
Abstract
Dynamic combinatorial chemistry applied to biological environments requires the exchange chemistry of choice to take place under physiological conditions. Thiol-disulfide exchange, one of the most popular dynamic combinatorial chemistries, usually needs long equilibration times to reach the required equilibrium composition. Here we report selenocystine as a catalyst mimicking Nature’s strategy to accelerate thiol-disulfide exchange at physiological pH and low temperatures. Selenocystine is able to accelerate slow thiol-disulfide systems and to promote the correct folding of an scrambled RNase A enzyme, thus broadening the practical range of pH conditions for oxidative folding. Additionally, dynamic combinatorial chemistry target-driven self-assembly processes are tested using spermine, spermidine and NADPH (casting) and glucose oxidase (molding). A non-competitive inhibitor is identified in the glucose oxidase directed dynamic combinatorial library.
Similar content being viewed by others
Introduction
Dynamic combinatorial chemistry (DCC) establishes molecular networks under thermodynamic control that responds to external stimuli1,2,3. We are interested in the application of DCC systems or Dynamic combinatorial libraries (DCLs) to biological environments where a protein or a biomolecule directs the assembly of the building blocks at dynamic equilibrium towards the synthesis of the best ligand or synthetic receptor in situ. Protein-directed DCC has proven its effectiveness as a hit identification strategy discovering enzyme inhibitors4,5,6,7.
Disulfide exchange8,9,10 is considered one of the most popular dynamic covalent chemistries applied to biological systems. This dynamic process is based on the thiol–disulfide equilibrium where the slow oxidation of the thiols competes with the disulfide exchange in aqueous solutions. Thiol/disulfide exchange is favored by highly nucleophilic thiolates attacking disulfide bonds constituted by good electrophiles and stable sulfur leaving groups. Therefore, to allow the mixture to self-correct for reaching the equilibrium composition, disulfide exchange must be faster than the oxidation of the thiols to avoid a kinetic trap11. Even though disulfide exchange proceeds smoothly at neutral or slightly basic pH, it usually requires several days to reach the required equilibrium12,13. To improve this limitation, several reaction conditions based on the use of a co-solvent such as DMSO14, glutathione redox buffer15, and high concentrations of selenol derivatives16,17 have been reported as alternative additives to speed up the exchange reaction from weeks to days. However, the application of certain additives in biological environments is limited. The addition of a suitable catalyst18 or additive was reported for acylhydrazone exchange to enable the exchange at neutral pH in 5 h even at low temperatures19. The possibility of conducting the thiol–disulfide exchange under a reasonable time-frame would benefit from its application in biotemplated-driven dynamic chemical systems and systems chemistry research20,21,22,23,24.
Inspired by Nature’s strategy to accelerate a thiol–disulfide exchange, we focus our attention on one of the major antioxidant systems in mammalian cells, the thioredoxin system which is formed by the mammalian thioredoxin reductase (TrxR), thioredoxin (Trx), and NADPH (nicotinamide adenine dinucleotide phosphate)25. Mammalian thioredoxin reductase (TrxR) is a pyridine nucleotide disulfide oxidoreductase that uses selenocysteine (Sec)26 in place of cysteine (Cys) in catalysis of the reduction of its target protein, thioredoxin (Trx) (Fig. 1)27,28. Sec can accelerate the rate of the thiol–disulfide exchange reactions at different stages of the TrxR mechanism. Selenium acts as a nucleophile initially attacking the disulfide bond of Trx (Fig. 1a), or as an electrophile accepting electrons from the redox center of TrxR as part of the selenosulfide bond (Fig. 1b).

a Selenium acting as a nucleophile. b Selenium acting as an electrophile.
The pH range influences the relative difference in reactivity between Cys and Sec in proteins. The pKa differences between Sec pKa ~ 5.4329, Cys pKa ~ 8.2230 contributes to the fact that at certain pHs the concentration of thiolates is low compared to the same reaction containing selenium. The higher polarizability of selenoates made them better nucleophiles and leaving groups than the corresponding sulfur derivatives. Additionally, the bond dissociation energy of Se–Se (46 kcal mol−1) compared to S–S (64 kcal mol−1) is considerably small31. Hence, the thiol oxidoreductase-like catalysis profits from the use of Sec whether it is due to the nucleophilicity, electrophilicity, pKa, or selenium leaving group ability. The exchange reactions of thiol–disulfide and selenol–diselenide share the nucleophilic addition mechanism (Fig. 2). Diselenide exchange has been reported as a DCC reversible reaction in water32. A comparative NMR kinetic study of thiol/disulfide and selenol/diselenide reactions showed that selenol–diselenide exchange reactions were ~107 fold faster than the thiol/disulfide exchange reactions (Fig. 2)33.

a Scheme of disulfide exchange. b Diselenide exchange. c Selenenylsulfide exchange in a thiol–disulfide DCC system.
In this work, we report the use of selenocystine (Secox) as a promoter of thiol–disulfide exchange at low temperatures and basic pH. Selenium generates selenenylsulfide intermediates which are more reactive towards thiolate nucleophilic addition accelerating the exchange between the different species. This chemistry is inspired by the operating mechanism proposed for the thioredoxin family of proteins. The catalytic efficiency of selenocystine is studied and applied to different thiol–disulfide DCLs including a slow DCC system constituted by alkyl thiols. Furthermore, we evaluate selenocystine as a promoter for the right formation of disulfide bonds during the folding of a scrambled RNase A even at acidic pH. DCC target-driven self-assembly (casting and molding) processes in selenocystine’s presence are tested. As a proof of concept, the casting approach where library members assemble around a template is tested using spermine, among other biomolecules (e.g., spermidine, NADPH). On the other hand, the molding approach in which the assembly of library members occurs inside the binding pockets of the template is studied with glucose oxidase (GOx) from Aspergillus niger. The affinity of the amplified molecule (4)2 is measured using fluorescence techniques and the glucose oxidase activity is evaluated in tandem with the horseradish-peroxidase system confirming the non-competitive inhibition of (4)2.
Results
Selenocystine based DCLs were prepared to mimic the mammalian thioredoxin reductase (TrxR) system which uses selenocysteine instead of cysteine in its catalysis of the reduction of Trx. DCLs were composed of dithiols (1–2)34,35,36 and monothiols (3–4) to form different architectures such as cyclic and linear oligomers, expanding the possibilities for the molecular recognition of different templates (Fig. 3b). Besides, the addition of carboxylate groups contributed to their solubility under the DCL conditions and the interaction with positively charged amines from the templates chosen through hydrogen bonding and ionic interactions. The aromatic rings may participate in hydrophobic interactions with the corresponding templates. Therefore, DCLs using thiols 1–4 were prepared in the absence and presence of cysteine and selenocystine derivatives (Fig. 3c). Their efficiency speeding up the thiol/disulfide exchange in a rich mixture of oligomers was tested at low temperatures and no stirring to maintain the stability of potential biological templates.

