and RuN3 entities are selectively fabricated by varying the pyrolysis temperature. In addition, the Ru–N distance also continuously decreases from 2.09 Å in Ru1/NC-700 to 2.03 Å in Ru1/NC-900–800NH3, indicating the enhanced Ru–N coordination strength.

a The normalized X-ray absorption near-edge spectra at the Ru K-edge. b The k2-weighted Fourier transform extended X-ray absorption fine structure spectra (EXAFS) in r-space. c, d Wavelet Transformation for the k2-weighted EXAFS signal of Ru foil and Ru1/NC-900-800NH3 sample. Red line: Ru1/NC-900-800NH3, Blue: Ru1/NC-900, Dark yellow: Ru1/NC-800, Wine red: Ru1/NC-700.
Catalytic performance of Ru1/NC-T catalysts in reductive amination reaction
The different coordination environments (i.e., RuN5, RuN4, and RuN3) of Ru single atoms will lead to significant differences in catalytic performances. Here, we choose the reductive amination of furfural (FAL) as the probe reaction considering that FAL is readily available from biomass and the target product furfuryl amine (FAM) is widely used in the manufacture of pharmaceuticals and pesticides19,44. It should be noted that neither Ru(acac)3 nor the mixture of Ru(acac)3 and N,C-precursor gives the target FAM; instead, oligomers arising from trimerization of imines are produced as the main product. Similar to the catalyst precursors, the Ru1/NC-700 does not provide any useful product other than oligomers (Table 2, entry 1), suggesting the negligible hydrogenation activity of the RuN5 structure. By contrast, with a decrease of the Ru–N coordination number, the target product FAM becomes the predominant one. For example, 43% yield of FAM is achieved on the Ru1/NC-800 catalyst having a RuN4 structure (Table 2, entry 2), whereas it is enhanced to 84% over the Ru1/NC-900 with a RuN3 structure (Table 2, entry 3). Notably, when the Ru1/NC-900 is further treated under NH3 atmosphere, the resultant Ru1/NC-900–800NH3 catalyst affords FAM yield as high as 97% (Table 2, entry 4), corresponding to a production rate of 170.7 gFAM.gRu−1 h−1. In contrast to the high efficiency of Ru1/NC-900 and Ru1/NC-900–800NH3 SACs, the Ru1/NC-1000 catalyst with predominance of Ru NPs provides FAM yield of only 53% (Table 2, entry 5), far inferior to the Ru single-atom catalysts. We also prepared other nanocatalysts including Ru/AC, Ru/Nb2O5, and Ru/HZSM-5 which were reported to be highly active for the reductive amination of furfural19,45,46. Under identical reaction conditions, the Ru/AC nanocatalyst gives an enhanced FAM yield (82%, Table 2, entry 6) than Ru1/NC-1000, which can be due to the smaller and more exposed Ru particles on the AC support (Supplementary Fig. 7), yet lower than that over the Ru1/NC-900-800NH3. Similarly, both the Ru/Nb2O5 and Ru/HZSM-5 nanocatalysts deliver an inferior yield of FAM to the Ru1/NC-900-800NH3 SAC even with extended reaction time (Table 2, entries 7–8). To further demonstrate the superiority of the Ru1/NC SAC, we synthesized the Ru1/NC SAC on the Nb2O5 support given that the electronic interaction between Ru and acidic and reducible Nb2O5 support would facilitate the reductive amination reaction19,25. To our delight, the as-prepared Ru1/NC/Nb2O5 SAC affords 97% yield in a shorter time of 3 h (Table 2, entry 9), resulting in a production rate of 227.6 gFAM·gRu−1 h−1, almost twofold higher than that on the Ru/Nb2O5 nanocatalyst. These strictly control experiments, together with the comparison with more heterogeneous and homogeneous Ru catalysts reported in the literature (Supplementary Table 3), unequivocally demonstrate the remarkable advantage of Ru1/NC-900–800NH3 SAC in maximizing the atom efficiency of Ru.
