
Abstract
Metal promotion in heterogeneous catalysis requires nanoscale-precision architectures to attain maximized and durable benefits. Herein, we unravel the complex interplay between nanostructure and product selectivity of nickel-promoted In2O3 in CO2 hydrogenation to methanol through in-depth characterization, theoretical simulations, and kinetic analyses. Up to 10 wt.% nickel, InNi3 patches are formed on the oxide surface, which cannot activate CO2 but boost methanol production supplying neutral hydrogen species. Since protons and hydrides generated on In2O3 drive methanol synthesis rather than the reverse water-gas shift but radicals foster both reactions, nickel-lean catalysts featuring nanometric alloy layers provide a favorable balance between charged and neutral hydrogen species. For nickel contents >10 wt.%, extended InNi3 structures favor CO production and metallic nickel additionally present produces some methane. This study marks a step ahead towards green methanol synthesis and uncovers chemistry aspects of nickel that shall spark inspiration for other catalytic applications.
Similar content being viewed by others

Introduction
In heterogeneous catalysis, numerous systems rely on metal promotion to maximize process throughput1,2. Since these additives might carry stand-alone activity for undesired reactions jeopardizing selectivity and/or contribute to catalyst deactivation in conventional forms such as supported nanoparticles, specific nanostructures need to be designed to stabilize metal speciations displaying tailored electronic and geometric properties that minimize drawbacks while preserving benefits3,4,5,6,7. This often is a challenging task since the uniform production and in-depth characterization of precise atom-resolved structures lie at the frontier of current technologies.
In the frame of mitigating global environmental changes and lessening our reliance on fossil feedstocks8,9,10,11, indium oxide was introduced as a breakthrough catalyst for CO2 hydrogenation to methanol12, exhibiting extraordinary high selectivity and superior activity and stability when supported on monoclinic ZrO213,14,15. Mechanistic investigations indicated that vacancies formed at a specific surface lattice position mediate CO2 activation and H2 heterolytic splitting16,17,18,19, the latter unlocking the preferential formation of methanol instead of CO via the reverse water-gas shift (RWGS) reaction18. Still, since heterolytic H2 activation is energetically demanding and limits the methanol synthesis rate, promotion with various hydrogenation elements was explored20,21,22,23,24,25,26,27,28,29,30.
Platinum and palladium nanoparticles were shown to boost catalyst performance by aiding H2 splitting, thus fostering CO2 hydrogenation and generating additional vacancies on In2O3, but led to inferior methanol selectivity due to intrinsic RWGS activity and substantial reduction-induced In2O3 sintering. These pitfalls were mitigated by anchoring low-nuclearity (ca. 3 atoms) clusters to the In2O3 lattice in the case of gold and palladium29,31. Ruthenium and cobalt, typical metals leading to methane, were successfully employed when the first was alloyed with indium and the second encapsulated with InxOy23,28. In view of these findings, the low-cost alternative nickel, also a prototypic methanation metal32, could find effective application in CO2 hydrogenation if a favorable structure is identified. A recent study showed that metallic nickel on In2O3 did not produce methane in CO2-based methanol synthesis, but clear support to its segregation from the oxide was not provided26. Another work indicated that CO2 hydrogenation on Ni catalysts can be driven to CO and methanol by doping with indium33. In methane dry reforming on InxNiy catalysts34,35, CO adsorption on nickel was fully suppressed upon doping with indium, suggesting their alloying. In CO2 dry reforming on InNi/SiO2, the surface of Ni–In alloy particles was progressively covered with InOxHy upon use36, as for cobalt-promoted In2O3. These radical deviations of the behavior of pure nickel from its characteristic chemistry call for a rationalization of the nanostructures underpinning them, in comparison to the other metal promoters.
Herein, the synergistic interaction of nickel with indium oxide in CO2-based methanol synthesis was explored through a comprehensive experimental and theoretical program. The nickel speciation was studied contrasting the behavior of coprecipitated and impregnated catalysts upon testing under industrially relevant conditions, with electronic effects being further evaluated on nickel deposited onto In–Al mixed oxides with distinct indium content. The nickel content was varied on the better performing impregnated catalysts identifying a clear impact on the product selectivity. In situ spectroscopy and diffraction methods along with thermal, volumetric, and microscopy analyses uncovered the nanometric construction of the selective nickel-poor and the unselective nickel-rich samples. Density functional theory (DFT) sheds light on the unique structural rearrangements of nickel deposited on In2O3 and the reactivity of complementary promoted surfaces, which was linked to experimental kinetic parameters. Alloying of nickel with In2O3 emerged as key to provide uncharged hydrogen atoms to active sites on In2O3 while curtailing the nickel-mediated detrimental methanation pathway. Overall, this work gathers a fundamental understanding of a relevant system for sustainable methanol production and unravels structural and electronic features at the basis of the tunable selectivity of nickel in CO2 hydrogenation routes.