a DCC general scheme. b Building blocks of DCL. c Sulfur and selenium Cys and Sec derivatives. The pKa corresponds to the thiol group. Estimated pKas data calculated with Epik as implemented in Schrödinger Suite Release 2020-254,55,56 and reported in the literature (Sec pKa29, Cys pKa30). See Supplementary Fig. 1 for pKa calculations.
Secox as a catalyst
We started our DCL by reacting building blocks 1–4 (Fig. 3b) in the absence and the presence of the oxidized and reduced forms of sulfur and selenium derivatives of cysteine (Cys), cystine (Cysox), and selenocystine (Secox) at basic pH and 6 °C. Sec was not used in the DCL since selenols are easily oxidized and thus more difficult to manipulate16.
The DCLs were set up at two different pHs 7.8 and 8.8 to ensure Cys deprotonation. Selenium and sulfur derivatives, Secox and Cysox (Fig. 3c) were added as the fifth building block in each of the experiments. Differences in equilibration time at pH 7.8 or 8.8 in tris buffer were negligible (Supplementary Fig. 3 and Supplementary Methods). Therefore, we set up the experiments at pH 7.8 and use diselenide Secox and disulfide Cysox for comparison (Fig. 4).

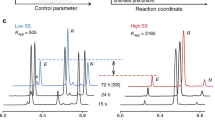
DCL conditions: building blocks (1–2) 95 μM each (3–4) at 190 μM concentration each. Tris buffer 20 mM, pH 7.8, 6 °C. a Control after 96 h. b Cysox (190 μM) after 72 h. c Secox (190 μM) after 24 h. Experiments were performed in triplicate.
The DCL that introduced Secox as a building block reached equilibrium after 24 h (Fig. 4c). Conversely, DCLs with its homolog Cysox needed 3 days to equilibrate (Fig. 4b) similar to the control library which required 4 days (Fig. 4a). Different oligomers were identified by HPLC-MS in the DCL (Supplementary Figs. 4–20 and Supplementary Discussion). Studies were performed to ensure the reversibility of the disulfide exchange in the DCLs. In the presence of Secox, consecutive DCLs were generated where an additional thiol was added each time the DCL has reached its equilibration point. The DCL started with thiol 1, followed by the addition after equilibration of thiols 3 and 4. Finally, the addition after equilibration of thiol 2 showed an identical distribution to Fig. 4 (Supplementary Fig. 35 and Supplementary Methods).
Optimal concentration of Secox in the DCL
The minimum concentration of Secox necessary for the exchange to occur in 24 h was studied. Several concentrations of Secox from 1 to 10% (mol) according to the total concentration of thiols in the DCL were evaluated. In the absence of selenocystine, the system took 96 h to reach equilibrium (Fig. 5a, (i)). As the concentration of Secox increased, the equilibration time was reduced from 96 to 72 h (1% mol), 48 h (2.5% mol), and 24 h (5 and 10% mol). The results showed that a concentration of 5% mol of Secox was required for the system to reach equilibrium within 24 h (Fig. 5a, (iv)). RPAs were calculated to confirm the library equilibration and to verify that the proportion of the species generated did not depend on the different Secox percentages. Selenyl-sulfide intermediates were detected through LC–MS supporting the existence of these reactive intermediates to promote the thiol–disulfide exchange (Supplementary Figs. 31–34).

a DCLs chromatograms showing different percentages of Secox. DCL conditions: building blocks (1–2) at 95 μM concentration each and building blocks (3–4) at 190 μM concentration each. Tris buffer 20 mM pH 7.8, 6 °C, (i) 0% mol as control DCL, 96 h (black), (ii) 1% mol (5.7 μM) (red) in 72 h, (iii) 2.5% mol (14.25 μM) (green) in 48 h, (iv) 5% mol (28.5 μM) (blue) in 24 h, (v) 10% mol (57 μM) (yellow) in 24 h. b Relative peak areas (RPAs) of the DCLs after equilibration in the absence and in the presence of different Secox percentages (Supplementary Table 1). Experiments were performed in triplicate.
Kinetic rate of Secox
To quantify the catalytic efficiency of Secox in thiol–disulfide exchange, kinetic studies were performed under our standard experimental conditions. The homodimerization of building block 3 to (3)2 (Fig. 6a) was followed by HPLC-MS in the absence and presence of the previously mentioned percentages of Secox.

a Homodimerization of building block 3 to (3)2. b Time course for the (3)2 formation using a concentration of 190 μM of (3), in 20 mM Tris buffer (pH 7.8), T = 6 °C, 0% (mol) Secox and 2.5% (v/v) DMSO (black dots), 5% (mol) TrxR and 2.5% (v/v) DMSO (light blue dots), 0% mol Secox and 20% (v/v) DMSO (purple dots), 1% (mol) Secox and 2.5% (v/v) DMSO (red dots), at 2.5% (mol) Secox and 2.5% (v/v) DMSO (green dots), 5% (mol) Secox and 2.5% (v/v) DMSO (blue dots) and 10% (mol) Secox and 2.5% (v/v) DMSO (orange dots). c Kinetic parameters. Reaction profiles were fitted to second-order reaction kinetic equations. Mean ± SD from three independent experiments. (Supplementary Fig. 38, Supplementary Methods, and Supplementary Discussion). Source data are provided as Source Data file.
The accelerating role of DMSO14 was discarded by running control experiments containing up to 20% (v/v) DMSO, which is much larger than the amount of this co-solvent used in our standard conditions (2.5% (v/v)). Furthermore, we have compared the catalytic activity of selenocystine to that of selenoenzyme TrxR.
Figure 6b shows the time course for the (3)2 homodimerization during the first 13 h and the derived kinetic parameters (Fig. 6c). In the presence of 5% (mol) Secox, the homodimerization reaction is completed in 24 h. The addition of 5% Secox (Kobs: 0.259 ± 0.009 M−1 s−1) led to a 18-fold acceleration over the uncatalyzed reaction (Kobs: 0.014 ± 0.001 M−1 s−1). The reaction rate obtained with 5% (mol) of selenocystine is superior to those observed with 20% (v/v) DMSO (Kobs: 0.068 ± 0.001 M−1 s−1) and 5% (mol) recombinant human TrxR 2 (Kobs: 0.035 ± 0.003 M−1 s−1). Of note, the kinetic rate observed with 5% (mol) selenoenzyme resulted lower than with 1% (mol) Secox (Kobs: 0.091 ± 0.002 M−1 s−1).
