
Abstract
The coexistence of single-crystallinity with a multidomain morphology is a paradoxical phenomenon occurring in biomineralization. Translating such feature to synthetic materials is a highly challenging process in crystal engineering. We demonstrate the formation of metallo-organic single-crystals with a unique appearance: six-connected half-rods forming a hexagonal-like tube. These uniform objects are formed from unstable, monodomain crystals. The monodomain crystals dissolve from the inner regions, while material is anisotropically added to their shell, resulting in hollow, single-crystals. Regardless of the different morphologies and growth mechanism, the crystallographic structures of the mono- and multidomain crystals are nearly identical. The chiral crystals are formed from achiral components, and belong to a rare space group (P622). Sonication of the solvents generating radical species is essential for forming the multidomain single-crystals. This process reduces the concentration of the active metal salt. Our approach offers opportunities to generate a new class of crystals.
Similar content being viewed by others
Introduction
The properties and functionalities of crystals are strongly affected by their shape and structure. The shape-based relationship is manifested in both biological and synthetic systems. Single crystalline scaffolds having hierarchical architectures and curved features are widely used for structural purposes by many plants and animals (e.g., mollusks, corals, echinoderms, and algae)1,2. Sea urchins sculpt their spines by producing single crystals of calcite having a complex fenestrated morphology with smooth and curved surfaces3. Among synthetic crystals, the shape of nanoparticles has been shown to affect optical properties4,5 and impact mechanical properties as well as cell membrane permeability6. The latter property is important for their efficacy for drug delivery and other therapeutic applications6,7. However, unlike bio-crystallization, where crystal shaping is under cellular control, mastering of crystallization chemistry and kinetics is required for synthetic materials. Various parameters such as solvents and additives can control the evolution of crystal facets by selective surface interactions8,9,10. Interfacial synthesis11, microemulsion12, and template-assisted growth13 are transversally used in colloidal chemistry to enhance the performance of both organic14,15 and inorganic materials16,17 such as drugs and functional nanoparticles18,19.
The synthesis of morphologically tailored metal-organic frameworks (MOFs) is still in its early stages20. Although several works have shown that control over crystal size and shape can affect porosity21, catalytic activity22, and cellular uptake23, to date, the majority of the efforts have been aimed at designing the crystal structures of MOFs24. This fact has confined much of the ongoing research to applications that exploit the porosity of the molecular frameworks25,26. Such crystals have rarely been utilized as three dimensional objects27. The shaping processes are mainly restricted to crystals with canonical frameworks and their fundamental aspects are poorly understood20. Our group has recently shown that a variability of uniform metallo-organic crystals with different morphologies can be obtained28,29,30. These studies include the formation of a unique yoyo-shaped, single crystal exhibiting both a multidomain and chiral morphology30. The coexistence of such a set of properties broke the axiom “morphological single unit–monocrystallinity” by reproducing a phenomenon previously observed in bio-minerals31,32.
In this work, we introduce an unknown crystal shape (Fig. 1). The entire crystals are hollow and have a multidomain appearance. Their single-crystallinity and chiral packing is evident from detailed crystallographic studies. A straightforward but unconventionally additive-free synthesis is described here, where the key step is the sonication of the reaction solvents to reduce the active concentration of the metal salt. Incipient monodomain, single crystals are sacrificial templates for the formation of the thermodynamically favored multidomain crystals; their instability results from internal structural defects generated by speed-up growth kinetics. The multidomain crystals have curved morphological features and retain single-crystallinity.

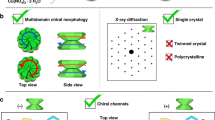
a Crystals consisting of six protruding units were obtained after 48 h by reacting the organic ligand (TPVA) and nickel(II) bromide at 105 °C in a mixture of DMF and CHCl3 (3:1 v/v). The solvents were first sonicated for 1.5 h prior to the addition of TPVA and the metal salt (TPVA:NiBr2 = 1:2). b Drawing of a X-ray diffraction pattern typical for single crystals. c Inside-out Ostwald ripening, resulting in large continuous channels and a multidomain morphology. d Isostructural crystals having a prismatic single domain (monodomain) morphology were formed without sonication.
Results
Formation of hollow crystals with a multidomain morphology
The tetrahydral organic ligand (TPVA) with nickel bromide (NiBr2) underwent a sonochemical–solvothermal treatment in a 1:2 molar ratio to form the hollow crystals. A mixture of dimethylformamide (DMF)/chloroform (v/v = 2:1) was sonicated for 1.5 h before the TPVA was added. Then, the solution containing the TPVA was mixed with a DMF solution of the metal salt in a glass pressure tube and heated at 105 °C for 48 h, resulting in the formation of a green precipitate (Fig. 1a). An excess of the metal salt was used: (1) to ensure the formation of a fully coordinative network (four pyridine units of four TPVA ligands are coordinated to one metal center, vide infra), and (2) to enhance morphological uniformity by surface saturation28. Sonication is known to result in the formation of radicals33,34, which affects crystal growth35. Indeed, electron paramagnetic resonance (EPR) measurements indicated the formation of radicals (vide infra, Supplementary Fig. 1) having a lifetime of ~45 min at room temperature, in the presence of the metal salt.
