
Abstract
Thermal-stimuli responsive nanomaterials hold great promise in designing multifunctional intelligent devices for a wide range of applications. In this work, a reversible isomeric transformation in an atomically precise nanocluster is reported. We show that biicosahedral [Au13Ag12(PPh3)10Cl8]SbF6 nanoclusters composed of two icosahedral Au7Ag6 units by sharing one common Au vertex can produce two temperature-responsive conformational isomers with complete reversibility, which forms the basis of a rotary nanomotor driven by temperature. Differential scanning calorimetry analysis on the reversible isomeric transformation demonstrates that the Gibbs free energy is the driving force for the transformation. This work offers a strategy for rational design and development of atomically precise nanomaterials via ligand tailoring and alloy engineering for a reversible stimuli-response behavior required for intelligent devices. The two temperature-driven, mutually convertible isomers of the nanoclusters open up an avenue to employ ultra-small nanoclusters (1 nm) for the design of thermal sensors and intelligent catalysts.
Similar content being viewed by others

Introduction
Stimuli-responsive materials are at the forefront of technological innovations to fabricate the next-generation devices with “intelligent” performance1,2,3. To meet the self-activation and -deactivation functionality requirements for smart materials, the stimuli-responsive behavior should be reversible. This can be satisfied by utilization of precise structural and phase transitions4,5,6,7,8. In particular, reversible conformational isomerism, a fundamental concept in molecular sciences that is commonly observed in organic molecules, offers a unique platform to prepare rotary motors at the nanoscale with atomistic precision.
Despite recent advances in controllable synthesis and structure determination of atomically precise nanoclusters9,10,11,12,13,14,15,16,17,18, the concept of reversible conformational isomerization for metal nanoclusters has not yet been explored. To date, irreversible structural isomerization of atomically precise nanoclusters has been reported in a few cases19,20,21,22,23,24. One case pertains to Au42(TBBT)26 (TBBT = 4-tert-butylbenzenethiolate) in the form of fcc and non-fcc frameworks that were synthesized via two different procedures19. However, the two isomeric structures of Au42 are “static” and cannot be reversibly transformed19. The same is true to the clusters prepared by Teo et al.20,21,22,23. One directional structural conversion from metastable Au38(SC2H4Ph)24 to thermodynamically stable biicosahedral Au38(SC2H4Ph)24 was reported to occur under thermal conditions, but unfortunately the reverse transformation could not occur24. Finally, while reversible transformation between Au28(SC6H11)20 and Au28(SPhC4H9)20 nanoclusters was realized by Chen et al., this case involves different surface-protecting ligands (-SC6H11 vs. -SC4H9), so it does not strictly meet the definition of isomeric transformation25 and cannot be utilized for construction of nanomotors. Therefore, breakthroughs are still needed to precisely modulate the bonding structure of ultra-small nanoparticles to make their frameworks flexible for a facile molecular rotary nanomotor.
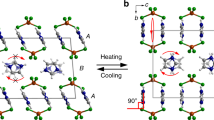
Herein, we report the discovery of a stimuli-responsive nanocluster that shows reversible conformational isomerism. The [Au13Ag12(PPh3)10Cl8]+(SbF6)ˉ nanocluster, hereafter abbreviated as Au13Ag12, presents two isomeric forms that can be selectively obtained by simply controlling the temperature (Fig. 1), hence forming the basis of a rotary nanomotor controlled by temperature. Through a combined approach of experiments, we have investigated the structure of the nanocluster and its stimuli-responsive isomerization. The isomers possess distinct optical properties, which can be utilized for advancing intelligent sensors, catalysts, and controlled drug release in biomedical applications.

E- eclipsed configuration, S- staggered configuration. Color code: Au: yellow; Ag: blue; Cl: green (C, H, P, and some Cl are omitted for clarity).
Results
Synthesis and characterization of Au13Ag12 isomers
The Au13Ag12 nanocluster was synthesized by reduction of a mixed solution of Ph3PAuCl and AgSbF6 using NaBH4 in an ice bath (see Supplementary Methods). The nanoclusters were analyzed by electrospray ionization mass spectrometry (ESI-MS) in a positive mode. A weak mass peak at m/z = 6762.10 and an intense one at m/z = 3362.56 were observed in the mass spectrum (Fig. 2a), with the spacing of their isotope patterns being 1 and 0.5 (Fig. 2a, inset), hence, 1+ and 2+ charges, respectively. The two mass peaks correspond to [Au13Ag12(PPh3)10Cl8]+ (theoretical m/z: 6761.46 Da, deviation: 0.62 Da) and [Au13Ag12(PPh3)10Cl7]2+ (theoretical m/z: 3363.00 Da, deviation: −0.44 Da), respectively, with the latter (2+ ion) being formed via loss of a Cl− from the intact 1+ cluster during the ESI-MS analysis. X-ray crystallographic analysis (vide infra) confirmed homogeneous [Au13Ag12(PPh3)10Cl8]+, with no observation of [Au13Ag12(PPh3)10Cl7]2+. It is worth noting that loss of a Cl− ligand often occurs in ESI-MS analysis26. Therefore, the molecular formula of the synthesized nanoclusters is [Au13Ag12(PPh3)10Cl8]+, and the product is highly pure.