Secox effect in a slow DCL
To broaden the scope of selenocystine we tested this catalyst in a slow disulfide-exchange system. The DCL comprised 4 building blocks (5–8)37, three of them being aliphatic thiols with pKa values around 9 to slow down the exchange reaction (Fig. 7a). Due to solubility issues, a 20% (v/v) DMSO was added and the reaction mixture was diluted. In the absence of 5% (mol) Secox (with respect to total thiol concentration), the system reached equilibrium after 192 h, whereas in the presence of selenocystine the system equilibrated after 60 h, i.e., three times faster. The different oligomers were identified by HPLC-MS (Supplementary Figs. 21–30).

a Slow thiol/disulfide exchange. Building block 5 (52,3 μM) and building blocks (6–8) at 104.6 μM concentration each. Tris buffer 20 mM pH 7.8 at 20% (v/v) DMSO and 6 °C. (i) 0% mol Secox as control after 192 h, (ii) 5% mol Secox (18.3 μM) after 60 h. Relative peak areas (RPAs) of the DCLs, see Supplementary Fig. 36 and Supplementary Table 2. pKa values estimated from Schrödinger Release 2020-254. See Supplementary Fig. 2 for pKa calculations. b Kinetics for scrambled RNase A folding at pH 7.8 and room temperature. Yellow circles symbolize disulfide bonds. General conditions: scrambled RNase A (5 μM), tris buffer 100 mM, pH 7.8, 2 mM EDTA. 0.2 mM GSSG/1 mM GSH (black dots), 1 mM Secox/5 mM GSH (red dots). Mean ± SD from two independent experiments. Source data are provided as Source Data file.
Folding of scrambled RNase A with Secox
The folding of a small protein such as RNase A was studied to explore Secox application in the formation of the correct disulfide bonds during protein folding. The scrambled RNase A, a mixture of oxidized forms of RNase A with a random distribution of four disulfide bonds38, was used to determine the refolding rate. Once the optimum concentration of GSH and Secox for protein folding was determined (Supplementary Fig. 40), the relative folding rates for (Secox/GSH) pair were measured and compared to the standard glutathione redox buffer (GSSG/GSH) pair (Fig. 7b)39,40. Aliquots were withdrawn from each of the experiments and immediately assayed for RNase A activity by following the hydrolysis of cyclic cytidine-2′,3′-monophosphate (cCMP) at pH 6 for 2 min in a spectrophotometric discontinuous assay41,42.
RNase A folding versus time was plotted where the percent activity was proportional to the activity of folded native RNase A (Fig. 7b). Improved refolding rates were observed replacing GSSG with Secox. Secox provided a 92% yield of the correctly folded enzyme during the first 65 min whereas GSSG/GSH reached the same yield (93%) in 341 min.
To confirm the benefit of using Secox over GSSG the same assays were performed at pH 5.4. Whereas GSSG/GSH only got half of the protein refolded, Secox reached a 75% yield of native RNase A in <2 h (Supplementary Fig. 41 and Supplementary Methods).
Secox in spermine-directed DCC system
To test the casting DCC target-driven self-assembly, the template spermine was introduced as a guest into a DCL36,43,44,45. As spermidine and its precursor, the putrescine, these polyamines play important roles in many cellular processes such as the regulation of the kinases’ activities, the protection from oxidative damage, the regulation of transcription and translation contributing to nucleic acid stability, modulation of ion channels activity and the preservation of membrane structure/function.
We chose the spermine two building blocks DCL reported by Vial et al.36 as a well-studied system to test that Secox did not interfere with the recognition process and that the same DCL with two building blocks was efficiently equilibrated in the presence of 5% (mol) selenocystine. Building blocks 2 and 3 equipped with carboxylate moieties to establish interactions with the protonated amine groups of the spermine were allowed to equilibrate at pH 7.8 and 6 °C (Fig. 8a). As expected in our control experiment, the major component in the absence of spermine was the linear tetramer (2)2(3)2. After the addition of spermine, the linear tetramer “decomposed” in order to form mainly the cyclic tetramer of (2)4, and the (3)2 dimer (Fig. 8a). As it was reported, several studies determined that the spermine was threaded through the macrocycle fixing the configuration to a highly symmetric stereoisomer of (2)436. The addition of 5% (mol) Secox not only speeded four times the DCL equilibration (from 96 to 24 h) but also did not interfere with the recognition event reaching the same result as our control experiment (absence of Secox), matching the previously reported data and amplifying mainly the cyclic tetramer (2)4 (Fig. 8a). The precise composition of the DCL (with and without spermine), was assessed by measuring the relative peak area (RPA). Indeed, the normalized change of RPA was used to quantify the spermine influence in the outcome (Supplementary Fig. 42)46. The same template (spermine) was tested in our reference DCL of four building blocks (1–4) in the absence and presence of Secox (Fig. 8b). The addition of selenocystine speeded the equilibration time of the DCL to 24 h and the normalized change of RPA showed the amplification of the cyclic tetramer (2)4 (Supplementary Fig. 43). Additionally, two different templates were tested to confirm the absence of conflict between selenocystine and the templating effect. Spermidine and NADPH were introduced in our 4 building blocks DCL (1–4) in the absence and presence of Secox, leading to the amplification of different species and getting the same results as the non-catalyzed DCLs (Supplementary Figs. 44, 45, Supplementary Tables 7–10, and Supplementary Methods).

a DCL chromatograms of building block 2 (95 μM) and 3 (190 μM) in the absence and presence of spermine (28.5 μM). Yellow circle (S–S), red square (CO2H), black hexagon (benzene ring). DCL conditions: Tris buffer 20 mM pH 7.8, 6 °C, 2.5% DMSO. (i) control in absence of spermine after 96 h (black), (ii) in the absence of spermine but with Secox (5% mol, 14.25 μM) 24 h (red), (iii) control in presence of spermine after 96 h (green), (iv) in the presence of spermine and 5% mol Secox 24 h (blue). b DCL chromatograms of building blocks 1–2 (95 μM each) and 3–4 (190 μM each), in the absence and presence of spermine (57 μM), Secox (5% mol, 28.5 μM), Tris buffer (20 mM, pH 7.8), T = 6 °C, 2.5% (v/v) DMSO. (i) control in absence of spermine 96 h (black), (ii) in the absence of spermine but with Secox (5%, mol 28.5 μM) 24 h (red), (iii) control in presence of spermine 96 h (green), (iv) in the presence of spermine and 5% mol Secox 24 h (blue). c Spermine template effect measured by the normalized change of RPA in the presence and absence of Secox for building blocks 2 and 3. d Spermine template effect measured by the normalized change of RPA in the presence and absence of Secox for building blocks 1–4. See Supplementary Tables 3–6. Mean ± SD from three independent experiments.