Not only possessing a higher production rate than the NPs counterparts but our Ru1/NC-900–800NH3 SAC also exhibits superior stability and resistance against being poisoned by CO or sulfur species. Figure 5a shows that the catalyst can be reused at least five times without any decay in catalytic performance. HAADF-STEM and XAS characterizations reveal that Ru single atoms and the local coordination environment remain unchanged after the reuse tests (Supplementary Fig. 8), demonstrating the excellent stability of the Ru1/NC-900–800NH3 SAC. Figure 5b compares the performances of Ru1/NC-900–800NH3 SAC and Ru/AC nanocatalyst in exposure to strongly adsorbed molecules such as CO and sulfur-containing species. When 1% CO is present in H2 gas, which is the common case when hydrogen comes from reforming natural gas47, the FAM yield over the Ru1/NC-900–800NH3 SAC goes from 97% slightly down to 88%, a <10% decrease; by contrast, it drastically goes down from 82 to 29% over the Ru/AC nanocatalyst, a more than 50% decrease. Sulfur species, which are frequently present in biomass as well as the biomass-derived platform compounds such as FAL, is another well-known poison to noble metal catalysts48. Here we use thiophene as a representative sulfur species to test the catalyst. We are delighted to see that the Ru1/NC-900–800NH3 SAC manifests high resistance against the sulfur poison; the FAM yield decreases by 18% in the presence of 500 ppm thiophene, which is in stark contrast to a decrease of 67% over the Ru/AC nanocatalyst. Similar to the Ru/AC, the Ru/Nb2O5 nanocatalyst also shows vulnerability towards being poisoned by CO and sulfur, which is in contrast to the robustness of the Ru1/NC-900–800NH3 SAC (Supplementary Fig. 9). Obviously, the Ru1/NC-900–800NH3 SAC is much superior to typical Ru nanocatalysts in terms of poison-resistance. The ultrarobustness of the Ru1/NC-900–800NH3 SAC is also demonstrated by high-temperature reduction treatment which has been reported to cause serious aggregation of both single atoms and nanoparticles49,50,51,52. As shown in Supplementary Fig. 10, NPs of Ru are observed only scarcely on the Ru1/NC-900–800NH3 SAC after H2 treatment at 600 °C, which is supported by the absence of Ru–Ru bonding in the EXAFS spectra, indicating the high resistance of Ru single atoms to aggregation. By contrast, serious aggregation takes place after the hydrogen treatment of Ru/AC catalyst, thus significantly reduces the number of active sites. The reaction test reveals that there is a 12% decrease in the FAM yield after the high-temperature hydrogen treatment, which is far less appreciable than the 49% yield decrease over the Ru/AC catalyst (Fig. 5b).

a Reusability tests at different conversion levels. b Resistance against CO, sulfur and H2 treatment of Ru1/NC-900–800NH3 and Ru/AC. Reaction conditions: 2 mmol furfural, 22 mg Ru1/NC-900–800NH3 or Ru/AC catalysts (0.25 mol% Ru, molar ratio of Ru:furfural = 1:400), 3 g methanol, 0.5 MPa NH3, 2 MPa H2, 100 °C. For reusability tests, data were taken at 1 h (left) and 10 h (right), respectively. For CO poisoning experiment, 2 MPa 1 vol% CO in H2 was used in place of pure H2; for sulfur-poisoning experiment, 500 ppm thiophene was added to the reaction mixture; for H2 treatment test, the catalysts were pre-reduced at 600 °C for 2 h before the reaction.
The Ru1/NC-900–800NH3 SAC can be applied to the reductive amination of a broad spectrum of substrates, and good to excellent yields of the corresponding primary amines are obtained. As shown in Table 3, for biomass-derived 5-methyl furfural and 5-hydroxymethyl furfural (Table 3, entries 1–2), high yields (93–98%) of the primary amines are reached. Even for the more challenging substrate, biomass-derived 2,5-diformylfuran, the desired diamine product 2,5-bis(aminomethyl)furan (BAMF) is still obtained with a yield of 63% (Table 3, entry 3), surpassing the earlier reported Raney Ni catalyst53. For the aromatic aldehydes with electron-donating/withdrawing groups (Table 3, entries 4–17), the yields of the primary amines are all above 80%. Sensitive functional groups including methoxyl, halides, esters as well as challenging amide and sulfo group are well tolerated. In particular, for heterocyclic aldehydes such as pyridinecarboxaldehyde and 2-thenaldehyde which are well-known poisons to noble metal catalysts, the reductive amination can still proceed smoothly (Table 3, entries 18–19). Aliphatic aldehydes with or without hydroxyl groups are also good substrates (Table 3, entries 20–23), although a mixture of primary amine and secondary amine is produced for hexaldehyde (Table 3, entry 23). Notably, the reductive amination of the more demanding molecules such as ketones generally requires a more active catalyst or more drastic conditions. Nevertheless, we are gratified to see that the more challenging ketones with various functional groups could also be smoothly converted to the corresponding amines (Table 3, entries 24–35). Finally, the kinetically more sluggish alcohols such as furfural alcohol could be transformed into FAM with a moderate yield at relatively harsh conditions (Table 3, entry 36). All these results demonstrated that the Ru1/NC-900–800NH3 SAC possessed high activity and selectivity, unrivalled robustness against various poisons and harsh hydrogen treating, as well as excellent tolerance to a wide spectrum of substrates, which is of great potential for practical applications.