a Positive-mode ESI mass spectrum of the as-synthesized Au13Ag12 nanocluster. (M = Au13Ag12(PPh3)10Cl8) b Thin-layer chromatography separation of the Au13Ag12 using CH2Cl2/CH3OH (2:1, v/v) as an eluent. c 31P NMR spectra of the S-Au13Ag12 and E-Au13Ag12 in CD2Cl2.
Surprisingly, two species were identified in the Au13Ag12 product by thin-layer chromatography (TLC) with CH2Cl2/CH3OH (2:1, v/v) as the eluent27. The two distinct bands (Fig. 2b) indicate two isomers of Au13Ag12 since they share the same composition, hereafter denoted as E- and S-Au13Ag12 (vide infra). The bands were cut out and extracted with CH2Cl2, and their optical spectra indeed show different profiles of the two isomers of Au13Ag12 (see Supplementary Fig. 1). The ultraviolet–visible (UV-vis) spectrum of the mixed isomers exhibit peaks at 324, 361, 420, 500, and 656 nm. After TLC separation, the E-Au13Ag12 nanocluster shows three peaks at 361, 418, and 497 nm, whereas the S-Au13Ag12 isomer shows four peaks at 330, 423, 510, and 657 nm. The energy gaps of the S- and E-Au13Ag12 isomers are 1.72 and 2.17 eV, respectively (see Supplementary Fig. 1, inset). Interestingly, the pure S- and E-Au13Ag12 isomers show different 31P nuclear magnetic resonance (NMR) signals: 54.80 ppm for E-Au13Ag12 and 57.36 ppm for S-Au13Ag12 using Au(PPh3)Cl (31P NMR: 33.13 ppm) as the internal reference (Fig. 2c and see Supplementary Fig. 2). It demonstrates that the E-Au13Ag12 and S-Au13Ag12 nanoclusters have different structures (vide infra).
Crystal structures
Furthermore, we found that keeping a fresh solution of the Au13Ag12 nanocluster containing both isomers at 25 °C for 4 weeks led to a solution containing only the S-Au13Ag12 isomer. In contrast, under −10 °C the same isomeric mixture completely transforms to the E-Au13Ag12 isomer after 6 weeks. Crystallizations of the S-Au13Ag12 and E-Au13Ag12 isomers were performed via a vapor diffusion method, followed by X-ray diffraction analysis. The total structures of S-Au13Ag12 and E-Au13Ag12 are displayed in Supplementary Figs. 3 and 4. For the core frameworks, each isomer is composed of two icosahedral Au7Ag6 units fused together by sharing a common vertex of Au (Fig. 3a). The two Au5 pentagons at the ends of the rod are ligated by ten phosphine ligands. Two Cl ligands bind with two apical Ag atoms. Six Cl ligands bridge the two icosahedra via bonding with 10 equatorial Ag atoms (see Supplementary Fig. 5).

a, b Two icosahedral Au7Ag6 units share one common vertex of Au to form the Au13Ag12 nanocluster. c Metal configuration of E-isomer. d Metal configuration of S-isomer. Color code: Au: yellow; Ag: blue. Other atoms are omitted for clarity.
The central Au atom (enlarged in Fig. 3b) connects the two Au7Ag6 icosahedra and serves as the pivot for the rotamerization of the metal configuration of the Au13Ag12. At −10 °C, the two Au7Ag6 units are in an eclipsed (E) configuration with D5h symmetry (i.e., E-Au13Ag12, Fig. 3c), whereas at 25 °C the two Au7Ag6 units rotate by about 36° to form a staggered (S) configuration with D5d symmetry (S-Au13Ag12, Fig. 3d). We note that, at the lower temperature (−10 °C), the Au13Ag12 nanocluster prefers to adapt the configuration with a higher symmetry. The six chloride ligands—which bridge the two Au7Ag6 units—serve as the “belt” to drive the rotamerization, Fig. 4. In the E-Au13Ag12 isomer, there are five Cl ligands with each being bonded with two silver atoms (d-mode). For the same isomer, there is also a Cl ligand that is bonded with four silver atoms (q-mode). In the S-Au13Ag12 isomer, four Cl ligands are in the d-mode bonding, and two Cl ligands are each bonded with three silver atoms (t-mode).