Secox in a glucose oxidase-directed DCC
Having established that Secox is an effective promoter for thiol/disulfide exchange, we next studied the introduction of the enzyme glucose oxidase (GOx) to the DCL. Glucose oxidase (β-d-glucose:oxygen-oxidoreductase, EC 1.1.3.4) from Aspergillus niger is a flavoprotein that catalyzes the oxidation of β-d-glucose to d-glucono-δ-lactone and hydrogen peroxide, using oxygen as an electron acceptor. GOx is well suited for DCC exploration, it is a robust and well-characterized protein with two identical subunits (molecular weight: 160 KDa)47,48,49. Besides, there are relatively few ligands reported for GOx being one of the most widely used enzymes.
DCC experiments involving thiols 1–4 were set up in the absence and the presence of GOx (Fig. 9). The stability of the protein under the experimental conditions (DMSO, Secox tolerance, and stability over time) was tested using the fluorescence technique (Supplementary Figs. 46, 47 and Supplementary Methods).

Conditions: building blocks 1–2 (95 μM each) and 3–4 (190 μM each), Secox (5% mol, 28.5 μM), GOx (57 μM), Tris buffer (20 mM, pH 7.8), T = 6 °C, 2.5% (v/v) DMSO. Yellow circle (S–S), black hexagon (benzene ring). a DCL chromatograms after 24 h (i) in absence, blank (ii) and presence of GOx, templated. b GOx templated effect relative peak area (RPA) and normalized change of RPA in the presence and absence of Secox (Supplementary Tables 11 and 12). DCC experiments were carried out in triplicate. Mean ± SD.
The equilibration of the DCL was completed after 24 h (Fig. 9) and the oligomers were identified by HPLC-MS (Supplementary Figs. 4–20). The precise composition of the DCL (with and without GOx), was assessed by measuring the relative peak area (RPA). The normalized change of RPA was used to quantify the protein influence in the final DCL showing the amplification of the dimer of compound 4, that is (4)2 (Fig. 9b). Control experiments were set up to verify that the amplification of the dimer was not due to unspecific binding. Firstly the GOx was replaced by the bovine serum albumin (BSA). A stability study of BSA under the DCL conditions was performed using a fluorescence technique confirming the BSA stability in the DCL experiment (Supplementary Fig. 48)50. The BSA-templated DCL did not amplify (4)2, but it modified the DCL distribution as it was previously reported for thiol–disulfide exchange (Supplementary Fig. 49 and Supplementary Methods)51. Therefore, we performed the DCL in the presence of a large excess of DTNB (5,5′-dithiobis-2-nitrobenzoic acid), an inhibitor of GOx, which prevented the amplification of any other ligands except DTNB (Supplementary Fig. 50), indicating that an inhibited GOx cannot influence the DCL equilibrium composition (4)2.
Glucose oxidase activity inhibited by compound (4)2
The amplified compound (4)2 was isolated and its activity towards GOx tested (Fig. 10). Affinity was measured by fluorescence-based experiments (Supplementary Fig. 52 and Supplementary Methods). The natural ligand for GOx, β-d-glucose (Fig. 10a), showed an apparent dissociation constant of 1.10 ± 0.02 mM similar to those reported for apo and holo GOx forms (Kd′ ≈ 10 mM)52. DTNB was chosen as an inhibitor reference compound. Compound (4)2 with Kd′ = 110 ± 10 µM, presented affinity values similar to DTNB (Kd′=64 ± 2 µM). Glucose oxidase activity was evaluated by the horseradish-peroxidase system using o-dianisidine as a chromogen53. Michaelis–Menten parameters KM and Vmax and the inhibition constants Ki were calculated for both inhibitors (Fig. 10c). Control experiments provided glucose conversion values of KM = 15 ± 2 mM and Vmax = 0.14 ± 0.01 min−1 53.

a Structures of substrate β-d-glucose and the studied inhibitors DTNB and (4)2 of glucose oxidase. b Lineweaver–Burk plots57 of DTNB and (4)2. Conditions: [glucose] from 0.2 to 4 mM, 37 °C at 50 mM sodium acetate buffer pH 5.15, [DTNB] 0.07 and 0.14 µM, [(4)2] 0.7 and 1.4 µM. Mean ± SD from four independent experiments. c Summary of the binding affinity and enzymatic activity parameters (Supplementary Methods). Source data are provided as Source Data file.
The results from Lineweaver–Burk plots determined that (4)2 and DTNB are non-competitive inhibitors (Fig. 10b). The Ki values of (4)2 (Ki = 1.7 ± 0.2 µM) and DTNB (Ki = 0.20 ± 0.02 µM) were obtained using different inhibitor concentrations and the data collected were fitted to a model of non-competitive inhibitor using the program Prism 8.3.4. Furthermore, IC50 values were performed at a final concentration of 10 mM of the substrate in presence of different concentrations of the inhibitors (1.4 nM to 2 mM). The results suggested that compound (4)2 is a less potent inhibitor compared to DTNB.
Discussion
Dynamic combinatorial chemistry applied to biological systems requires that the reversible chemistry of choice can be performed in practical timescales and under physiological conditions. Even though thiol–disulfide exchange is one of the most popular chemistries for DCC as it proceeds smoothly at neutral or slightly basic pH, it usually needs long equilibration times to reach its final composition.
Inspired by the way the natural enzymes thioredoxin reductases accelerate the rate of thiol–disulfide reactions, we report the use of the biocompatible selenocystine as a catalyst for a number of thiol–disulfide exchange reactions. The operating principle of this chemistry is that selenenylsulfide intermediates are more reactive towards thiolate nucleophilic additions, thus accelerating the exchange between the different species.
5% (mol) Secox is found to be optimal to reach DCL equilibration in 24 h. To study the catalytic performance of Secox, kinetic studies on the homodimerization of compound 3 are performed. We show that the addition of 5% (mol) selenocystine produces an 18-fold acceleration on the homodimerization reaction of compound 3 over the uncatalyzed reaction. The use of DMSO as a co-solvent is known to accelerate the disulfide exchange. Nevertheless, the catalytic efficiency of 5% (mol) Secox clearly outcompetes that of 20% (v/v) DMSO. Moreover, Secox is catalytically more effective than selenoenzyme TrxR 2 under identical conditions. We have shown that the presence of selenocystine produced a positive effect on the thiol/disulfide exchange in a dynamic combinatorial library, being four times faster than conventional DCC even at low temperatures and physiological pH.
To broaden its scope of application as a reagent in biological redox events, the performance of Secox in the refolding of scrambled RNase A is compared to the standard glutathione redox buffer. The refolding rate increases five times by replacing GSSG with Secox. Unlike GSSG, Secox is highly active even at acidic pH (5.4), expanding the range of conditions for protein refolding.