SEM images of the precipitate show the formation of hollow, multidomain structures (Fig. 2a–d and Supplementary Fig. 2). These crystals consist of six-connected half-rods that are positioned circularly. Regardless of their complex and highly unusual morphology, their uniformity is high (l = 35.7 ± 5.1 μm, ⌀out = 13.6 ± 2.3 μm). Morphologically, these half-rods are thinner at their terminal rounded edges and their external surfaces seem to exhibit three structurally similar subdomains. High-resolution SEM images of these rounded termini show large, microsized cavities having a fine-textured, inner surface (Figs. 2b and 3). The textures resemble the main veins of leaves. The entire structures are hollow, as proven by full volume reconstruction and cross-section analysis of micro-computed tomography (micro-CT) data (Figs. 2e and 4 and Supplementary Movie 1). The continuous channel is double-cone shaped; the diameter at the edges is substantially larger (~10 times) than that at the core. Relatively low electron-dense regions are present at the interfaces that connect the half-rods.

a–d Scanning electron microscopy (SEM) images and e micro-computed tomography (Micro-CT) volume rendering of the crystals (sonochemical–solvothermal conditions, 105 °C, t = 48 h). The color legend in e shows the Hounsfield units values, which are proportional to the amount of material.

a–e A series of ex situ scanning electron microscopy (SEM) images showing the stages in the temporal evolution of MOF-NiBr2 (sonochemical–solvothermal conditions, 105 °C). Left: zoom-out view and zoom-in micrographs (central, right).

Series of ex situ micro-computed tomography (Micro-CT) volume renderings (a, d, f) and scanning electron microscopy (SEM) images (b, c, e) showing snapshots of the formation of MOF-NiBr2 with time (sonochemical–solvothermal conditions, 105 °C). The cross-sections shown in b, c, e obtained with the focused ion beam (FIB) microscope are denoted by red lines and are numbered.
Single-crystallinity and its retention
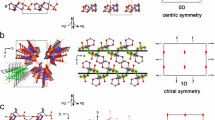
For diffraction analysis, isolated, entire crystals (sonochemical–solvothermal conditions, t = 48 h) were subjected to a 100 μm X-ray beam of a home-source diffractometer. This relatively large beam exposes the entire crystal. The observed diffraction patterns were characteristic of a typical single crystal, with well-defined lattice spacings as can be seen from the Ewald’s sphere projection (Fig. 5a). X-ray diffraction data were also obtained from synchrotron radiation (Fig. 5b), albeit, with a 30 μm beam. Again, the reflections could be indexed by a single domain orientation. These measurements did not indicate multiplicity or twinning. The single-crystallinity was consistent with diffraction patterns obtained from transmission electron microscopy (TEM). These crystals and initial crystals isolated after 1.5 h reaction time were analyzed by both powder X-ray diffraction (PXRD) (Fig. 5c and Supplementary Figs. 3a and 4) and by single crystal X-ray diffraction for structural determination at the molecular level (Fig. 5d, Supplementary Figs. 5 and 6, and Supplementary Table 1). The PXRD patterns of both crystals (t = 1.5 h and t = 48 h) are very similar, indicating that the final crystallographic structure is already established after a relatively short reaction time and is retained during the growth process (Fig. 5c, Supplementary Figs. 3a and 4b, c, and Supplementary Note 1). The diffraction collected from these entire crystals (obtained after 1.5 h) was also typical for single-crystallinity. Moreover, the crystallographic isostructurality is evident by the superposition of the TEM nanobeam electron diffraction pattern (t = 48 h) and the single-crystal diffraction pattern in the corresponding viewing direction based on the X-ray structure (t = 1.5 h) (Fig. 5e–g). Both crystals are isomorphous and their structures were solved by single-crystal X-ray crystallography (hexagonal space group, P622 with unit cell parameters t = 1.5 h: a = b = 25.719 Å, c = 17.870 Å versus t = 48 h: a = b = 25.961 Å, c = 17.818 Å). The uncommon space group, P622, is one of the 65 Sohncke groups; it indicates that the molecular components have chiral packing (Supplementary Table 1). The crystal structures were solved and refined to an atomic resolution of 1.19 Å (t = 1.5 h) and 1.10 Å (t = 48 h) with a final R factor of 0.0762 (t = 1.5 h) and 0.0941 (t = 48 h) for [I > 2σ(I)]. The Flack parameters are similar 0.11(9) (t = 1.5 h) and 0.13(8) (t = 48 h), and indicate that a small amount of enantiomeric twinning could be present in this crystal. Circular dichroism measurements of dispersions of these two crystals in ethanol show a zero signal, indicating there is no enantiomeric excess in these bulk samples (Supplementary Fig. 7). Apparently, the bulk samples consist of a racemic mixture. The achiral components (TPVA and the nickel bromide) are arranged in a complex, continuous network consisting of different helicoids forming supramolecular homochiral channels (Supplementary Figs. 5 and 6). The diameters of these hexagonal (⌀ ≈ 9.1 Å) and trigonal (⌀ ≈ 11.6 Å) channels can be observed along the c-axis. The bivalent nickel centers have an octahedral geometry, with four pyridine moieties of four TPVA ligands in the equatorial positions (t = 1.5 h: Ni–N = 2.021(15) Å and 2.037(14) Å versus t = 48 h: Ni–N = 2.100(8) Å and 2.08(11) Å). Two water molecules are bound with their oxygen atoms in the axial positions (t = 1.5 h: Ni–O = 2.451(0.007) Å versus t = 48 h: Ni–O = 2.427(0.006)). The Ni–N bond distances and lengths are within the range commonly found for coordinately saturated nickel complexes. However, the axial position is longer than usual (the Ni–O distances for analog pyridine-based complexes is ~2.08 Å)36. The four pyridine moieties around the metal center are arranged in a chiral, propeller-type conformation, which is most likely the origin of the chirality (Supplementary Fig. 8). The unit cell contain six of these propeller arrangements with the same handedness.