Results
Impact of nickel content and synthesis method on activity and selectivity
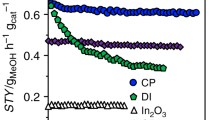
Nickel was incorporated into In2O3 by dry impregnation (DI, 1–20 wt.%, coded as xNi-In2O3, x = 1–20) and coprecipitation (CP, 1–2.5 wt.%) aiming at a deposition on the oxide surface and formation of solid solutions to appreciate the role of metals intermixing and of the nickel oxidation state and chemical environment. N2 sorption and X-ray fluorescence spectroscopy (XRF) indicated that all catalysts featured sufficiently high surface areas and nominal metal contents were closely matched (Supplementary Table 1). Assessing the samples containing 1 wt.% of nickel in CO2 hydrogenation evidenced a higher methanol space-time yield (STY) compared to pure In2O3 (STY = 0.16 gMeOH h−1 gcat−1, Fig. 1a), which, after 65-h equilibration, remained at a ca. twofold higher value (0.34 gMeOH h−1 gcat−1) for the DI catalyst and levelled to a 25% higher value (0.20 gMeOH h−1 gcat−1) for the CP catalyst. The stable behavior of the former contrasts palladium-promoted In2O3 prepared through DI, which experienced fast deactivation (Supplementary Fig. 1)31. The inferior performance of the CP catalyst is likely due to the synthesis approach burying a substantial portion of the promoter within the bulk of In2O3, but the presence of more nickel added by this method did not yield materials superior to those produced by DI (Supplementary Fig. 2). Considering the more favorable DI synthesis (Fig. 1b), methanol formation was progressively lowered in favor of the RWGS reaction up to a nickel content of 20 wt.%, while the CO2 conversion was practically unchanged at ca. 8%, suggesting that sites activating CO2 are unlikely located on the nickel phase. Methane formation was observed only at higher promoter contents (SCH4 = 6 and 13% at 15 and 20 wt.% nickel, respectively). This hints that In2O3 exerts a strong influence on the nickel properties and, only for high contents, a fraction of promoter remains unperturbed and can express its intrinsic behavior. To further address the selectivity switch, the amount of indium required to trigger it was explored by supporting 5 wt.% nickel on mixed indium-aluminum oxides with variable indium content (0–100 mol% In, Supplementary Fig. 3). While nickel on pure alumina was highly selective to methane (SCH4 > 98%), 25 mol% of indium in the support sufficed to suppress methanation almost entirely (SCH4 = 3%). At the same time, the CO2 conversion significantly dropped (from \(X_{{\rm{CO}}_{2}}=18\) to 4%), corroborating that indium-modulated nickel species are significantly less active compared to pure nickel.

a Space-time yield (STY) of methanol as a function of time-on-stream (TOS) over Ni-In2O3 containing 1 wt.% of nickel incorporated by dry impregnation (DI) and coprecipitation (CP) and unpromoted In2O3 as a reference. b CO2 conversion and product selectivity of catalysts produced by DI as a function of the nominal nickel content on In2O3. Reaction conditions: T = 553 K, P = 5 MPa, molar H2:CO2 = 4, and WHSV = 24,000 cmSTP3 h−1 gcat−1.
Characterization of nickel 'speciation
In-depth investigations were conducted to rationalize the behavior of the DI systems. Concerning catalyst reducibility, temperature-programmed reduction with hydrogen (H2-TPR, Fig. 2a, Supplementary Table 2) evidenced that NiO conversion into metallic nickel occurs at ca. 340 K in all samples, preceding surface In2O3 reduction (370 K), which is substantially facilitated compared to the pure oxide (521 K). Based on the signal intensity in the profile of the 5Ni-Al2O3 reference, nickel was fully reduced in all cases. In 1Ni-In2O3, some In2O3 still reduced at its standard temperature, likely because the nickel amount was insufficient to facilitate reduction of the entire In2O3 surface. Diffuse reflectance infrared Fourier transform spectroscopy of adsorbed carbon monoxide (CO-DRIFTS) evidenced a weak signal specific to linearly bound CO (2176 cm−1) only for 10Ni-In2O3 and 15Ni-In2O3 (Fig. 2b), while pronounced bands of linear, bridged, (2119 cm−1), and three-fold (2066 cm−1) adsorbed CO was detected for the 5Ni-Al2O3 reference. Temperature-programmed desorption of carbon monoxide (CO-TPD, Fig. 2c) corroborated that the contribution of nickel to the adsorption of this molecule is significantly inferior when this metal is in contact with In2O3. These findings suggest remarkable electronic effects, i.e., metal-support interactions and/or alloying of nickel and indium37, implying high dispersion, for the majority of nickel present, and small particle size for nickel unaffected by indium oxide adsorbing CO.

a H2-TPR at 5 MPa of pressure, b CO-DRIFTS, and c CO-TPD of Ni-In2O3 catalysts prepared by DI containing 1–15 wt.% nickel, with unpromoted In2O3 and 5Ni-Al2O3 serving as references. Deconvoluted signals specific to nickel reduction and oxygen vacancy formation on In2O3 are colored in green and blue in (a). a.u. = arbitrary units.
To shed further light on the promoter’s features, catalysts were analyzed by additional methods. Scanning transmission electron microscopy coupled to energy-dispersive X-ray spectroscopy (STEM-EDX, Fig. 3a) revealed highly and almost homogeneously dispersed nickel in fresh lower-content specimens (1 and 5 wt.% Ni) and more agglomerated structures in fresh higher-content samples (15 and 20 wt.% Ni). Only in 20Ni-In2O3, some of the promoters appeared fully segregated from indium oxide. Slight nickel agglomeration is evident for all samples upon use in the reaction. Investigations by high-resolution transmission electron microscopy (HRTEM) of used materials did not visualize any nickel-based phases in 1Ni-In2O3 (Fig. 3b), although the magnification was chosen such that, based on the STEM-EDX results, some nickel must have been present within the imaged areas. In the 5 wt.% Ni sample, some amorphous islands can be found on In2O3, which might tentatively correspond to nickel-rich structures, due to their lower contrast compared to In2O3. In 15Ni-In2O3, a similar phase forms a ca. 1-nm thick layer covering many of the imaged In2O3 particles, which are additionally accompanied by poorly crystalline nickel-based particles in the 20 wt.% catalyst.