Establishing the structure–performance relationship
In situ 1H nuclear magnetic resonance (1HNMR) experiments, kinetic studies, and microcalorimetric measurements were performed to get insight into the structure–performance relationship of the series of Ru1/NC-T catalysts. First, the in- situ 1HNMR detected the formation of primary imine upon condensation of FAL with NH3 in the absence of H2, while in the presence of H2, both Schiff base (i.e., secondary imine) and FAM were produced associated with the disappearance of primary imine (Supplementary Fig. 11), suggesting both of them come from the hydrogenation of highly unstable intermediate primary imine. Second, conversion-time kinetic profile (Supplementary Fig. 12) reveals that at the beginning stage of the reaction, a large proportion of Schiff base (i.e., secondary imine) was produced, which was then followed by the gradual increase of FAM at the expense of Schiff base. This result indicates that Schiff base is the key intermediate during the reductive amination of FAL toward FAM. As Schiff base is yielded from the condensation between primary amine and aldehyde (or primary imine), its rapid accumulation indicates at least the hydrogenation of primary imine to yield primary amine must proceed fast and therefore cannot be a rate-determining step. Instead, the conversion-time profile shows that after the Schiff base yield reaches the maximum, its subsequent conversion to primary amine proceeds slowly, suggesting the transimination of Schiff base being most likely a rate-determining step in the reductive amination, as shown in Fig. 1.
According to the above reaction mechanism, it is obvious that the catalytic performances of Ru1/NC-T SACs are governed by their transimination and hydrogenation capabilities. To probe into the adsorption behavior of NH3 and H2 on Ru1/NC-T catalysts with different coordination environments (i.e., RuN5, RuN4, and RuN3), microcalorimetric measurements were conducted. As shown in Fig. 6a, all the catalysts have strong adsorption to NH3 with initial adsorption heat of ~160 kJ/mol and appear not sensitive to the coordination structure of Ru. By contrast, the adsorption of H2 is highly dependent on the coordinative structure, and both of the initial adsorption heat and uptake increase with a decrease of Ru–N coordination number (Fig. 6a and Supplementary Table 4). Moreover, the Ru1/NC-900–800NH3 with the same Ru–N CN as the Ru1/NC-900 according to our EXAFS fitting result (Table 1) exhibits much stronger adsorption for H2 than the latter, suggesting that the electronic structure, in addition to geometric structure, significantly affects the adsorption properties of Ru single atoms. Based on XANES and XPS results, the Ru single atoms in the Ru1/NC-900–800NH3 catalyst are more electron-rich than those in the Ru1/NC-900 structure, which facilitates the electron transfer to the antibonding orbital of H2 molecules and thus activate hydrogen. In our previous work33, Pt1/Fe2O3 SACs with different Pt–O CNs were fabricated and a similar trend was found: the lower the Pt–O CN is, the higher the electron density of Pt single atoms gets, and the higher hydrogenation activity is.

a Initial adsorption heat of NH3 as well as H2 before and after NH3 pre-adsorption as a function of the Ru–N coordination structure. b Differential adsorption heat as a function of coverage for hydrogen adsorption. For guiding the structure–performance relationship, FAM yield is also plotted as a function of Ru–N coordination structure.