Selenocystine has proven not to interfere with DCC target-driven self-assembly processes (casting and molding) using spermine, spermidine, NADPH, and the enzyme glucose oxidase as templates. In spermine-templated DCL the equilibration time is reduced four times using 5% (mol) Secox compared to the DCL control and the cyclic tetramer (2)4 is amplified as expected in different DCLs. The same catalytic effect is observed with the spermidine and NADPH as templates. Glucose oxidase (GOx) has confirmed to be an excellent DCL template directing the library to the synthesis of compound (4)2. Accordingly, this compound presents affinity values in the micromolar range similar to the reference GOx inhibitor DTNB. As an additional proof for specific binding of (4)2 to GOx, glucose oxidase activity is evaluated and results suggest that compound (4)2 is a non-competitive inhibitor with a moderate inhibitory activity compared to the reported inhibitor DTNB.
In summary, the use of selenocystine as a catalyst broadens the scope of dynamic combinatorial chemistry in biological environments. It accelerates the DCLs equilibration time even at low temperatures without interfering with the templating effect. Furthermore, selenocystine has proven its effectiveness as a catalyst promoting the formation of the right disulfide bonds in protein folding expanding the pH range conditions.
Methods
Cysox and Secox DCL study
Control DCL of dithiols 1–2 (2 × 1.8 μL, 25 mM, 4.5·10−8 mol per monomer, DMSO) and monothiols 3–4 (2 × 1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO), DMSO (4.5 μL) in 20 mM Tris buffer pH 7.8 (466 μL). DMSO percentage is 2.5% v/v. Secox-catalyzed DCL: Dithiols 1–2 (2 × 1.8 μL, 25 mM, 4.5·10−8 mol per monomer, DMSO) and monothiols 3–4 (2 × 1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO), Secox (1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO with 4% (v/v) 1 M NaOH). DMSO (2.7 μL) in 20 mM Tris buffer pH 7.8 (466 μL). Cysox-catalyzed DCL: dithiols 1–2 (2 × 1.8 μL, 25 mM, 4.5·10−8 mol per monomer, DMSO) and monothiols 3–4 (2 × 1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO), Cysox (1.8 μL, 50 mM, 9.0·10−8 mol per monomer, H2O), DMSO (4.5 μL) in 20 mM Tris buffer pH 7.8 (464.2 μL). DCLs were analyzed by HPLC-MS until equilibration without stirring at 6 °C.
Percentage (% mol) of Secox
Control DCL. Dithiols 1–2 (2 × 1.8 μL, 25 mM, 4.5·10−8 mol per monomer, DMSO) and monothiols 3–4 (2 × 1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO), DMSO (4.5 μL) in 20 mM Tris buffer pH 7.8 (466 μL). DMSO percentage is 2.5% v/v. Regarding Secox catalyzed DCL, different stocks of Secox were carried out in order to study the percentages of Secox. The general catalyzed DCL was the addition of dithiols 1–2 (2 × 1.8 μL, 25 mM, 4.5·10−8 mol per monomer, DMSO), monothiols 3–4 (2 × 1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO), Secox (1.8 μL, corresponding concentration, DMSO with 4% (v/v) 1 M NaOH), and DMSO (2.7 μL) in 20 mM Tris buffer pH 7.8 (466 μL). Concentration according to the percentage of Secox: 10% mol—15 mM stock (2.7·10−8 mol), 5% mol—7.5 mM stock (1.35·10−8 mol), 2.5% mol—3.75 mM (6.7·10−9 mol), 1% mol—1.5 mM stock (2.7·10−9 mol). The DCLs were prepared at 6 °C without stirring and analyzed in several hours until the complete stabilization.
Kinetic studies
The reaction started by the addition of the thiol 3 (3.8 μL, 50 mM, 1.9·10−7 mol, in DMSO) over the mixture of Secox (3.8 μL, at different concentrations according to the percentage, in DMSO with 4% (v/v) 1 M NaOH) or DMSO in control DCL (3.8 μL), DMSO (17.4 μL) and buffer Tris 20 mM pH 7.8 (975 μL) up to a total volume of 1 mL, and a final percentage of 2.5% (v/v) DMSO. T = 6 °C without stirring. Concentration according to the percentage of Secox: 10% mol—5 mM stock (1.9·10−8 mol), 5% mol—2.5 mM stock (9.5·10−9 mol), 2.5% mol—1.25 mM (4.75·10−9 mol), 1% mol—0.5 mM stock (1.9·10−9 mol). For the 20% (v/v) DMSO-reaction, 3 was added over the solution of DMSO (196.2 μL) and buffer Tris 20 mM pH 7.8 (800 μL) following the same parameters described above. Besides, for TrxR-catalyzed reaction, 3 (3.8 μL, 50 mM, 1.9·10−7 mol, in DMSO) was added over a solution of TrxR (40 μL, 237.5 μM, 9.5·10−9 mol, in buffer Tris), DMSO (21.2 μL) and buffer Tris 20 mM pH 7.8 (935 μL). For the data treatment, see Supplementary Methods and Supplementary Discussion.
Slow DCL system
Control DCL. Dithiol 5 (1.0 μL, 25 mM, 2.5·10−8 mol, DMSO) and monothiols 6–8 (3 × 1.0 μL, 50 mM, 5·10−8 mol per monomer, DMSO), DMSO (91.6 μL) in 20 mM Tris buffer pH 7.8 (382 μL), affording a final DMSO percentage of 20% (v/v). Secox-catalyzed: Dithiol 5 (1.0 μL, 25 mM, 2.5·10−8 mol, DMSO) and monothiols 6–8 (3 × 1.0 μL, 50 mM, 5·10−8 mol per monomer, DMSO), Secox (1.0 μL, 8.75 mM, 8.75·10−9 mol, DMSO with 4% (v/v) 1 M NaOH), DMSO (90.6 μL) in 20 mM Tris buffer pH 7.8 (382 μL), DMSO is 20% (v/v) due to the solubility issues, at 6 °C and without stirring. The DCLs were analyzed until stabilization.
RNase A refolding by Secox
The activity of folded scrambled RNase was determined by the selective hydrolysis of cCMP. The folding experiment was carried out by the addition of scrambled RNase A at a final concentration of 5 µM in presence of two redox pairs, GSSG/GSH (0.2 mM/1 mM) and Secox/GSH (1 mM/5 mM) in buffer 20 mM Tris 2 mM EDTA pH 7.8 at room temperature. The concentration of the properly folded protein was determined by the calibration curve based on the initial velocity vs. the concentration. Aliquots (30 μL) of the refolding solution were withdrawn to measure the hydrolysis of cCMP for 2 min at 25 °C spectrophotometrically (292 nm). The experiment was evaluated for 3 days at prescribed times. Refolding experiments at pH 5.4 are detailed in Supplementary Methods and Supplementary Discussion.