a STEM-EDX maps of Ni-In2O3 catalysts containing 1–20 wt.% Ni in fresh and used forms. b HRTEM images, with phases identified by fringe analysis and features of interest indicated, and c k-weighted Ni K-edge XANES, with spectra of NiO and metallic nickel serving as references, for the same used catalysts. d XRD patterns collected in monochromatic light and e EXAFS with model fit and an indication of neighbors’ number along a specific scattering path for selected fresh and used samples. f Percentage of In2O3 surface not covered by nickel as a function of the nominal nickel content, determined by volumetric CO2 chemisorption. The qualitative trend respective to nanoparticle formation and the coverage predicted by DFT is indicated. a.u. = arbitrary units.
X-ray absorption near-edge structure spectra (XANES, Fig. 3c) of fresh 15 and 20Ni-In2O3 closely resemble that of Ni2+ in NiO, while some discrepancies are observed for the lower-content solids, which can be explained by higher dispersion and stronger interaction with In2O3. In all but the highest nickel content catalyst, cationic nickel completely transformed into metallic species different from those in pure nickel metal upon use in the reaction, strongly suggesting alloying with indium. In 20Ni-In2O3, nickel is present in both metallic and oxidic states, likely because some NiO particles do not fully reduce due to their large size32.
High-resolution X-ray diffraction (XRD, Fig. 3d, Supplementary Fig. 4) measured in monochromatic light (λ = 0.49292 Å) indicated that NiO, present in the fresh materials, reduced indeed forming InNi3 alloys. Their average crystal size is <1, 2.7 ± 0.7, and 7.3 ± 0.9 nm for the catalysts containing 1, 5, and 15 wt.% nickel, respectively, in good agreement with the microscopy analyses. Alloy formation with indium shall be key to anchor the intermetallic phase strongly to the catalyst surface, preventing significant sintering. Only for 15Ni-In2O3, weak reflections specific to metallic nickel nanoparticles (ca. 1.2 nm) were also detected, confirming the origin of methane production over this catalyst. Since this analysis necessitated catalyst operation directly within the capillary used for diffraction analysis and the consequent application of significantly lower flow rates than in the reactor, some NiO is still detected in the used solids. However, other characterization techniques mirroring the reaction environment more closely exclude that this phase exists upon reaction for these catalysts.
Analysis of the extended X-ray absorption fine structure (EXAFS, Fig. 3e, Supplementary Table 3) shows Ni–Ni and Ni–In bonds in the first coordination shell of nickel. The total number of first neighbors around nickel is 5.9, 6.5, and 8.6 for 1, 5, and 15Ni-In2O3, respectively. Since 12 neighbors are expected for nickel atoms in the bulk metal and the InNi3 alloy, the alloy on all catalysts shall be well dispersed. Moreover, the number of Ni-Ni bonds progressively increases with higher nickel contents, i.e., 1.4, 2.4, and 3.7 for 1, 5, and 15Ni-In2O3, respectively. This is indicative of stronger nickel interaction with indium and suggests a two-dimensional layer-like morphology for the for the catalysts with lower nickel contentst, whereas particles additionally exist in higher-content samples.
Volumetric CO2 chemisorption enabled to determine a linear decrease of the exposed In2O3 surface at increasing nickel content, consistent with a Stranski–Krastanov film growth (layer followed by nanoparticle formation)38. Indeed, an asymptotic decrease would be expected if nickel species formed agglomerates following a Volmer-Weber film growth39 (Fig. 3f), as previously observed for the Pd-In2O3 system31. Although In2O3 should be fully covered at a nickel content of 12.5 wt.% (Supplementary Table 4), it is likely that inhomogeneous precursor distribution upon impregnation and the large anisotropy of the In2O3 surface prevented the formation of a perfectly uniform layer. Hence, ca. 40% of the In2O3 surface remained still exposed at a nickel content of 15 wt.%.
With a sound understanding of the DI systems, their structure was further investigated by first principles density functional theory. To represent the two-dimensional nickel phase, Ni(111) layers were placed on top of In2O3(111), identified previously and herein as the most abundant indium oxide termination (Supplementary Table 5)18. Based on the stability of nickel atoms with six neighbors each and to commensurate the oxide lattice (Supplementary Table 6), a slightly compressed nickel layer with 36 atoms per unit cell of In2O3 was considered more relevant than uncompressed layers with fewer nickel atoms (25–27). Interestingly, upon relaxation, nickel atoms in this discrete layer became more densely packed and extracted oxygen atoms from the underlying In2O3 (Fig. 4, Supplementary Movie 1). The driving force for this reconstruction can be traced back to the relative bond strengths, since Ni–In affinities are higher with respect to those of In-In and Ni-Ni (Supplementary Table 7), and is in line with other hydrogenation systems for which distinct but pronounced metal-support interactions were described, such as Cu-ZnO, Pt-TiO2, Pt-CeO2, and Pd-In2O3 (Supplementary Figs. 5 and 6, Supplementary Tables 8 and 9). The oxygen atoms extracted emerge to the outermost catalyst surface and are readily stripped as water by the hydrogen present in the reaction environment, which rationalizes the alloying of nickel with indium. Considering these dynamics, the oxygen atoms on the In2O3 termination were removed prior to placing the nickel layer. The most stable structure was found to be Ni36-In2O3−v, which presents the maximal number of oxygen vacancies (v) on the oxide and nickel atoms (12 and 36, respectively, Supplementary Fig. 7). Still, since it cannot be excluded that a small fraction of the oxygen atoms that emerged at the outermost surface is dynamically stored on the InNi3 patches upon CO2 hydrogenation, the effect of residual four oxygen atoms at the most stabilized positions was also explored (Ni36O4-In2O3−v). Finally, since the presence of isolated nickel species can also not be discarded, nickel atoms were alternatively deposited onto In2O3(111), i.e., Nix-In2O3, x = 1–4. These Nix-In2O3 structures are less stable than the nickel layer but more stable than NiO under a reducing atmosphere such as upon CO2 hydrogenation. For instance, Ni2-In2O3 is less stable than Ni36-In2O3−v by + 0.36 eV per nickel atom. Overall, the catalysts with up to 10 wt.% nickel are better represented by a combination of Ni36-In2O3−v, InNi3(111), and In2O3(111), while for higher-content materials InNi3(111) and Ni(111) terminations, along with potentially Ni2-In2O3 (vide infra), are additionally significant40.