As both NH3 and H2 participate in the reductive amination reaction, we also investigate the adsorption of H2 after pre-adsorption of NH3. In agreement with that reported in other catalyst systems21, the adsorption of H2 is suppressed to a great extent upon pre-adsorption of NH3 (Fig. 6b and Supplementary Table 4), indicating the competitive adsorption of the two reactants on the active sites. Nevertheless, the suppressing degree is quite different depending on the coordination and electronic structure of Ru single atoms. For Ru1/NC-700 catalyst with the RuN5 structure, the hydrogen adsorption is totally prevented by NH3 adsorption, which may account for its inactivity in the reductive amination reaction. By contrast, hydrogen molecules can still be activated on the other three catalysts with measurable uptakes, and the adsorption strength and uptakes increase in the order of Ru1/NC-800 < Ru1/NC-900 < Ru1/NC-900-800NH3. This experimental result is consistent with the density functional theory (DFT) calculations reported by Zhou et al.54, which reveals that the smaller the difference between the adsorption energy of NH3 and H2 is, the higher the activity for reductive amination is. Indeed, when the FAM yield is correlated with the RuNx structure, a similar increasing trend can be found as that of hydrogen adsorption heat (Fig. 6a), which suggests that moderate hydrogenation capability, as well as strong transimination activity of the catalyst, is highly desirable for the selective synthesis of primary amines. Kinetic isotope effect (KIE) experiment by using D2 to replace H2 in the reductive amination reveals KIE value of 1.1, 1.7, and 2.1 for the Ru1/NC-800, Ru1/NC-900, and Ru1/NC-900-800NH3 catalysts (Supplementary Table 5), respectively, proving that hydrogen activation is an important factor determining the product selectivity although it is not involved in the rate-determining step.
To get more insight into the effect of the relative strength between hydrogenation and transimination capabilities on the product selectivity, we modulate the hydrogenation/transimination activities by varying the pressure of H2/NH3. As shown in Supplementary Fig. 13a, when the H2 pressure decreases from 2 to 1 MPa, the FAM yield is correspondingly reduced to 41%, while the yield of oligomer increases to 18% due to the reduced hydrogenation capability. By contrast, when the NH3 pressure falls from 0.5 to 0.2 MPa (Supplementary Fig. 13b), 9% secondary amine is yielded at the expense of primary amine (yield: 68%). Lower pressure of NH3 results in weakened transimination activity yet enhanced hydrogenation capability due to competitive adsorption between NH3 and H2, which leads to the hydrogenation of Schiff base to give secondary amine.
Since the reaction direction (selectivity) is steered by the relative adsorption strength between NH3 and H2, and this factor can be modulated by the Ru–Nx coordination sphere, it is important to elucidate how H2 is activated by the Ru–Nx. Based on our previous studies on the Ni–Nx36 and Pt–Ox33 SACs, we suppose the central Ru and the neighboring N atoms form Frustrated Lewis Pairs (FLPs)55,56, which is able to dissociate H2 following a heterolysis manner. According to this FLP mechanism, pyrrole and potassium thiocyanate (KSCN) were selected as poisons of Lewis basic and Lewis acid sites, respectively. As shown in Supplementary Fig. 14, upon the addition of pyrrole or KSCN, the yield of primary amine decreases significantly from 97 to 21% or 1%, respectively, which provides strong evidence that FLP is required for hydrogen dissociation. In addition, CO2 temperature-programmed desorption (CO2-TPD) experiments (Supplementary Fig. 15) and CO2 adsorption measurements (Supplementary Fig. 16) show that both of the adsorption strength and uptake of CO2 are greatly enhanced after NH3 treatment of Ru1/NC-900, suggesting the higher basicity of the Ru1/NC-900–800NH3 catalyst, which is in line with that reported previously57,58. Also, this is in agreement with the XPS result that reveals the increased electron density on the N atoms coordinating to Ru. The increased basicity in FLPs facilitates the heterolytic cleavage of H2 according to our previous DFT calculations33 and thus contributes to the high hydrogenation activity.
In summary, we have fabricated highly active, selective and robust Ru SACs supported on N-doped carbons using a simple pyrolysis approach. By raising the pyrolysis temperature from 700 to 900 °C, the Ru–N coordination number decreases from RuN5, RuN4 to RuN3, meanwhile, the electron density of the Ru single atoms increases in the order of RuN5 < RuN4 < RuN3. By further treatment with NH3 at 800 °C, the electron density on the RuN3 site is further increased. As a consequence of the fine-tuning in both the coordination structure and the electron density of the Ru single atoms, the catalytic performance is finely modulated in the reductive amination of aldehydes/ketones. Among them, the Ru1/NC-900–800NH3 SAC affords the highest activity and selectivity to primary amines thanks to its moderate capability for H2 activation in excess of NH3. Moreover, the catalyst exhibits excellent stability in reuse tests and tolerance to a wide spectrum of biomass-derived aldehydes/ketones substrates. In particular, it shows superior robustness against being poisoned by CO, sulfur, or harsh hydrogen treatment which, by contrast, drastically deactivate the control Ru/AC nanocatalyst. The structure–performance relationship established in this work will open an avenue toward the rational design of highly selective and robust SACs for other demanding transformations.