Secox catalyzing Spermine-directed DCLs
Methodology of two building blocks DCLs
Control DCL: dithiol 2 (1.8 μL, 25 mM, 4.5·10−8 mol, DMSO) and monothiol 3 (1.8 μL, 50 mM, 9.0·10−8 mol, DMSO), DMSO (8.1 μL) in 20 mM tris buffer pH 7.8 with 2.5% (v/v) DMSO. The DCL was stabilized at 96 h. Control DCL-Secox catalyzed: dithiol 2 (1.8 μL, 25 mM, 4.5·10−8 mol, DMSO) and monothiol 3 (1.8 μL, 50 mM, 9.0·10−8 mol, DMSO), Secox (1.8 μL, 3.75 mM, 6.75·10−9 mol, DMSO with 4% (v/v) 1 M NaOH), DMSO (6.3 μL) in 20 mM tris buffer pH 7.8 (466 μL) with 2.5% (v/v) DMSO. The mixture was stabilized after 24 h. Spermine-directed DCL: dithiol 2 (1.8 μL, 25 mM, 4.5·10−8 mol, DMSO) and monothiol 3 (1.8 μL, 50 mM, 9.0·10−8 mol, DMSO), DMSO (8.1 μL), Spermine solution in 20 mM tris buffer pH 7.8 (5 μL, 2.7 mM, 1.35·10−8 mol), and 20 mM tris buffer pH 7.8 with 2.5% (v/v) DMSO (461 μL). The DCL was stabilized at 96 h. Spermine-directed-Secox catalyzed DCL: Dithiol 2 (1.8 μL, 25 mM, 4.5·10−8 mol, DMSO) and monothiol 3 (1.8 μL, 50 mM, 9.0·10−8 mol, DMSO), Secox (1.8 μL, 3.75 mM, 6.75·10−9 mol, DMSO with 4% (v/v) 1 M NaOH), DMSO (6.3 μL), Spermine solution in 20 mM tris buffer pH 7.8 (5 μL, 2.7 mM, 1.35·10−8 mol), and 20 mM tris buffer pH 7.8 with 2.5% (v/v) DMSO (461 μL). The DCL was analyzed in 96 h after equilibration. DCLs were performed without stirring at 6 °C and analyzed by HPLC-MS.
Methodology of four building blocks DCLs
Control DCL: dithiols 1–2 (2 × 1.8 μL, 25 mM, 4.5·10−8 mol per monomer, DMSO), monothiols 3–4 (2 × 1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO), DMSO (4.5 μL) in 20 mM tris buffer pH 7.8 with 2.5% (v/v) DMSO. The mixture was stabilized in 96 h. Control DCL-Secox catalyzed: dithiols 1–2 (2 × 1.8 μL, 25 mM, 4.5·10−8 mol per monomer, DMSO) and monothiols 3–4 (2 × 1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO), Secox (1.8 μL, 7.5 mM, 1.35·10−8 mol, DMSO with 4% (v/v) 1 M NaOH), DMSO (2.7 μL) in 20 mM tris buffer pH 7.8 (466 μL) with 2.5% (v/v) DMSO. The mixture was stabilized after 24 h. Spermine-directed DCL: dithiols 1–2 (2 × 1.8 μL, 25 mM, 4.5·10−8 mol per monomer, DMSO) and monothiols 3–4 (2 × 1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO), DMSO (4.5 μL), Spermine solution in 20 mM tris buffer pH 7.8 (10 μL, 2.7 mM, 2.7·10−8 mol) and 20 mM tris buffer pH 7.8 with 2.5% v/v DMSO (456 μL). It was stabilized in 24 h. Spermine-directed-Secoxcatalayzed DCL: dithiols 1–2 (2 × 1.8 μL, 25 mM, 4.5·10−8 mol per monomer, DMSO), monothiols 3–4 (2 × 1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO), Secox (1.8 μL, 7.5 mM, 1.35·10−9 mol, DMSO with 4% (v/v) 1 M NaOH), DMSO (2.7 μL), Spermine solution in 20 mM tris buffer pH 7.8 (10 μL, 2.7 mM, 2.7·10−8 mol) and 20 mM tris buffer pH 7.8 with 2.5% v/v DMSO (456 μL). DCL was equilibrated in 96 h. DCLs were performed without stirring at 6 °C and analyzed by HPLC-MS.
See Supplementary Methods for more templated-DCLs (e.g., Spermidine and NADPH).
Glucose oxidase-directed DCL
Blank DCL: Dithiols 1–2 (2 × 1.8 μL, 25 mM, 4.5·10−8 mol per monomer, DMSO) and monothiols 3–4 (2 × 1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO), Secox (1.8 μL,7.5 mM, 1.35·10−8 mol, DMSO), DMSO (2.7 μL) in 20 mM tris buffer pH 7.8 with 2.5% (v/v) DMSO (466 μL). GOx-directed DCL: Dithiols 1–2 (2 × 1.8 μL, 25 mM, 4.5·10−8 mol per monomer, DMSO) and monothiols 3–4 (2 × 1.8 μL, 50 mM, 9.0·10−8 mol per monomer, DMSO), Secox (1.8 μL,7.5 mM, 1.35·10−8 mol, DMSO with 4% (v/v) 1 M NaOH), DMSO (2.7 μL), GOx (135 μM, 200 μL, 2.7·10−8 mol, 10% mol) in 20 mM tris buffer pH 7.8 with 2.5% (v/v) DMSO (266 μL). The DCL was stabilized for 24 h at 6 °C. Then, GOx was removed by ultracentrifugation through an Amicon ultra-filter (100 KDa). HPLC analysis was performed.
Supplementary Methods: BSA-directed DCL and GOx-directed DCL in presence of DTNB.
Reversibility study
See Supplementary Methods and Supplementary Discussion.
Synthesis of building blocks 1, 2, and 5
See Supplementary Methods and Supplementary Discussion.
Synthesis of compounds (3)2 and (4)2
See Supplementary Methods and Supplementary Discussion.
Protein stability experiments
Fluorescence emission experiments
GOx activity assays
Enzymatic assay to determine Michaelis–Menten constant Km and maximum velocity Vmax, IC50, and inhibition constant Ki of the ligands in presence of compounds were performed by glucose oxidase-horseradish peroxidase-coupled system using o-dianisidine as chromogen, with a 2.5% (v/v) of DMSO.