The sketches show the restructuring of the interface between a nickel layer and the underlying In2O3. The pristine oxide surface is covered with a commensurate nickel layer, in this case containing 25 atoms. This structure is relaxed following a standard energy minimization algorithm. Given the corrugation of the In2O3 surface and the Ni-O and Ni–Ni relative binding energies, some oxygen atoms are stripped from the oxide (colored purple) and isolated hexagonally arranged Ni-patches are formed upon the relaxation. The dashed lines in the models indicate the border of the unit cell of In2O3 along with the (111) termination. The relaxation process is shown in Supplementary Movie 1.
Reaction mechanism and kinetics
To explain the reactivity of the DI catalysts, the adsorption energies (Ea) of reactants on the distinct models were calculated (>200 adsorption calculations, Fig. 5). Ni(111) is associated with a mildly endothermic CO2 adsorption, which can still be overcome (0.22 eV, Fig. 5), and barrierless hydrogen splitting and will readily transform carbon-based adsorbates into methane. In contrast, CO2 adsorption is significantly weakened on clean InNi3(111) (0.74 eV, Fig. 5), while hydrogen splits more exothermically and without an energy barrier. The chemical behavior of all layered nickel structures simulated is very similar to InNi3(111) (Supplementary Fig. 8). Hence, any alloy type shall be virtually inert in converting CO2 into any product on its own, but can provide hydrogen to the active site of In2O3(111) for coverages lower than one monolayer18,31. Nevertheless, the bulk alloy is expected to provide hydrogen radicals at a faster rate than the layer, due to its overall more metallic character. Even considering the presence of residual oxygen atoms upon catalyst operation, CO2 adsorption remains far weaker on the nickel layers than on In2O3(111) or Ni(111). The structure retaining some oxygen atoms (Ni36O4-In2O3−v) features inhibited CO adsorption compared to Ni36-In2O3−v. Considering the suppressed CO adsorption ability of the catalysts in CO-DRIFTS and CO-TPD analyses, it is conceivable that some oxygen or hydroxide species populate the alloy film during reaction.

Visualization of the structures of Ni-In2O3 catalysts with 1 and 15 wt.% nickel content loading (top), surfaces representative of the different catalyst constituents generated by DFT (middle), and adsorption energies (Eads) of species relevant to the reaction associated with them (bottom). The multiple energy levels for individual species for models containing nickel layer relate to the presence of a distinct number of oxygen atoms. The subscript v stands for vacancy. CO2 adsorption at the boundary between unpromoted In2O3(111) and a strip of Ni(111) (layer equivalent to Ni36-In2O3−v) is shown in Supplementary Fig. 11.
When considering low-content samples, the CO2 hydrogenation performance can be seen as the convolution of that of a (multi)layer of InNi3, pure In2O3, and nickel dimers on In2O3. The unpromoted In3O5 ensembles adsorb CO2 and activate H2 heterolytically18. Then, hydrides and protons are transferred to CO2 forming methanol. On InNi3(111) and Ni36-In2O3−v, CO2 and CO adsorption are either endothermic or weaker than on In2O3, whereas homolytic H2 adsorption is exothermic and CO2 activated at the In3O5 ensemble on clean In2O3(111) shall be hydrogenated both with hydrogen split on the same active site and with hydrogen spilled from nickel layers16, whereby sites at the periphery of the patches will be most relevant in the latter process (Supplementary Fig. 9). The fact that methanol selectivity is higher in nickel-poor catalysts than in nickel-rich ones suggests that hydrides and protons generated on In2O3 are still quite strongly utilized, since hydrogen radicals produced on the alloy favor both methanol and CO formation. In contrast to alloyed phases, low-nuclearity nickel clusters at the In3O5 ensemble, in particular Ni2-In2O3, are expected to be highly active in the competing RWGS (Supplementary Figs. 10 and 11), in striking contrast to low-nuclearity palladium clusters anchored to the same ensemble.