Methods
Sample preparation
Typically, a mixture of dicyandiamide (C2H4N4) (12 g), L-cysteine (C3H7NO2S) (3 g) and Ru(III) acetylacetonate (Ru(acac)3) (40 mg) were added to a ball mill pot, and were ground at a speed of 400 rpm for 2.0 h for twice at room temperature. The obtained fine powder was then subjected to temperature-programmed pyrolysis in a tubular furnace under N2 atmosphere with a flow rate of 60 ml min–1. First stage: from 25 to 600 °C at a ramping rate of 3 °C min–1, maintained at 600 °C for 2 h; second stage: from 600 to 900 °C at a ramping rate of 2 °C min–1, maintained at 900 °C for 1 h. The as-made sample was denoted as Ru1/NC-900. Other Ru1/NC samples were synthesized by the same procedure except for different maximum temperatures. Ru1/NC-900-800NH3 was prepared by subjecting Ru1/NC-900 to pyrolysis at 800 °C for 30 min (ramping rate: 5 °C min–1) under NH3/He mixture atmosphere (30 ml min–1 He and 20 ml min–1 NH3).
More sample preparation details are described in the Supplementary Methods section.
Sample characterization
The actual Ru loadings were determined by inductively coupled plasma spectroscopy (ICP-OES) on an IRIS Intrepid II XSP instrument (Thermo Electron Corporation). X-ray diffraction (XRD) patterns were recorded on a PANalytical X’ pert diffractometer with a Cu–Kα radiation source (40 kV and 40 mA). A continuous mode was used to record data in the 2θ range from 10° to 80°. N2 adsorption-desorption experiments were conducted on a Micromeritics ASAP-2010 physical adsorption apparatus. The specific surface area was calculated using a Brunauer–Emmett–Teller (BET) method.
Scanning transmission electron microscopy (STEM) and energy-dispersive X-ray spectroscopy (EDS) experiments were performed on a JEOL JEM-2100F microscope operated at 200 kV, equipped with an Oxford Instruments ISIS/INCA energy-dispersive X-ray spectroscopy (EDS) system with an Oxford Pentafet Ultrathin Window (UTW) Detector. The aberration-corrected high-angle annual dark-filed scanning transmission electron microscopy (AC-HAADF-STEM) analysis was performed on a JEOL JEM-ARM200F equipped with a CEOS probe corrector, with a guaranteed resolution of 0.08 nm. The Electron Energy Loss Spectroscopy (EELS) analysis of Ru1/NC-900-800NH3 was performed on a Field Emission HF5000 Microscope (200 kV accelerating voltage; aberration corrector, 0.078-nm spatial resolution). Before microscopy examination, the sample was ultrasonically dispersed in ethanol for 15–20 min, and then a drop of the suspension was dropped on a copper TEM grid coated with a thin holey carbon film.
X-ray photoelectron spectroscopy (XPS) spectra were obtained on a Thermo ESCALAB 250 X-ray photoelectron spectrometer equipped with Al Kα excitation source and with C as internal standard (C 1 s = 284.6 eV).
The X-ray absorption spectra (XAS) including X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) at Ru K-edge of the samples were measured at the beamline 14 W of Shanghai Synchrotron Radiation Facility (SSRF) in China. The output beam was selected by Si(311) monochromator, and the energy was calibrated by Ru foil. The data were collected at room temperature under transmission mode. Athena software package was employed to process the XAS data.
Microcalorimetric measurement was performed by a BT2.15 heat-flux calorimeter, which was connected to a gas handling and a volumetric system employing MKS Baratron Capacitance Manometers for precision pressure measurement. The ultimate dynamic vacuum of the microcalorimetric system was 10−7 Torr by calculation. First, the fresh sample was treated in a special tube in H2 at 100 °C for 1 h and then high pure He at 200 °C for 1 h to eliminate the adsorption. Then, the tube was transferred into the high vacuum system and stabilized for (6–8 h). After thermal equilibrium was reached, the H2-microcalorimetric data were collected by sequentially introducing small doses (10−6 mol) of H2 (CO2 or NH3) into the system until it became saturated (5–6 Torr). Simultaneously, the differential heat versus adsorbate coverage plots and adsorption isothermals can be obtained after a typical microcalorimetric experiment.
More characterization performance details are described in the Supplementary Methods section.