The data shown are for crystals obtained by sonochemical–solvothermal conditions (105 °C). a Ewald sphere projections of MOF-NiBr2 (t = 48 h) down the a*, b*, and c* axes, b X-ray experimental diffraction 1 degree frame of the entire crystal of MOF-NiBr2 (t = 48 h). c Powder X-ray diffraction (PXRD) of MOF-NiBr2, t = 1.5 h (red line) and t = 48 h (blue line). d Single-crystal X-ray structure of MOF-NiBr2 (t = 1.5 h). e Nanobeam electron diffraction of MOF-NiBr2 (t = 48 h). f Kinematical zone axis diffraction patterns calculated from the single-crystal X-ray data shown in d of MOF-NiBr2 (t = 1.5 h), using electron scattering factors. g Comparison between the diffraction data shown in e and f. Deviating intensities are related to dynamical scattering into reflections that are a sum of reflections in the same zone axis pattern (Supplementary Fig. 16d).
Elemental analyses were performed on the crystals showing that these materials have a similar composition (Supplementary Table 2; sonochemical–solvothermal and solvothermal conditions). Only traces (0.32–0.65%) of bromide (Br−) have been observed. The positive charges of the metal centers are balanced by chloride (Cl−), as indicated by an average ratio of Cl/Ni = 2.2. These anions (from chloroform)29,30,34,37,38,39 are not resolved as part of the crystallographic structures. The reaction with NiCl2 and TPVA under solvothermal conditions resulted in uniform, sub-micron-sized crystals having a different morphology than observed in this study with NiBr239. This observation indicate that the bromide–chloride anion exchange happened after coordination of the TPVA to the NiBr2.
From mono- to multidomain single crystals
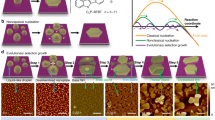
In order to follow the morphological transformation, a series of ex situ scanning electron microscopy (SEM) measurements and micro-CT measurements were carried out by stopping the solvothermal reaction at different reaction times (Figs. 3 and 4). Cross-sectional samples were prepared using a focused ion beam (FIB) instrument (Fig. 5b, c, e). Small parallelogram-shaped structures (side length 2.4 ± 0 .2 μm, 4.1 ± 0.3 μm) were observed after 30 min of reaction time (Fig. 3a). However, these structures were not present in samples analyzed after longer reaction times and that contained larger and elongated objects. These morphological changes were also accompanied by a structural rearrangement at the molecular level. Raman spectroscopy of the parallelogram-shaped structures (t = 30 min) and the final crystals (t = 48 h) showed clear differences (both shifts and signal shapes) in the ranges: ν = 950–1050 cm−1 (pyridine), ν = 1590–1620 cm−1 (pyridine), and ν = 1620–1650 cm−1 (C = C) (Supplementary Fig. 10). IR spectra show a signal intensity decrease at ν = 2750–3000 cm−1 (pyridine); during the course of the reaction a marked intense band appears at ν = 1660 cm−1 (pyridine) (Supplementary Fig. 11). These observations are consistent with Ostwald ripening40: small kinetically favored particles dissolve to form larger and thermodynamically favored structures. After 1.5 h, cylindrical-like crystals (vide supra) were formed having an apparent defect-free monodomain morphology and hexagonal-shaped termini (l = 29.9 ± 4.8 μm, ⌀ = 8.2 ± 2.0 μm) (Fig. 3b and Supplementary Fig. 9). The presence of a central concavity was visible at the termini. Micro-CT reconstruction revealed inner areas with relatively low electron densities and even empty regions (Fig. 4a and Supplementary Movie 2). After 3 h, the dimensions of the crystals increased both in length and diameter while maintaining a high level of uniformity (Fig. 3c and Supplementary Fig. 12). Enlarged cavities having diameters of ~1 μm were evident at the extremities. SEM analysis of the cross-sections, as well as the micro-CT data, showed that these cavities initially involved only the termini (Fig. 4b). Upon increasing the reaction times, the cavities propagated inside the cylindrical-like crystals, forming one double-cone-shaped channel (Fig. 4c, d). The diameter of these channels at the termini were ~15 μm after 48 h. Another striking morphological transformation was observed after 3 h: the channels were formed concurrently with the change in the external surface texture (Fig. 3b–e center images and Supplementary Figs. 2, 12, 13). The smooth surface gradually evolved into protruding areas by layered addition of materials (Supplementary Fig. 14), eventually resulting in six-connected half-rods after 48 h. These half-rods developed first in those areas that are closest to the termini via preferential deposition of material on the lateral edges (Supplementary Fig. 12, morphological feature highlighted by cyan and yellow lines).