Michaelis–Menten parameters were set up using different final concentrations of glucose from 0.2 to 4 mM. Glucose stocks from 0.5 to 10 mM (100 µL, 50 mM sodium acetate buffer pH 5.1), o-dianisidine (127.8 µL, 3.5 mM, MilliQ water), DMSO (6.2 µL), horseradish peroxidase (8 µL, 207 µM, 50 mM sodium acetate buffer pH 5.1) and GOx (8 µL, 42.7 nM, 50 mM sodium acetate buffer pH 5.1) were added to a final volume of 250 µL in a 2.5% (v/v) DMSO. GOx activity was measured spectrophotometrically at 500 nm 20 min at 37 °C. The data were fitted to a Lineweaver–Burk model.
IC50 was determined using different concentrations of inhibitors DTNB and (4)2. The solution containing glucose (100 µL, 25 mM, 50 mM sodium acetate buffer pH 5.1), o-dianisidine (127.8 µL, 3.5 mM, MilliQ H2O), DTNB, or (4)2 at different concentrations (6.2 µL, from 1.4 nM to 2 mM, DMSO), horseradish peroxidase (8 µL, 207 µM, 50 mM sodium acetate buffer pH 5.1) and GOx (8 µL, 42.7 nM, 50 mM sodium acetate buffer pH 5.1) was analyzed spectrophotometrically at 500 nm in a plate reader, 20 min at 37 °C. The data were fitted to a dose–response model for three parameters using Prism 8.3.4.
Inhibition constant Ki was determined by the use of several ligands and substrate concentrations. Glucose (100 µL, 0.5−10 mM, in 50 mM sodium acetate buffer pH 5.1), o-dianisidine (127.8 µL, 3.5 mM, MilliQ H2O), DMSO, several concentrations of DTNB (35−140 nM) or (4)2 (0.07–1.4 µM) in DMSO (6.2 µL), horseradish peroxidase (8 µL, 207 µM, 50 mM sodium acetate buffer pH 5.1) and GOx (8 µL, 42.7 nM, 50 mM sodium acetate buffer pH 5.1). Plates were measured spectrophotometrically at 500 nm 20 min at 37 °C. The data were fitted to a non-competitive inhibition model by Prism 8.3.4. For more details, see Supplementary Methods.
Data availability
A reporting summary for this Article is available as a Supplementary Information file. The rest of the data are available in the Supplementary Information File. Source data are provided with this paper.
References
Li, J., Nowak, P. & Otto, S. Dynamic combinatorial libraries: from exploring molecular recognition to systems chemistry. J. Am. Chem. Soc. 135, 9222–9239 (2013).
Lehn, J.-M. Dynamic combinatorial chemistry and virtual combinatorial libraries. Chem. Eur. J. 5, 2455–2463 (1999).
Cougnon, F. B., Sanders, J. K. M. Evolution of dynamic combinatorial chemistry. Acc. Chem. Res. 45, 2211–2221 (2012).
Canal-Martín, A. & Pérez-Fernández, R. Protein-directed dynamic combinatorial chemistry: an efficient strategy in drug design. ACS Omega. 5, 26307–26315 (2020)
Huang, R. & Leung, I. K. H. Protein-directed dynamic combinatorial chemistry: a guide to protein ligand and inhibitor discovery. Molecules 21, 910–928 (2016).
Frei, P., Hevey, R. & Ernst, B. Dynamic combinatorial chemistry: a new methodology comes of age. Chem. Eur. J. 25, 60–73 (2019).
Mondal, M. & Hirsch, A. K. H. Dynamic combinatorial chemistry: a tool to facilitate the identification of inhibitors for protein targets. Chem. Soc. Rev. 44, 2455–2488 (2015).
Ponnuswamy, N., Cougnon, F. B. L., Clough, J. M., Pantos, G. D. & Sanders, J. K. M. S. Discovery of an organic trefoil knot. Science 338, 783–785 (2012).
Au-Yeung, H. Y., Cougnon, F. B. L., Otto, S., Pantos, G. D. & Sanders, J. K. M. Exploiting donor–acceptor interactions in aqueous dynamic combinatorial libraries: exploratory studies of simple systems. Chem. Sci. 1, 567–574 (2010).
Komáromy, D., Nowak, P., & Otto, S. in Dynamic Covalent Chemistry: Principles, Reactions and Applications (eds Zhang, W. & Jin, Y.) 31–119 (Wiley, 2017).
Corbett, P. T. et al. Dynamic combinatorial chemistry. Chem. Rev. 106, 3652–3711 (2006).
Ramström, O. & Lehn, J.-M. In situ generation and screening of a dynamic combinatorial carbohydrate library against concanavalin A. ChemBioChem 1, 41–48 (2000).
Milanesi, L., Hunter, C. A., Sedelnikova, S. E. & Waltho, J. P. Amplification of bifunctional ligands for calmodulin from a dynamic combinatorial library. Chem. Eur. J. 12, 1081–1087 (2006).
Atcher, A. & Alfonso, I. The effect of DMSO in the aqueous thiol-disulfide dynamic covalent chemistry of model pseudopeptides. RSC Adv. 3, 25605–25608 (2013).
Scott, D. E., Dawes, G. J., Ando, M., Abell, C. & Ciulli, A. A fragment-based approach to probing adenosine recognition sites by using dynamic combinatorial chemistry. ChemBioChem 10, 2772–2779 (2009).
Singh, R. & Whitesides, G. M. Selenols catalyze the interchange reactions of dithiols and disulfides in water. J. Org. Chem. 56, 6931- (1991).
Caldwell, K. A. & Tappel, A. L. Acceleration of sulfhydryl oxidations by selenocystine. Arch. Biochem. Biophys. 112, 196–200 (1965).
Bhat, V. T. et al. Nucleophilic catalysis of acylhydrazone equilibration for protein-directed dynamic covalent chemistry. Nat. Chem. 2, 490–497 (2010).
Canal-Martín, A. et al. Insights into real-time chemical processes in a calcium sensor protein directed dynamic library. Nat. Commun. 10, 2798–2805 (2019).
Malakoutikhah, M., Schaeffer, G., Monreal Santiago, G., Yang, S., Marić, I. & Otto, S. Cross-catalysis between self-replicators of different handedness. J. Sys. Chem. 7, 9–18 (2019).
Liu, B. et al. Complex molecules that fold like proteins can emerge spontaneously. J. Am. Chem. Soc. 141, 1685–1689 (2019).
Wagner, N. & Ashkenasy, G. Systems chemistry: logic gates, arithmetic units and network motifs in small networks. Chem. Eur. J. 15, 1765–1775 (2009).
Sadownik, J. W., Mattia, E., Nowak, P. & Otto, S. Diversification of self-replicating molecules. Nat. Chem. 8, 264–269 (2016).