Catalytic test
The reductive amination reaction was conducted in a 50-mL stainless-steel autoclave (Parr Instrument Company, America). Typically, the substrate (2 mmol), Ru1/NC catalyst (molar ratio of Ru:substrate was 1:400), dodecane (internal standard) and 3 mL methanol (solvent) were added to a teflon lining. Then the autoclave was sealed and purged by N2 for three times, followed by charging with 0.5 MPa NH3 and 2 MPa H2. The reaction mixture was stirred at 100 °C for 10 h. The products were identified using a Varian G450/320 GC/MS system and were quantitatively analyzed using an Agilent 7890 A GC system equipped with a HP-5 capillary column and an FID detector.
The furfural conversion (1) and furfural amine selectivity (2) for furfural reductive amination reaction were calculated as follows:
Data availability
All data are available within the article, and its supplementary information file is available from the authors upon request.
References
Wang, A., Li, J. & Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 3, 634–641 (2018).
Liu, L. & Corma, A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 118, 4981–5079 (2018).
Böhme, D. K. & Schwarz, H. Gas-phase catalysis by atomic and cluster metal ions: the ultimate single-site catalysts. Angew. Chem. Int. Ed. 44, 2336–2354 (2015).
Cui, X., Li, W., Ryabchuk, P., Junge, K. & Beller, M. Bridging homogeneous and heterogeneous catalysis by heterogeneous single-metal-site catalysts. Nat. Catal. 1, 385–397 (2018).
Thomas, J. M., Raja, R. & Lewis, D. W. Single-site heterogeneous catalysts. Angew. Chem. Int. Ed. 44, 6456–6482 (2005).
Li, J. et al. Highly active and stable metal single-atom catalysts achieved by strong electronic metal-support interactions. J. Am. Chem. Soc. 141, 14515–14519 (2019).
DeRita, L. et al. Structural evolution of atomically dispersed Pt catalysts dictates reactivity. Nat. Mater. 18, 746–751 (2019).
Zhang, L., Zhou, M., Wang, A. & Zhang, T. Selective hydrogenation over supported metal catalysts: from nanoparticles to single atoms. Chem. Rev. 120, 683–733 (2019).
Wei, H. et al. FeOx-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes. Nat. Commun. 5, 5634 (2014).
Sun, X. et al. Single cobalt sites in mesoporous N-doped carbon matrix for selective catalytic hydrogenation of nitroarenes. J. Catal. 357, 20–28 (2018).
Yan, H. et al. Atomic engineering of high-density isolated Co atoms on graphene with proximal-atom controlled reaction selectivity. Nat. Commun. 9, 3197 (2018).
Sun, Q. et al. Zeolite-encaged single-atom Rhodium catalysts: highly-efficient hydrogen generation and shape-selective tandem hydrogenation of nitroarenes. Angew. Chem. Int. Ed. 58, 18570–18576 (2019).
Yang, F. et al. Atomically dispersed Ni as the active site towards selective hydrogenation of nitroarenes. Green Chem. 21, 704–711 (2019).
Irrgang, T. & Kempe, R. Transition-metal-catalyzed reductive amination employing hydrogen. Chem. Rev. 120, 9583–9674 (2020).
Murugesan, K. et al. Catalytic reductive aminations using molecular hydrogen for synthesis of different kinds of amines. Chem. Soc. Rev. 49, 6273–6328 (2020).
Wang, Y., Furukawa, S., Fu, X. & Yan, N. Organonitrogen chemicals from oxygen-containing feedstock over heterogeneous catalysts. ACS Catal. 10, 311–335 (2019).
Pelckmans, M., Renders, T., Van de Vyver, S. & Sels, B. F. Bio-based amines through sustainable heterogeneous catalysis. Green Chem. 19, 5303–5331 (2017).
Deng, W. et al. Catalytic amino acid production from biomass-derived intermediates. Proc. Natl Acad. Sci. USA 115, 5093–5098 (2018).
Komanoya, T., Kinemura, T., Kita, Y., Kamata, K. & Hara, M. Electronic effect of Ruthenium nanoparticles on efficient reductive amination of carbonyl compounds. J. Am. Chem. Soc. 139, 11493–11499 (2017).
Wang, T. et al. Rational design of selective metal catalysts for alcohol amination with ammonia. Nat. Catal. 2, 773–779 (2019).
Qi, H. et al. Modulating trans-imination and hydrogenation towards the highly selective production of primary diamines from dialdehydes. Green Chem. 22, 6897–6901 (2020).
Senthamarai, T. et al. Simple Ruthenium-catalyzed reductive amination enables the synthesis of a broad range of primary amines. Nat. Commun. 9, 4123 (2018).