Mechanistically some of the structural features (i.e., inner texture, a double-cone-shaped channel) suggest that material was redissolved during the growth process (Figs. 3 and 4). We assume that supramolecular objects undergo reorganization. The bond dissociation energy of nickel(II)–pyridine is ~43 kcal/mol and is prone to dissociation during heating41. The formation of the morphology seems strongly influenced by the balance between dissociation of weak bonds and redeposition. Plausible explanations are the presence of higher reactive regions (due to crystalline defects at the edges) and a higher degree of supersaturation (due to the diffusion of TPVA and metal salt from the dissolving crystal termini). The formation of the half-rods gradually progressed toward the center of the structure in parallel with the development of the double-coned-shaped channel. This multidomain shaping resulted in three structurally similar subdomains along each half-rod (clearly observed after 48 h). Linear engravings in the form of the main veins of leaves were evident after 24 h at the internal surfaces of the cavities close to the termini. Such features are typical of crystal erosion42,43. The crystal growth bears clear hallmarks of inside-out Ostwald ripening. The regular Ostwald ripening (~3 h) is followed by a relatively slow process whereby the initially formed defective monodomain crystals serve as both sacrificial templates and feedstock for the multidomain, single crystals.
Sonication as a means to control crystal morphology and reactivity
We found that under solvothermal conditions, without sonication, monodomain hexagonal prism are formed that have a morphology and crystallographic structure that closely resembled the initial, sacrificial crystals (Supplementary Figs. 9 and 15). The diffraction (single-crystal X-ray, PXRD, TEM-single-crystal X-ray comparison, Supplementary Figs. 3 and 16 and Supplementary Note 2) and optical properties (Raman and FT-IR, Supplementary Figs. 10 and 11) were nearly identical39. Although these monodomain, single crystals are isomorphous to the crystals obtained using sonochemical–solvothermal conditions (t = 1.5 h), they did not develop further into the multidomain structures. Micro-CT measurements of the hexagonal prisms (l = 9.9 ± 0.5 μm, ⌀ = 4.3 ± 0.4 μm) having six slightly bent facets (t = 48 h) indicated the formation of defect-free, solid crystals (Supplementary Fig. 17). The density distribution is nearly constant and no empty regions are observable. Ex situ SEM measurements of these solid crystals were carried out by stopping the solvothermal reaction at different reaction times (Supplementary Fig. 18). The measurements indicated an Ostwald ripening from parallelogram to prismatic structures and highly uniform crystals—akin to the formation of the initial crystals formed under sonication–solvothermal conditions. The main mechanistic difference between the solvothermal and sonication–solvothermal conditions that we observed was the reactivity of the parallelogram-shaped structures formed at the beginning of the reactions (t = 30 min).
Sonication–solvothermal conditions accelerate the dissolution of parallelogram-shaped crystals to form defective cylindrical crystals as observed by micro-CT (Fig. 4a and Supplementary Movie 2) that further transferred into the hollow and multidomain single crystals. At a much earlier stage, the formation of elongated crystals was observed by using sonicated solvent (sonication–solvothermal: ~1.5 h versus solvothermal: ~24 h) (Fig. 3 and Supplementary Figs. 18–20). Sonication of the solvent prior to the solvothermal reaction is the key to the formation of the multidomain, single crystals. To further verify this observation, isolated parallelogram crystals obtained under solvothermal conditions were added to the sonicated solvent and converted to the hollow and multidomain single crystals (albeit also other structures were observed; Supplementary Fig. 21). Likewise, isolated parallelogram crystals obtained under sonochemical–solvothermal conditions were added to non-sonicated solvent and formation of the monodomain, hexagonal prism were observed (Supplementary Fig. 22).
EPR measurements were carried out after the sonication of the solvent mixture; they showed the formation of DMF radicals having a lifetime of ~45 min at room temperature, even in the presence of NiBr2 (Supplementary Fig. 1)33,34. Such metal salts are known to interact with analogous radical species44. In order to further elucidate the role of the sonication pretreatment, we hypothesized that radicals (or products therefore) could reduce the amount of reactive NiBr2. Indeed, the formation of hollow, multidomain crystals have been observed by SEM when lowering the concentration of NiBr2 by 50% or more under solvothermal conditions after 48 h (Supplementary Fig. 23). An excess of metal salt is known to stabilize and direct the formation of organic crystals with ligands that are structurally similar to TPVA29. Inactivating pyridine coordinating sites by excess of metal salts can be the cause of the here observed differences in growth kinetics (Fig. 3 and Supplementary Figs. 18–20). The faster growth kinetics of the unstable, cylindrical structures (sonochemical–solvothermal conditions, t = 1.5 h) is consistent with the presence of low density inner regions. Saturation of the crystal surface by the metal salt can have a stabilizing effect, hence the Ostwald ripening (excess of metal) versus the inside-out Ostwald ripening pathways.
Thermogravimetric analysis (TGA) under nitrogen reveals similar thermal stabilities of the prismatic crystals separated under the solvothermal conditions and the hollow and multidomain single crystals synthesized under sonication–solvothermal conditions (for both crystals, t = 48 h). These observations indicate that the stability of the isostructural crystals is related to the 3D metal-organic networks, and not affected by the different morphology or defects (Supplementary Fig. 24).