To corroborate the DFT findings, kinetic analyses were carried out over all catalysts to experimentally assess the mechanistic origin of the promotional effect (Fig. 6a). The apparent activation energies for both methanol synthesis and the RWGS reaction, extracted from catalytic tests conducted at variable temperature (Supplementary Fig. 12), are significantly lower already upon incorporation of the smallest nickel amount in comparison to pure indium oxide (from 101 to 83 and from 116 to 92 kJ mol−1, respectively). The smaller difference between activation barriers for methanol and CO synthesis over 1Ni-In2O3 rationalizes its lower methanol selectivity compared to the unpromoted oxide (from 15 to 9 kJ mol−1). A further increase of the nickel content has little impact on the activation energies, which reach values of 84 and 80 kJ mol−1 for the RWGS and methanol synthesis for 20Ni-In2O3 respectively. The activation barriers for methanation on the materials active for this reaction (15 and 20Ni-In2O3) were determined at ca. 90 kJ mol−1, in good agreement with literature on pure nickel catalysts (85–94 kJ mol−1)35,41,42 and corroborating the feasibility of methane formation on metallic nickel sites in these systems. Based on the weak dependence of the apparent activation energies for methanol and CO formation on the nickel content, the mechanisms leading to these products are likely highly similar throughout the materials. Hence, the progressive decay of methanol selectivity in favor of CO at higher loadings (Fig. 1b) has a kinetic origin. Since DFT calculations pointed to facilitated hydrogen activation as the origin of the promotional effect, apparent reaction orders respective to this reactant were determined from experiments at variable H2 concentrations in the feed (Fig. 6b, Supplementary Fig. 13). For methanol synthesis, they decreased by equal amounts for all systems relative to bulk In2O3, from 0.8 to 0.5, in line with the stoichiometric coefficient of hydrogen splitting on the InNi3 layers (½H2 → H*). At higher partial pressures of H2, the chemical potential of H* species adsorbed on the alloy layers increases as well, thus promoting H* spillover towards the In3O5 active site. The 0.5 reaction order suggests that the spillover mechanism dominates over the In3O5 on-site splitting at working conditions. For the RWGS reaction, the reaction orders increased from −0.7 to −0.3 for 1 and 5Ni-In2O3, i.e., the systems which contain mainly flat InNi3 structures. On these catalysts, both methanol and CO are produced on unpromoted In3O5 ensembles with the two paths competing for H* donated by the alloy patches. The negative reaction orders for the RWGS are explained by its first step (CO2 + H*→COOH) being kinetically unfavored compared to the first of methanol production (CO2 + H*→HCO2, Supplementary Fig. 11a). The progressive, decrease in apparent activation energy for the RWGS reaction upon increasing Ni content might be explained based on the formation of additional metastable Ni1-In2O3 and Ni2-In2O3 ensembles selective to this competitive reaction (Supplementary Figs. 10 and 11). On Nix-In2O3 ensembles, H2 splitting is barrierless, and thus the net reaction is not controlled by the partial pressure of H2. At Ni contents of 15–20 wt.%, the RWGS reaction shall be mostly performed on these ensembles rather than on those free of nickel, and the reaction order with respect to H2 decreases to zero. Overall, the kinetic data are in good agreement with earlier and above-presented investigations. Specifically, when H2 is activated on pure In2O3, it is split into polarized species (Hδ– and Hδ+) which are adsorbed on the In3O5 ensemble on an In3δ+ substructure and Oδ– atom respectively18. The subsequent transfer to adsorbed CO2 is energetically disfavored due to the strong polar interaction between adsorbed H2 and the In2O3 surface. Neutral hydrogen atoms, provided by the alloy phases, do not have to overcome this energy barrier, thus leading to lower activation energies for methanol synthesis and the RWGS reaction32. However, consecutive proton and hydride supply to adsorbed CO2 are highly selective towards methanol, whereas uncharged species foster both methanol and CO formation. Consequently, methanol synthesis cannot be enhanced beyond a certain threshold even if more homolytically split hydrogen is provided by more abundant alloy phases. On the contrary, the RWGS reaction is boosted to a greater extent in the presence of high nickel contents, presumably as a consequence of an excessive supply of hydrogen atoms (Fig. 6c).

a Apparent activation energies (Ea) and b reaction orders respective to H2 (\(n_{{\rm{H}}_2}\)) for methanol synthesis and the RWGS reaction over In2O3 catalysts as a function of their nickel content. c Graphical representation of the structures of In2O3 and Ni-In2O3 catalysts with low and high nickel loadings and indications where products shall be predominantly formed on them. The rings surrounding the molecules represent the respective product selectivity. Reaction conditions: T = 553 K, P = 5 MPa, WHSV = 24,000 cmSTP3 h−1 gcat−1.
Discussion
Herein, the use of nickel as an economically attractive promoter for In2O3 in the direct hydrogenation of CO2 to methanol was studied in fundamental and applied terms. Considering facile synthetic strategies, dry impregnation led to more stable and active catalysts than coprecipitation. Methanol synthesis was boosted along with the RWGS reaction to some extent and no methane was formed below a nickel content of 10 wt.%, despite the known high CO2 methanation activity of nickel nanoparticles. In-depth characterization revealed a two-dimensional InNi3 phase highly dispersed on In2O3 in nickel-lean samples, which is accompanied by nanoparticles of the same alloy as well as metallic nickel at progressively higher promoter contents. The formation of layered structures rather than agglomerated particles, due to peculiar wetting properties of nickel on In2O3 fostering film growth, and their strong anchoring on the oxide via alloying emerged as key contributors to the high catalyst stability. DFT simulations elucidated that indium-modulated nickel layers easily provide homolytically split hydrogen to In2O3, enhancing oxygen vacancy formation and contributing to CO2 hydrogenation, while barely activating CO2 on their own, which overall explains the beneficial effects and the lack of methane generation. Hydrogen radicals spilled from the InNi3 phase can concomitantly support methanol and CO formation, while hydrides and protons produced on In2O3 preferably mediate methanol production. The variable relevance of the former species at distinct contents rationalizes the product distribution and kinetic parameters experimentally determined across all samples. The catalyst comprising 1 wt.% of nickel offers an optimal balance between charged and radical hydrogen atoms, reaching a doubled methanol STY compared to unpromoted indium oxide. Overall, this study identified key structural and electronic features controlling the performance of the classical hydrogenation metal nickel in contact with indium oxide relevant to attain a stable promoted system for a sustainable application. It also highlights that the atomic engineering of a promoter for In2O3 is strongly metal-specific, even when similarity in behavior is expected for elements belonging to the same group in the periodic table.