Wang, Y.-G., Mei, D., Glezakou, V.-A., Li, J. & Rousseau, R. Dynamic formation of single-atom catalytic active sites on ceria-supported gold nanoparticles. Nat. Commun. 6, 6511 (2015).
Matsubu, J. C. et al. Adsorbate-mediated strong metal-support interactions in oxide-supported Rh catalysts. Nat. Chem. 9, 120–127 (2016).
Liang, G. et al. Production of primary amines by reductive amination of biomass-derived aldehydes/ketones. Angew. Chem. Int. Ed. 56, 3050–3054 (2017).
Liang, G., Zhou, Y., Zhao, J., Khodakov, A. Y. & Ordomsky, V. V. Structure-sensitive and insensitive reactions in alcohol amination over nonsupported Ru nanoparticles. ACS Catal. 8, 11226–11234 (2018).
Chandra, D. et al. A high performance catalyst of shape-specific Ruthenium nanoparticles for production of primary amines by reductive amination of carbonyl compounds. Chem. Sci. 9, 5949–5956 (2018).
Murugesan, K., Beller, M. & Jagadeesh, R. V. Reusable Nickel nanoparticles-catalyzed reductive amination for selective synthesis of primary amines. Angew. Chem. Int. Ed. 58, 5064–5068 (2019).
Hahn, G., Kunnas, P., de Jonge, N. & Kempe, R. General synthesis of primary amines via reductive amination employing a reusable nickel catalyst. Nat. Catal. 2, 71–77 (2018).
Jagadeesh, R. V. et al. MOF-derived cobalt nanoparticles catalyze a general synthesis of amines. Science 358, 326–332 (2017).
Wang, X. et al. Regulation of coordination number over single Co sites: triggering the efficient electroreduction of CO2. Angew. Chem. Int. Ed. 57, 1944–1948 (2018).
Resasco, J. et al. Uniformity is key in defining structure-function relationships for atomically dispersed metal catalysts: the case of Pt/CeO2. J. Am.Chem. Soc. 142, 169–184 (2019).
Ren, Y. et al. Unraveling the coordination structure-performance relationship in Pt1/Fe2O3 single-atom catalyst. Nat. Commun. 10, 4500 (2019).
Vilé, G. et al. A stable single-site Palladium catalyst for hydrogenations. Angew. Chem. Int. Ed. 54, 11265–11269 (2015).
Liu, W. et al. Single-atom dispersed Co-N-C catalyst: structure identification and performance for hydrogenative coupling of nitroarenes. Chem. Sci. 7, 5758–5764 (2016).
Liu, W. et al. A durable Nickel single-atom catalyst for hydrogenation reactions and cellulose valorization under harsh conditions. Angew. Chem. Int. Ed. 57, 7071–7075 (2018).
Yang, H. B. et al. Atomically dispersed Ni(I) as the active site for electrochemical CO2 reduction. Nat. Energy 3, 140–147 (2018).
Liu, W. et al. Discriminating catalytically active FeNx species of atomically dispersed Fe-N-C catalyst for selective oxidation of the C-H bond. J. Am. Chem.Soc. 139, 10790–10798 (2017).
Yin, P. et al. Single cobalt atoms with precise N-coordination as superior oxygen reduction reaction catalysts. Angew. Chem. Int. Ed. 55, 10800–10805 (2016).
Folkesson, B. ESCA studies on charge-distribution in some dinitrogen complexes of Rhenium, Iridium, Ruthenium and Osmium. Acta Chem. Scand. 27, 287–302 (1973).
Feng, Q. et al. Isolated single-atom Pd sites in intermetallic nanostructures: high catalytic selectivity for semihydrogenation of alkynes. J. Am.Chem.Soc. 139, 7294–7301 (2017).
DeRita, L. et al. Catalyst architecture for stable single atom dispersion enables site-specific spectroscopic and reactivity measurements of CO adsorbed to Pt atoms, oxidized Pt clusters, and metallic Pt clusters on TiO2. J. Am. Chem.Soc. 139, 14150–14165 (2017).
Ha, Y. et al. Atomically dispersed Co-pyridinic N-C for superior oxygen reduction reaction. Adv. Energy Mater. 10, 2002592 (2020).
Chatterjee, M., Ishizaka, T. & Kawanami, H. Reductive amination of furfural to furfurylamine using aqueous ammonia solution and molecular hydrogen: an environmentally friendly approach. Green Chem. 18, 487–496 (2016).