Discussion
Single-crystallinity emerges first, whereas the multidomain and curved appearance occurs afterwards. The complexity of the formation includes different processes that are dominant during specific time periods. Prismatic crystals formed at relatively early stages undergo a series of transformations (dictated by factors such as metal salt concentration, structural defects, and local supersaturation changes), finally resulting in the multidomain, single crystals. Despite the complex molecular packing, the crystallographic structure neither changes nor develops polycrystallinity properties during the single-to-multidomain morphological transformation. Our new findings confirm the idea that such chiral, metallo-organic crystals are not oddities, but rather are among the first examples of a new class of paradoxical and isomorphous materials with concurrently extraordinary shapes and sought-after crystalline properties (e.g., single-crystallinity, porosity, and chirality)30. Our previously reported yoyo-shaped, single crystals are formed from a copper nitrate salt and a poorly soluble ligand30, structurally similar to the here reported tetrahedral achiral ligand (having carbon–carbon triple bonds instead of double bounds). Despite differences in bond order and solubility, these crystals are isostructural and belong to the rare P622 space group. These findings might indicate that the octahedral molecular geometry around the metals center having four pyridine groups in plane is a key factor to the molecular packing of such crystals, whereas their complex and highly unusual morphologies are determined mainly by other factors including metal-to-ligand ratios, reaction time, and growth kinetics. Sonochemistry has been used for the formation of MOFs45,46. Typically, the organic constituent and metal salt are mixed together in a solvent and exposed to high-energy ultrasounds. The product formation has been related to local increases of temperature and pressure by implosion of cavitation vacuum bubbles. Our finding indicates that sonication can also change the concentration and ratios of the active components resulting in morphologically different structures.
Methods
Materials
Chloroform (CHCl3, ≥99.8%) and DMF (≥99.8%) were obtained from Sigma Aldrich and J.T. Baker, respectively. NiBr2 (>98.0%) was obtained from Sigma Aldrich. Reagents were used without further purification. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) (>98.0%) was obtained from Cayman Chemical Co. and used as a spin trap. Glass pressure tubes (Ace Glass, Inc., pressure tubes #15 with a plunger valve, PTFE Bushing and FETFE® O-Ring, volume 50 ml) were cleaned by immersion in a H2SO4/30% H2O2 piranha solution (7:3 v/v) for 10 min. Subsequently, they were washed with deionized water and dried in an oven for 24 h at 130 °C. Caution: piranha is an extremely dangerous oxidizing agent and should be handled with care using appropriate personal protection. 1,3,5,7-Tetrakis{4-[(E)-2-pyridin-4-yl-vinyl]phenyl} adamantane (TPVA) was prepared according to a literature procedure47.
Preparation of the MOFs
Sonochemical–solvothermal crystallization: a mixture of CHCl3 (1.0 ml) and DMF (2.0 ml) was sonicated in a glass vial (20 ml) with a DCG-120H MRC professional ultrasonic cleaner in normal mode (33–40 KHz frequency) for 1.5 h in an ice-bath. Subsequently, this solution was used immediately to dissolve TPVA (3.0 mg, 3.5 mmol, conc. 1.2 mM). The metal salt was dissolved in DMF (5.0 mg of NiBr2 in 3.3 ml DMF, metal salt conc. = 7.0 mM). The solution of TPVA (3.0 ml, 3.5 mmol) and 1.0 ml of the solution of the metal salt (7.0 mmol) were mixed in a glass pressure tube (the final concentrations of TPVA and the metal salt in the solution are 0.9 and 1.8 mM, respectively). Then, the tube was sealed and heated in an oven at 105 °C for 48 h without stirring and with the exclusion of light. Next, the tube was removed from the oven and left at room temperature for 5 min before opening. A green precipitate was formed; it was isolated by centrifugation and washing with ethanol.
Solvothermal crystallization: this procedure is identical to the above-described sonochemical–solvothermal crystallization process but without sonication of the solvents for 1.5 h in an ice-bath.
Scanning electron microscopy (SEM) and focused ion beam (FIB)-assisted cutting
SEM measurements were performed using HRSEM ULTRA-55 ZEISS and SIGMA 500 ZEISS instruments at an EHT voltage of 1.5 kV. Images were collected in secondary mode by using an Everhart–Thornley detector. Samples were prepared by placing a drop of the reaction mixture on a silicon substrate and allowing the solvent to evaporate. A Helios 600 FIB/SEM dual beam microscope was used for the assisted cutting and the subsequent imaging of the structures. In order to immobilize the structures during the cut, beam-assisted deposition of platinum layers was performed between the sides of the structures and the silicon substrate before milling.
Transmission electron microscopy (TEM)
TEM samples were prepared by placing 5 µl drops of the reaction mixture on 400-mesh carbon-coated copper grids (SPI-GridsTM, 3 mm) followed by blotting after 10 s. TEM nanobeam scanning electron diffraction was recorded under low-dose conditions with a FEI Tecnai F20 Twin TEM in STEM microprobe mode with a defocused beam of ~10 nm diameter and at a beam current of 10 pA. Undersampling was applied in order to avoid beam damage causing lattice damage during the exposure of 0.1 s per raster point. For a comparison with the crystal structure obtained by single-crystal X-ray diffraction, electron diffraction patterns were simulated using SingleCrystal (CrystalMaker Software Ltd., UK).