Methods
Catalyst preparation
Unpromoted and nickel-promoted (1–2.5 wt.% nickel) In2O3 catalysts were prepared via a (co)precipitation (CP) synthesis similar to one reported earlier31. In addition, 1–20 wt.% and 5 wt.% nickel was added to pure indium oxide and to mixed indium-aluminum oxide supports with variable stoichiometry (0–100 mol% indium), respectively, by a dry impregnation (DI) method. The nickel-containing catalysts are labeled with the amount of promoter in wt.%, separated by a hyphen from the carrier, i.e., 5Ni-In2O3 indicates 5 wt.% nickel on In2O3. Details to all syntheses applied are provided in the Supplementary Methods.
Catalyst characterization
The metal content in the catalysts was determined by XRF, and porous properties of the catalysts were assessed by N2 sorption. Catalyst reducibility was monitored by temperature-programmed reduction in hydrogen (H2-TPR). CO adsorption was assessed by diffuse-reflectance Fourier transform infrared spectroscopy (CO-DRIFTS) and temperature-programmed desorption (CO-TPD). Nickel speciation, coordination geometry, and dispersion were accessed via X-ray absorption spectroscopy (XAS), X-ray diffraction in monochromatic light (XRD), STEM-EDX, and HRTEM. The surface area of In2O3 accessible to reactants was determined using volumetric chemisorption of CO2. Details to all characterization techniques are available in the Supplementary Methods.
Catalytic evaluation
The experimental setup used for catalytic testing is described in detail elsewhere18. Briefly, all experiments were performed in a high-pressure continuous-flow fixed-bed reactor with an inner diameter of 2.1 mm surrounded by an electric furnace. In a typical experiment the reactor was loaded with 100 mg of catalyst with a particle size of 75–100 µm, which was held in place by a bed of quartz wool and heated from ambient temperature to 553 K (5 K min−1) at 5 MPa under a flow of He (20 cm3STP min−1). After 3 h, the gas flow was switched to the reactants mixture (40 cm3STP min−1) comprising H2 and CO2 in a molar ratio of 4:1. To determine apparent activation energies, the reaction was initiated at 473 K and the temperature stepwise increased to 553 K (increments of 20 K). Reaction orders with respect to H2 were acquired applying a constant flow of CO2 (8 cm3STP min−1) and increasing the flow of H2 (from 20–32 cm3STP min−1, increments of 3 cm3STP min−1), while using He to balance the total flow to 40 cm3STP min−1. Ethane (0.5 cm3STP min−1, Messer, >99.9%) was added to the effluent stream to serve as an internal standard before the stream was sampled every 20 min and analyzed by online gas chromatography. The evaluation procedure of gas chromatography data is reported in the Supplementary Methods. Materials were tested for 16 h for performance comparison, catalyst stability was established over 72 h on stream, and, during kinetic tests, data were collected for 3 h at each condition and averaged. The absence of intra- and extraparticle diffusion limitation during kinetic tests were corroborated by the fulfillment of the Weisz-Prater and Carberry criteria.
Computational methods
DFT simulations were conducted with the Vienna ab initio simulation package (VASP) using the Perdew-Burke-Ernzerhof (PBE) density functional43,44,45. Core electrons were described by projector augmented-wave pseudopotentials (PAW)46, while valence electrons were expanded from a plane-wave basis set with a kinetic energy cutoff of 500 eV and a reciprocal grid size narrower than 0.025 Å–1. Bulk metal, intermetallic, and oxide structures relevant to investigate In2O3 promotion by nickel were modeled from their stable structures at ambient conditions. All bulk structures were fully relaxed and formation energies were obtained taking the bulk elements and gas-phase O2 as reference. Spin-polarization was considered for Ni-containing systems.
The most abundant termination of bixbyite In2O3, the (111) surface16,18, was modeled as a p(1 × 1) slab containing five O-In-O trilayers. The two outermost layers were allowed to relax and the three bottommost layers were fixed in their bulk positions. This surface is 14.56 Å wide, corrugated, and highly anisotropic. It features a protrusion, which is the active site for CO2 hydrogenation to methanol. To represent In2O3 catalysts with low nickel contents, a nickel atom was adsorbed on the pristine In2O3(111) surface between three oxygen atoms of the protrusion at symmetrically inequivalent positions. This process was repeated for low-nuclearity clusters containing 2–4 nickel atoms. Finally, 1–3 oxygen vacancies were created to check the ability of these clusters to favor oxygen abstraction. Besides, different nickel layers were accommodated onto In2O3 deriving from the 5 × 5, 3\(\sqrt 3\) × 3\(\sqrt 3\), and 6 × 6 expansions of a Ni(111) monolayer, containing 25, 27, and 36 Ni atoms, respectively. Each layer was placed on In2O3(111) surfaces with 0, 1, 2, 3, 6, 9, and 12 vacancies considering three different translations. To describe In2O3 catalysts with high nickel contents, the InNi3(111) and Ni(111) surfaces were also tested. The mechanism and energetics of CO2 hydrogenation were investigated considering the adsorption of relevant species and full reaction paths18,31. Transition states were obtained from the climbing image nudged elastic band (CI-NEB)47 and improved dimer method (IDM)48. Details to the calculations specific to surfaces containing low-nuclearity clusters and to metal-support interactions are provided in the Supplementary Methods.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information file. The DFT data are accessible at the ioChem-BD database at https://doi.org/10.19061/iochem-bd-1-183. All other relevant source data are available from the corresponding author upon reasonable request.