Ashkenasy, G., Hermans, T. M., Otto, S. & Taylor, A. F. Systems chemistry. Chem. Soc. Rev. 46, 2543–2554 (2017).
Mustacich, D. & Powis, G. Thioredoxin reductase. Biochem. J. 346, 1–8 (2000).
Günzler, W. A. et al. The Amino-Acid Sequence of Bovine Glutathione Peroxidase Hoppc-Seyler’s Z. Physiol. Chem. 365, 195–21 (1984).
Lothrop, A. P. et al. Selenium as an electron acceptor during the catalytic mechanism of thioredoxin reductase. Biochemistry 53, 654–663 (2014).
Hondal, R. J., Marino, S. M. & Gladyshev, V. N. Selenocysteine in thiol/disulfide-like exchange reactions. Antiox. Red. Signal. 18, 1675–1689 (2013).
Huber, R. E. & Criddle, R. S. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch. Biochem. Biophys. 122, 164–173 (1967).
Perrin, D. D. & Sayce, I. G. Complex formation by nickel and zinc with penicillamine and cysteine. J. Chem. Soc. A 53–57 (1968).
Senning, A. (ed.) Sulfur in Organic and Inorganic Chemistry 37–69 (Marcel Dekker, New York, 1972).
Ramussen, B., Sorensen, A., Gotfredsen, H. & Pittelkow, M. Dynamic combinatorial chemistry with diselenides and disulfides in water. Chem. Commun. 50, 3716–3718 (2014).
Pleasants, J. C., Guo, W. & Rabenstein, D. L. A comparative study of the kinetics of selenol/diselenide and thiol/disulfide exchange reactions. J. Am. Chem. Soc. 111, 6553–6558 (1989).
Carnall, J. M. et al. Mechanosensitive self-replication driven by self-organization. Science 327, 1502–1506 (2010).
Matysiak, B. M. et al. Antiparallel dynamic covalent chemistries. J. Am. Chem. Soc. 139, 6744–6751 (2017).
Vial, L., Ludlow, R. F., Leclaire, J., Pérez-Fernández, R. & Otto, S. Controlling the biological effects of spermine using a synthetic receptor. J. Am. Chem. Soc. 28, 10253–10257 (2006).
Otto, S. I., Furlan, R. L. E. & Sanders, J. K. M. Dynamic combinatorial libraries of macrocyclic disulfides in water. J. Am. Chem. Soc. 122, 12063–12064 (2000).
Hillson, D. A., Lambert, N. & Freedman, R. B. Formation and isomerization of disulfide bonds in proteins: protein disulfide isomerase. Methods Enzymol. 107, 281–292 (1984).
Lyles, M. M. & Gilbert, H. F. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry 30, 613–619 (1991).
Litt, M. The kinetics of ribonuclease action on cytidine-2′-3′-cyclic phosphate. J. Biol. Chem. 236, 1786–1790 (1961).
Beld, J., Woycechowsky, J. & Hilvert, D. Catalysis of oxidative protein folding by small-molecule diselenides. Biochemistry 47, 6985–6987 (2008).
Gough, J. D., Williams, R. H., Donofrio, A. E. & Lees, W. J. Folding disulfide-containing proteins faster with an aromatic thiol. J. Am. Chem. Soc. 124, 3885–3892 (2002).
Pegg, A. E. The function of spermine. Int. Union Biochem. Mol. Biol. 66, 8–18 (2014).
Saggiomo, V., Hristova, Y. R., Ludlow, R. F. & Otto, S. Systems chemistry: using thermodynamically controlled networks to assess molecular similarity. J. Syst. Chem. 4, 2 (2013).
Pérez-Fernández, R. et al. Two-phase dynamic combinatorial discovery of a spermine transporter. Chem. Commun. 3708–3710 (2009).
Frei, P. et al. Target-directed dynamic combinatorial chemistry: a study on potentials and pitfalls as exemplified on bacterial target. Chem. Eur. J. 23, 11570–11577 (2017).
Bankar, S. D., Bule, M. V., Singhal, R. S. & Ananthanarayan, L. Glucose oxidase—an overview. Biotechnol. Adv. 27, 489–501 (2009).
Tsuge, H., Natsuakai, O. & Ohashi, K. Purification, properties and molecular features of glucose oxidase from Aspergillus niger. J. Biochem 78, 835–843 (1975).
Meng, Y. et al. Production and characterization of recombinant glucose oxidase from Aspergillus niger expressed in Pichia pastoris. Lett. Appl. Microbiol. 58, 393–400 (2013).
Pabbathi, A., Patra, S. & Samanta, A. Structural transformation of bovine serum albumin induced by dimethyl sulfoxide and probed by fluorescence correlation spectroscopy and additional methods. Chem. Phys. Chem. 14, 2441–2449 (2013).
Danieli, B. et al. Thiocolchicine-podophyllotoxin conjugates: dynamic libraries based on disulfide exchange reaction. J. Org. Chem. 71, 2848–2853 (2006).
D’Auria, S., Herman, P., Rossi, M. & Lakowicz, J. R. The fluorescence emission of the apo-glucose oxidase from Aspergillus niger as probe to estimate glucose concentrations. Biochem. Biophys. Res. Commun. 263, 550–553 (1999).
Odebunmi, E. O. & Owalude, S. O. Kinetic and thermodynamic studies of glucose oxidase catalysed oxidation reaction of glucose. J. Appl. Sci. Environ. Manag. 11, 95–100 (2007).
Epik. Schrödinger Suite Release 2020-2 (Schrödinger LLC, New York (NY), 2020).
Greenwood, J. R., Calkins, D., Sullivan, A. P. & Shelley, J. C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comp. Aided Mol. Des. 24, 591–604 (2010).
Shelley, J. C. et al. Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J. Comp. Aided Mol. Des. 21, 681–691 (2007).
Lineweaver, H. & Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 56, 658–666 (1934).
Acknowledgements
We thank Prof. J. Elguero (IQM-CSIC) and Dr. G. Jiménez-Osés (CICbioGUNE) for helpful discussions on the paper and Dr. F. J. Medrano (CIB-CSIC) for his assistance with the folding studies. This work was supported by the Spanish Ministry of Science and Innovation with Grant PID2019-108587RB-I00 (R.P.F.)
Author information
Authors and Affiliations
Contributions
A.C.-M. and R.P.-F. designed the experiments. A.C.-M. performed the DCC, enzymatic assays, fluorescence, and folding studies. R.P.-F. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Ignacio Alfonso and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Canal-Martín, A., Pérez-Fernández, R. Biomimetic selenocystine based dynamic combinatorial chemistry for thiol-disulfide exchange. Nat Commun 12, 163 (2021). https://doi.org/10.1038/s41467-020-20415-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-20415-6
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