Micro-computed tomography (Micro-CT)
The MOFs were separated from the mother liquor by centrifugation and dried overnight under vacuum by a vacuum pump (Edwards RV12). A plastic pipette tip was used as a sample container: the narrowest extremity of the pipette tip was melted and sealed using a flame. Subsequently, the dried crystals were placed into the tip. Micro-CT data of the MOF obtained by the sonochemical–solvothermal crystallization were acquired with a Micro-XCT400 Zeiss X-ray microscope (Peasanton, California, USA). The tomographic images were obtained by taking 1200 projections over 180 deg at 40 KV and 200 µA. The final pixel sizes were 0.33 µm. Micro-CT data of the MOFs obtained by the solvothermal crystallization were acquired with a Xradia 520 Versa Zeiss X-ray microscope. The tomographic images were obtained by taking 2401 projections over 360 deg at 100 KV and 90 µA. The final pixel size was 0.39 µm. 3D images of the samples were collected for all the analyzed systems. Finally, several individual structures were analyzed in detail by using the Avizo 9.5 software (Thermo Fisher Scientific Inc, USA).
Single-crystal X-ray diffraction (SXRD)
SXRDs were collected both by a synchrotron source at the Beamline ID-29 of the European Synchrotron Radiation Facility and by a Rigaku XtaLabPro X-ray home-source diffractometer. The XtaLabPro X-ray diffractometer is equipped with a four-circle Kappa goniometer, a Dectris S200K detector, and a micro-focus sealed tube with microCMF-VHF. The data were collected with λ = 0.700 Å (synchrotron) and for CuKα1 radiation, λ = 1.5418 Å (Rigaku XtaLabPro diffractometer). The crystals were placed in Hampton Paratone oil, mounted on a MiTeGen loop, and plunged into liquid nitrogen to flash freeze them. The data were collected at 100 K with Oxford Cryostream. The crystals analyzed at the synchrotron were transported frozen in a Taylor-Wharton CX100 dry shipper. Data collection and reduction for the synchrotron data were done using MXCube, and using the EDNA automated data processing pipeline with XDS. Data collection, reduction, and analysis for the XtaLabPro laboratory data were performed with the CrysAlisPro software package (version 1.171.39.22a). The crystal structures were solved by direct methods using SHELXT 2016/448. All non-hydrogen atoms were further refined by SHELXL with anisotropic displacement coefficients. Hydrogen atoms were assigned isotropic displacement coefficients, and their coordinates were allowed to ride on the respective carbon atoms. The Platon SQUEEZE protocol was applied for all the structures49,50. Mercury CSD 3.10.2 and PLATON software were used for graphics. The reported Ewald sphere’s projections were generated by the Rigaku XtaLabPro instrument with CrysAlisPro, from diffraction data taken with a 100 μm beam; such a beam is large enough to expose the entire crystal. Details of the crystal structure analysis are presented in Supplementary Table 1.
Powder X-ray diffraction (PXRD)
Diffraction measurements were carried out by reflection geometry using an Ultima III (Rigaku, Japan) diffractometer equipped with a sealed Cu anode X-ray tube operating at 40 kV and 40 mA. A bent graphite monochromator and a scintillation detector were aligned in the diffracted beam. Next, θ/2θ scans were performed under specular conditions in the Bragg–Brentano mode with variable slits. The samples were scanned from 5 to 30 degrees in step mode with a step size of 0.025 degrees and a collection time of 12 s per step. The phases the XRD pattern were analyzed using Jade 2010 software (Materials Data, Inc.). The lattice parameters were refined using the Whole Pattern Fitting/Rietveld refinement module of Jade 2010.
Micro-raman spectroscopy
Raman measurements were performed on a LabRAM HR Evolution instrument (Horiba, France), equipped with an 800 mm spectrograph and a CCD detector (1024 pixels × 256 pixels open electrode front illuminated CCD camera, cooled to −60 °C). The system was based on an open confocal microscope (BX-FM Olympus, Japan). A 632.8 nm HeNe laser, with 600 grooves/mm grating and a ×100 objective (with spatial resolution better than 1 μm) were used for the measurements. The pixel spacing was 1.3 cm−1.
Electron paramagnetic resonance (EPR)
EPR spectra were recorded on a Bruker ELEXSYS 500 X-band spectrometer equipped with a Bruker ER4102ST resonator in a Wilmad flat cell for aqueous solutions (WG-808-Q) at room temperature. The experimental conditions were 1024 points, with a microwave power of 20 mW, 0.1 mT modulation amplitude, and 100 kHz modulation frequency. The sweep range was 10 mT. The spectrum was simulated using a custom-written MATLAB program based on EasySpin subroutines.
Elemental analyses
Elemental analyses (C, H, N, Cl, Ni, Cl, Br) were performed at Kolbe Laboratorium, Mulheim, Germany. The oxygen is calculated as the leak to 100%. The errors are ±0.01% for C, H, N; ±0.015% for Ni, and ±0.005% for Cl and Br.
Thermogravimetric analysis (TGA)
TGA was performed by a SDT Q600 V8.3 Build 101 Instrument. MOF-NiBr2 obtained under sonochemical–solvothermal and solvothermal conditions (t = 48 h, 105 °C) were dried under vacuum overnight and put in an alumina sample pans under a stream of nitrogen for the analysis (30–800 °C).
Circular dichroism (CD)
CD and relative UV spectra were collected by a Chirascan™—plus Auto CD Spectrometer. The measurements were performed over a wavelength range of 200−600 nm, by using a 1 mm path length cuvette. The spectral resolution was 1 nm.