References
van Santen, R. A. Chemical basis of metal catalyst promotion. Surf. Sci. 251-252, 6–11 (1991).
Bartholomew, C. H. & Farrauto, R. J. Fundamentals of Industrial Catalytic Processes. (Wiley, Hoboken, 2006).
Hutchings, G. J. Promotion in heterogeneous catalysis: A topic requiring a new approach? Catal. Lett. 75, 1–12 (2001).
Zaera, F. Shape-controlled nanostructures in heterogeneous catalysis. ChemSusChem 6, 1797–1820 (2013).
Pérez-Ramírez, J. & López, N. Strategies to break linear scaling relationships. Nat. Catal. 2, 971–976 (2019).
Dong, C. et al. Supported metal clusters: Fabrication and application in heterogeneous catalysis. ACS Catal. 10, 11011–11045 (2020).
Zhang, Z., Zandkarimi, B. & Alexandrova, A. N. Ensembles of metastable states govern heterogeneous catalysis on dynamic interfaces. Acc. Chem. Res. 53, 447–458 (2020).
Kondratenko, E. V., Mul, G., Baltrusaitis, J., Larrazábal, G. O. & Pérez-Ramírez, J. Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 6, 3112–3135 (2013).
Alvarez, A. et al. Challenges in the greener production of formates/formic acid, methanol, and DME by heterogeneously catalyzed CO2 hydrogenation processes. Chem. Rev. 117, 9804–9838 (2017).
González-Garay, A. et al. Plant-to-planet analysis of CO2-based methanol processes. Energy Environ. Sci. 12, 3425–3436 (2019).
Dias, V. et al. Energy and economic costs of chemical storage. Front. Mech. Eng. 6, 21 (2020).
Sun, K. et al. Hydrogenation of CO2 to methanol over In2O3 catalyst. J. CO2 Util. 12, 1–6 (2015).
Martin, O. et al. Indium oxide as a superior catalyst for methanol synthesis by CO2 hydrogenation. Angew. Chem., Int. Ed. 55, 6261–6265 (2016).
Frei, M. S. et al. Role of zirconia in indium oxide-catalyzed CO2 hydrogenation to methanol. ACS Catal. 10, 1133–1145 (2020).
Tsoukalou, A. et al. Operando X-ray absorption spectroscopy identifies a monoclinic ZrO2:In solid solution as the active phase for the hydrogenation of CO2 to methanol. ACS Catal. 10, 10060–10067 (2020).
Albani, D. et al. Semihydrogenation of acetylene on indium oxide: Proposed single-ensemble catalysis. Angew. Chem., Int. Ed. 56, 10755–10760 (2017).
Zhang, S. et al. Solid frustrated-lewis-pair catalysts constructed by regulations on surface defects of porous nanorods of CeO2. Nat. Commun. 8, 15266 (2017).
Frei, M. S. et al. Mechanism and microkinetics of methanol synthesis via CO2 hydrogenation on indium oxide. J. Catal. 361, 313–321 (2018).
Wang, L. et al. In2O3 nanocrystals for CO2 fixation: atomic-level insight into the role of grain boundaries. iScience 16, 390–398 (2019).
Ye, J., Ge, Q. & Liu, C.-J. Effect of PdIn bimetallic particle formation on CO2 reduction over the Pd-In/SiO2 catalyst. Chem. Eng. Sci. 135, 193–201 (2015).
Rui, N. et al. CO2 hydrogenation to methanol over Pd/In2O3: Effects of Pd and oxygen vacancy. Appl. Catal., B 218, 488–497 (2017).
Snider, J. L. et al. Revealing the synergy between oxide and alloy phases on the performance of bimetallic In-Pd catalysts for CO2 hydrogenation to methanol. ACS Catal. 9, 3399–3412 (2019).
Bavykina, A. et al. Turning a methanation Co catalyst into an In-Co methanol producer. ACS Catal. 9, 6910–6918 (2019).
Chou, C. Y. & Lobo, R. F. Direct conversion of CO2 into methanol over promoted indium oxide-based catalysts. Appl. Catal., A 583, 117144 (2019).
Han, Z., Tang, C., Wang, J., Li, L. & Li, C. Atomically dispersed Ptn+ species as highly active sites in Pt/In2O3 catalysts for methanol synthesis from CO2 hydrogenation. J. Catal. https://doi.org/10.1016/j.jcat.2020.06.018 (2020).
Jia, X., Sun, K., Wang, J., Shen, C. & Liu, C.-J. Selective hydrogenation of CO2 to methanol over Ni/In2O3 catalyst. J. Energy Chem. 50, 409–415 (2020).