Jiang, S. et al. Direct catalytic conversion of furfural to furan-derived amines in the presence of Ru-based catalyst. ChemSusChem. 13, 1699–1704 (2020).
Dong, C. et al. Ru/HZSM-5 as an efficient and recyclable catalyst for reductive amination of furfural to furfurylamine. Mol. Cat. 482, 110755 (2020).
Lin, L. et al. A highly CO-tolerant atomically dispersed Pt catalyst for chemoselective hydrogenation. Nat. Nanotechnol. 14, 354–361 (2019).
Marshall, S. T. et al. Controlled selectivity for palladium catalysts using self-assembled monolayers. Nat. Mater. 9, 853–858 (2010).
Hansen, T. W., DeLaRiva, A. T., Challa, S. R. & Datye, A. K. Sintering of catalytic nanoparticles: particle migration or ostwald ripening? Acc. Chem. Res. 46, 1720–1730 (2013).
Qin, R., Liu, P., Fu, G. & Zheng, N. Strategies for stabilizing atomically dispersed metal catalysts. Small Methods 2, 1700286 (2018).
Tian, C. et al. A new trick for an old support: stabilizing gold single atoms on LaFeO3 perovskite. Appl. Catal. B 261, 118178 (2020).
Qin, R. et al. Alkali ions secure hydrides for catalytic hydrogenation. Nat. Catal. 3, 703 (2020).
Le, N.-T., Byun, A., Han, Y., Lee, K.-I. & Kim, H. Preparation of 2,5-bis(aminomethyl)furan by direct reductive amination of 2,5-diformylfuran over Nickel-Raney catalysts. Green Sustainable Chem. 5, 115–127 (2015).
Zhou, K. et al. A comprehensive study on the reductive amination of 5-hydroxymethylfurfural into 2,5-bisaminomethylfuran over Raney Ni through DFT calculations. ChemCatChem 11, 2649–2656 (2019).
Stephan, D. W. Frustrated Lewis pairs. J. Am. Chem.Soc. 137, 10018–10032 (2015).
Zhang, S. et al. Solid frustrated-Lewis-pair catalysts constructed by regulations on surface defects of porous nanorods of CeO2. Nat. Commun. 8, 15266 (2017).
Zitolo, A. et al. Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials. Nat. Mater. 14, 937–942 (2015).
Li, J. et al. Identification of durable and non-durable FeNx sites in Fe-N-C materials for proton exchange membrane fuel cells. Nat. Catal. https://doi.org/10.1038/s41929-020-00545-2 (2020).
Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFB1501602), the National Natural Science Foundation of China (21690080, 21690084, 21673228, 21721004, and 21878289), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB17020100), and the Dalian National Laboratory for Clean Energy (DNL) Cooperation Fund, the CAS (DNL 180303). We are grateful to Dr. Wentao Wang and Mr. Yiqi Ren for their kind help in products isolation and NMR discussion.
Author information
Authors and Affiliations
Contributions
H.Q. and J.Y. prepared the catalysts. H.Q. performed reaction tests and most of the characterizations. F.L. performed a part of characterizations and helped to revise the manuscript. J.Y.Y. and R.H. helped to do XPS and NMR data analysis. X.L. helped to do XAS measurement and data fitting. L.L. helped to do microcalorimetric measurements. Y.S. and Y.L. helped to do HAADF-STEM and EELS characterization. A.W. and T.Z. conceived the idea and directed the project. H.Q., L.Z., and A.W. co-wrote the manuscript. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Jagadeesh Rajenahally, Rhett Kempe and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qi, H., Yang, J., Liu, F. et al. Highly selective and robust single-atom catalyst Ru1/NC for reductive amination of aldehydes/ketones. Nat Commun 12, 3295 (2021). https://doi.org/10.1038/s41467-021-23429-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-23429-w
This article is cited by
-
Performance Regulation of Single-Atom Catalyst by Modulating the Microenvironment of Metal Sites
Topics in Current Chemistry (2023)
-
Catalytic reductive amination of furfural to furfurylamine on robust ultra-small Ni nanoparticles
Nano Research (2023)
-
Enriching the surface oxygen as efficient anchoring site of highly dispersed Ru for enhanced hydrogenolysis activity
Nano Research (2023)
-
Mechanochemical-Assisted Synthesis of Nitrogen-Doped Carbon Supported Cobalt Catalysts for Efficient and Selective Hydrogenation of Furfural
Catalysis Letters (2023)
-
Potassium-promoted single-atom Co-N-C catalyst for direct dehydrogenation of ethylbenzene
Nano Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