Data availability
The data supporting the findings of this study are available within the paper and its Supplementary information files. The crystallographic data have also been submitted to the database at the Cambridge Crystallographic Data Centre. The CCDC numbers are 1996729 and 2016190. CIF V296 corresponds to Fig. 5d and Supplementary Figs. 5 and 8, left panel. CIF V394 corresponds to Supplementary Figs. 6 and 8, right panel.
References
Rimer, J. D. Inorganic ions regulate amorphous-to-crystal shape preservation in biomineralization. Proc. Natl Acad. Sci. USA 117, 3360–3362 (2020).
Aizenberg, J. et al. Skeleton of euplectella sp.: structural hierarchy from the nanoscale to the macroscale. Science 309, 275–278 (2005).
Politi, Y., Arad, T., Klein, E., Weiner, S. & Addadi, L. Sea urchin spine calcite forms via a transient amorphous calcium carbonate phase. Science 306, 1161–1164 (2004).
Murphy, C. J. et al. Gold nanoparticles in biology: beyond toxicity to cellular imaging. Acc. Chem. Res. 41, 1721–1730 (2008).
Jiang, W. et al. Emergence of complexity in hierarchically organized chiral particles. Science 368, 642–648 (2020).
Blanco, E., Shen, H. & Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 33, 941–951 (2015).
Chithrani, B. D., Ghazani, A. A. & Chan, W. C. W. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 6, 662–668 (2006).
Jiang, W. et al. Chiral acidic amino acids induce chiral hierarchical structure in calcium carbonate. Nat. Commun. 8, 1–13 (2017).
Shtukenberg, A. G., Ward, M. D. & Kahr, B. Crystal growth with macromolecular additives. Chem. Rev. 117, 14042–14090 (2017).
Orme, C. A. et al. Formation of chiral morphologies through selective binding of amino acids to calcite surface steps. Nature 411, 775–779 (2001).
Ciesielski, A., Piot, L., Samorì, P., Jouaiti, A. & Hosseini, M. W. Molecular tectonics at the solid/liquid interface: controlling the nanoscale geometry, directionality, and packing of d coordination networks on graphite surfaces. Adv. Mater. 21, 1131–1136 (2009).
Ganguli, A. K., Ganguly, A. & Vaidya, S. Microemulsion-based synthesis of nanocrystalline materials. Chem. Soc. Rev. 39, 474–485 (2010).
Kageyama, K., Tamazawa, J. I. & Aida, T. Extrusion polymerization: catalyzed synthesis of crystalline linear polyethylene nanofibers within a mesoporous silica. Science 285, 2113–2115 (1999).
Nie, Z., Xu, S., Seo, M., Lewis, P. C. & Kumacheva, E. Polymer particles with various shapes and morphologies produced in continuous microfluidic reactors. J. Am. Chem. Soc. 127, 8058–8063 (2005).
Aida, T., Meijer, E. W. & Stupp, S. I. Functional supramolecular polymers. Science 335, 813–817 (2012).
Melillo, J. M. et al. Shape-controlled synthesis of gold and silver nanoparticles. Science 298, 2176–2180 (2002).
Noorduin, W. L., Grinthal, A., Mahadevan, L. & Aizenberg, J. Rationally designed complex, hierarchical microarchitectures. Science 340, 832–837 (2013).
Prasetyanto, E. A. et al. Breakable hybrid organosilica nanocapsules for protein delivery. Angew. Chem. Int. Ed. 55, 3323–3327 (2016).
Sacanna, S., Irvine, W. T. M., Chaikin, P. M. & Pine, D. J. Lock and key colloids. Nature 464, 575–578 (2010).
Wang, S., McGuirk, C. M., d’Aquino, A., Mason, J. A. & Mirkin, C. A. Metal–organic framework nanoparticles. Adv. Mater. 30, 1–14 (2018).
Yang, J. M., Liu, Q. & Sun, W. Y. Shape and size control and gas adsorption of Ni(II)-doped MOF-5 nano/microcrystals. Micropor. Mesopor. Mater. 190, 26–31 (2014).
Xu, X., Zhang, Z. & Wang, X. Well-defined metal-organic-framework hollow nanostructures for catalytic reactions involving gases. Adv. Mater. 27, 5365–5371 (2015).
Horcajada, P. et al. Metal-organic frameworks in biomedicine. Chem. Rev. 112, 1232–1268 (2012).
Furukawa, H., Cordova, K. E., O’Keeffe, M. & Yaghi, O. M. The Chemistry and applications of metal-organic frameworks. Science 9, 1230444 (2010).
Murray, L. J., Dincă, M. & Long, J. R. Hydrogen storage in metal-organic frameworks. Chem. Soc. Rev. 38, 1294–1314 (2009).
Zarra, S., Wood, D. M., Roberts, D. A. & Nitschke, J. R. Molecular containers in complex chemical systems. Chem. Soc. Rev. 44, 419–432 (2015).
Tang, J. & Yamauchi, Y. Carbon materials: MOF morphologies in control. Nat. Chem. 8, 638–639 (2016).