Sun, K. et al. A highly active Pt/In2O3 catalyst for CO2 hydrogenation to methanol with enhanced stability. Green. Chem. 22, 5059–5066 (2020).
Wang, J., Sun, K., Jia, X. & Liu, C.-J. CO2 hydrogenation to methanol over Rh/In2O3 catalyst. Catal. Today https://doi.org/10.1016/j.cattod.2020.05.020 (2020).
Rui, N. et al. Hydrogenation of CO2 to methanol on a Auδ+-In2O3–x catalyst. ACS Catal. 11307–11317 (2020).
Li, M. M.-J. et al. Methanol synthesis at a wide range of H2/CO2 ratios over a Rh-In bimetallic catalyst. Angew. Chem., Int. Ed. 59, 16039–16046 (2020).
Frei, M. S. et al. Atomic-scale engineering of indium oxide promotion by palladium for methanol production via CO2 hydrogenation. Nat. Commun. 10, 3377 (2019).
Vogt, C. et al. Unravelling structure sensitivity in CO2 hydrogenation over nickel. Nat. Catal. 1, 127–134 (2018).
Hengne, A. M. et al. Ni-Sn-supported ZrO2 catalysts modified by indium for selective CO2 hydrogenation to methanol. ACS Omega 3, 3688–3701 (2018).
Károlyi, J. et al. Carbon dioxide reforming of methane over Ni–In/SiO2 catalyst without coke formation. J. Ind. Eng. Chem. 58, 189–201 (2018).
Jalama, K. Carbon dioxide hydrogenation over nickel-, ruthenium-, and copper-based catalysts: review of kinetics and mechanism. Catal. Rev. 59, 95–164 (2017).
Németh, M., Somodi, F. & Horváth, A. Interaction between CO and a coke-resistant NiIn/SiO2 methane dry reforming catalyst: a DRIFTS and CO pulse study. J. Phys. Chem. C. 123, 27509–27518 (2019).
Bergwerff, J. A. & Weckhuysen, B. M. in Handbook of Heterogeneous Catalysis Vol. 1 (eds. Ertl, G. et al.) 1188–1197 (Wiley-VCH, 2008).
Baskaran, A. & Smereka, P. Mechanisms of Stranski-Krastanov growth. J. Appl. Phys. 111, 044321 (2012).
Ernst, B. Phänomenologische Theorie der Kristallabscheidung an Oberflächen. Z. Kristallogr. 110, 372–394 (1958).
Göltl, F., Murray, E. A., Tacey, S. A., Rangarajan, S. & Mavrikakis, M. Comparing the performance of density functionals in describing the adsorption of atoms and small molecules on Ni(111). Surf. Sci. 700, 121675 (2020).
Van Herwijnen, T., Van Doesburg, H. & De Jong, W. A. Kinetics of the methanation of CO and CO2 on a nickel catalyst. J. Catal. 28, 391–402 (1973).
Weatherbee, G. D. & Bartholomew, C. H. Hydrogenation of CO2 on group VIII metals: II. Kinetics and mechanism of CO2 hydrogenation on nickel. J. Catal. 77, 460–472 (1982).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Heyden, A., Bell, A. T. & Keil, F. J. Efficient methods for finding transition states in chemical reactions: comparison of improved dimer method and partitioned rational function optimization method. J. Chem. Phys. 123, 224101 (2005).
Acknowledgements
Total Research & Technology Feluy is thanked for sponsoring this project. Dr. S. Mitchell and Dr. F. Krumeich are thanked for the electron microscopy measurements, and the Scientific Center for Optical and Electron Microscopy (ScopeM) at the ETH Zurich for the use of their facilities. We are grateful to Dr. Nicola Casati for performing the in situ XRD analyses. Dr. Marcos Rellán-Piñeiro is thanked for his input in the theoretical calculations. The Spanish Ministry of Science and Innovation RTI2018-101394-B-I00 project is acknowledged for financial support and the Barcelona Supercomputing Center – MareNostrum (BSC-RES) for providing generous computer resources.
Author information
Authors and Affiliations
Contributions
J.P.-R. and C.M. conceived and coordinated all stages of this research. M.F. and M.P. prepared and characterized the catalysts and conducted the catalytic tests. O.S. coordinated acquisition and performed the evaluation of X-ray absorption spectroscopy data. R.G.-M., J.M.-V., and N.L. conducted computational studies. J.A.S. and D.C.F. contributed setting industrial targets for the experimental program. All authors contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Dequan Xiao, Bert Weckhuysen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Frei, M.S., Mondelli, C., García-Muelas, R. et al. Nanostructure of nickel-promoted indium oxide catalysts drives selectivity in CO2 hydrogenation. Nat Commun 12, 1960 (2021). https://doi.org/10.1038/s41467-021-22224-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-22224-x
This article is cited by
-
Low-nuclearity CuZn ensembles on ZnZrOx catalyze methanol synthesis from CO2
Nature Communications (2024)
-
A bibliometric analysis of CO2 methanation: research trends and comprehension of effective catalysts
Journal of the Iranian Chemical Society (2024)
-
Nickel-modified In2O3 with inherent oxygen vacancies for CO2 hydrogenation to methanol
Science China Chemistry (2024)
-
Investigation of In Promotion on Cu/ZrO2 Catalysts and Application in CO2 Hydrogenation to Methanol
Catalysis Letters (2023)
-
Switching of CO2 hydrogenation selectivity via chlorine poisoning over Ru/TiO2 catalyst
Nano Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