Shankar, S. et al. Metal-organic microstructures: from rectangular to stellated and interpenetrating polyhedra. J. Am. Chem. Soc. 137, 226–231 (2015).
di Gregorio, M. C. et al. Metal-coordination-induced fusion creates hollow crystalline molecular superstructures. J. Am. Chem. Soc. 140, 9132–9139 (2018).
di Gregorio, M. C. et al. Emergence of chirality and structural complexity in single crystals at the molecular and morphological levels. Nat. Commun. 11, 380 (2020).
Donnay, G. & Pawson, D. L. X-ray diffraction studies of Echinoderm plates. Science 166, 1147–1150 (1969).
Mann, S. & Sparks, N. H. C. Single crystalline nature of coccolith elements of the marine alga Emiliania huxleyi as determined by electron diffraction and high-resolution transmission electron microscopy. Proc. R. Soc. Lond. Ser. B. Biol. Sci. 234, 441–453 (1988).
Mišík, V. & Riesz, P. Peroxyl radical formation in aqueous solutions of N,N-dimethylformamide, N-methylformamide, and dimethylsulfoxide by ultrasound: Implications for sonosensitized cell killing. Free Radic. Biol. Med. 20, 129–138 (1996).
Riesz, P., Berdahl, D. & Christman, C. L. Free radical generation by ultrasound in aqueous and nonaqueous solutions. Environ. Health Perspect. 64, 233–252 (1985).
Feng, G. et al. Accelerated crystallization of zeolites via hydroxyl free radicals. Science 351, 1188–1191 (2016).
Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. The Cambridge structural database. Acta Crys. B72, 171–179 (2016).
Zhu, L. & Bozzelli, J. W. Kinetics and mechanism for the thermal chlorination of chloroform in the gas phase: Inclusion of HCl elimination from CHCl3 Int. J. Chem. Kinet. 35, 647–660 (2003).
MiŠík, V. & Riesz, P. Free radical formation by ultrasound in organic liquids: a spin trapping and EPR study. J. Phys. Chem. 98, 1634–1640 (1994).
Wen, Q. et al. Chiral and SHG-active metal-organic frameworks in solution and on surfaces: Uniformity, morphology control, oriented growth and post-assembly functionalization. J. Am. Chem. Soc. 142, 14210–14221 (2020).
Yec, C. C. & Zeng, H. C. Synthesis of complex nanomaterials via Ostwald ripening. J. Mater. Chem. A 2, 4843–4851 (2014).
Nose, H. & Rodgers, M. T. Influence of the d orbital occupation on the structures and sequential binding energies of pyridine to the late first-row divalent transition metal cations: A DFT study. J. Phys. Chem. A 118, 8129–8140 (2014).
Konrad-Schmolke, M. et al. Mineral dissolution and reprecipitation mediated by an amorphous phase. Nat. Commun. 9, 1637 (2018).
Wangen, M. & Munz, I. A. Formation of quartz veins by local dissolution and transport of silica. Chem. Geol. 209, 179–192 (2004).
Kaizaki, S. Coordination effects of nitroxide radicals in transition metal and lanthanide complexes. Coord. Chem. Rev. 250, 1804–1818 (2006).
Vaitsis, C., Sourkouni, G. & Argirusis, C. Metal organic frameworks (MOFs) and ultrasound: a review. Ultrason. Sonochem. 52, 106–119 (2019).
Stock, N. & Biswas, S. Synthesis of metal-organic frameworks (MOFs): routes to various MOF topologies, morphologies, and composites. Chem. Rev. 112, 933–969 (2012).
Vasylyev, M., Popovitz-Biro, R., Shimon, L.J.W. & Neumann, R. Inorganic-organic hybrid materials based on keggin type polyoxometalates and organic polyammonium cations. J. Mol. Struct. 656, 27–35 (2003).
Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. C71, 3–8 (2015).
Spek, A. L. CheckCIF validation ALERTS: what they mean and how to respond. Acta Cryst. E76, 1–11 (2020).
Spek, A. L. PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. C71, 9–18 (2015).
Acknowledgements
This research was supported by the Irving and Cherna Moskowitz Center for Nano and Bioimaging at the Weizmann Institute of Science, the Israel Science Foundation (ISF), the Minerva Foundation, the Weizmann-Yale collaborative program, and the Helen and Martin Kimmel Center for Molecular Design. Dr. I. Pinkas, Dr. R. Carmeli, Dr. Y. Fieldman, and K. Rechav are acknowledged for carrying out the Raman, EPR, PXRD, and FIB-SEM measurements, respectively. Lior Snarsky is acknowledged for his help in TGA measurements. We thank the scientists of ID-29 beamline at ESRF (Grenoble, France) for their technical support. We would also like to thank Prof. M. Lahav (Weizmann Institute of Science) and Prof. J. M. McBride (Yale University) for their fruitful discussions. M.E.v.d.B. holds the Bruce A. Pearlman Professional Chair in Synthetic Organic Chemistry.
Author information
Authors and Affiliations
Contributions
M.C.d.G., M.L., and M.E.v.d.B. designed the experiments. M.C.d.G., M.E., Q.W., L.J.W.S., V.B., and L.H. performed the experiments and analyzed the data. M.C.d.G., M.L., and M.E.v.d.B. prepared this manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Benlai Wu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
di Gregorio, M.C., Elsousou, M., Wen, Q. et al. Molecular cannibalism: Sacrificial materials as precursors for hollow and multidomain single crystals. Nat Commun 12, 957 (2021). https://doi.org/10.1038/s41467-021-21076-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-21076-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