Doubly (d), triply (t), and quadruply (q) binding patterns of six equatorial Cl ligands bridging the two icosahedra. Color codes: Au: yellow; Ag: blue; Cl: green.
Reversible transformation between the E- and S- isomer
The E-Au13Ag12 and S-Au13Ag12 isomers can be 100% selectively obtained at −10 and 25 °C, respectively. To test the reversibility of the isomeric transformation, we monitored the process by UV-vis absorption spectroscopy using the crystal sample of E-Au13Ag12 nanoclusters as the starting material. We first studied the E-Au13Ag12 nanocluster in a dichloromethane solution at 25 °C. The characteristic peak at 361 nm gradually decreased over time and simultaneously peaks at 330, 423, and 657 nm gradually increased. The peak at 418 nm also red-shifted to 510 nm during a period of 28 days, Fig. 5a. The 31P NMR test of the sample gave a doublet-splitting peak centered at 57.36 ppm (see Supplementary Figs. 6 and 7). These results indicate that the E-Au13Ag12 isomer slowly converts to the S-Au13Ag12 isomer at 25 °C. We next studied the transformation of the as-obtained S-Au13Ag12 isomer at −10 °C. The characteristic peaks at 330 and 657 nm disappeared after 42 days. Meanwhile, the peak at 361 nm increased and the peak at 510 nm blue-shifted to 418 nm, demonstrating that the S-Au13Ag12 isomer transforms to the E-Au13Ag12 counterpart (Fig. 5b). This is further supported by 31P NMR analysis, as the NMR peak appears at 54.80 ppm (see Supplementary Figs. 8 and 9). Of note, no byproduct was formed during the reversible transformation, evidenced by the ESI-MS tests (see Supplementary Figs. 10 and 11). These results imply that both isomers are stable, and the isomeric transformation process is reminiscent of typical phase transitions.

a E-Au13Ag12 conversion to S-Au13Ag12 at 25 °C. b The as-obtained S-Au13Ag12 conversion to E-Au13Ag12 at −10 °C.
Differential scanning calorimeter (DSC) method was used to investigate the isomeric transformation process and the corresponding enthalpy28. Starting from the S-Au13Ag12 isomer, a negative peak centered at 79.6 °C was observed during the DSC test, which points to endothermicity of the isomeric transformation of the S-Au13Ag12 to E-Au13Ag12 (Fig. 6a). Both UV-vis and 31P NMR analyses indicate that the S-Au13Ag12 isomer was partially converted to the E-Au13Ag12 isomer (see Supplementary Figs. 12 and 13). Furthermore, thermogravimetric analysis (TGA) revealed that the removal of Cl ligands from the nanoclusters occur at 175 °C (weight loss of ~3.7 wt%, consistent with the expected value of 4.0 wt%), Fig. 6b. Subsequently, the phosphine ligands began to desorb at ~210 °C, consistent with the desorption temperature reported for the phosphine-protected Au nanocluster28,29. The TGA results indicate that the Au13Ag12 nanoclusters are intact below 100 °C (see Supplementary Figs. 14 and 15); thus the observed endothermic process in the DSC analysis is solely associated with the isomeric transformation, rather than with any ligand loss.

a DSC analysis for the conversion of the S-Au13Ag12 to E-Au13Ag12 under a N2 atmosphere. b TGA test of the Au13Ag12 nanoclusters under a N2 atmosphere.
Owning to the higher symmetry of the E-Au13Ag12 (D5h) than the S-Au13Ag12 (D5d), the nanocluster is a potential prototype of thermal molecular motor where Gibbs free energy serves as the driving force. In a previous work, Teo et al.20,21,22,23 synthesized two isomers of Au13Ag12(PPh3)10Br8 nanoclusters; however, no isomeric rotamerization was observed, which can be attributed to the stronger Ag–Br bonds than Ag–Cl. Our study shows that employing the –Cl ligand, instead of –Br, leads to higher flexibility on Ag–halide bonds, which is essential and critical to achieve a more flexible cluster framework for the thermally responsive, reversible transformation between the E-Au13Ag12 and S-Au13Ag12 isomers. Such a Cl-ligand-induced rotary nanomotor is simulated based on the experimental structures (see Supplementary Movie 1). For practical applications, the temperature will serve as the driving force.
In summary, two isomers of the biicosahedral [Au13Ag12(PPh3)10Cl8]+ (counterion: [SbF6]−) nanocluster (i.e., S-Au13Ag12 and E-Au13Ag12) are discovered, and these two isomers are reversibly transformable by controlling the temperature. The metal configuration of the E-Au13Ag12 isomer possesses a higher symmetry (D5h) than that of the S-Au13Ag12 isomer (D5d) and is preferably formed at low temperature (−10 °C). As the temperature increases to 25 °C, the S-Au13Ag12 isomer (lower symmetry) is exclusively formed. This study shows that the alloying and ligand engineering (i.e., Ag–halide bond) provide a rational strategy to make the framework of metal nanoclusters more flexible for achieving the conformational isomerism, which has the potential to be applied in designing intelligent molecular engines with Gibbs free energy as the driving force of the activity.
Methods
Synthesis of Au13Ag12 nanoclusters
Au(I)PPh3Cl (25 mg, dissolved in 2 mL chloromethane/methanol with v:v = 1:1) was mixed with AgSbF6 (17.2 mg, dissolved in 2 mL chloromethane/methanol with v:v = 1:1). The solution was stirred in dark and air atmosphere. Then the solution was cooled using an ice bath for 30 min, followed by dropwise addition of NaBH4 solution (2 mg, dissolved in 4 mL ice cold methanol). The mixture was kept in the dark and stirred for another 24 h. Next, the temperature was increased slowly to 25 °C. The mixture was then dried via vacuum evaporation and washed with hexane (2 × 1 mL), leaving a black solid. Finally, the black solid was dissolved in 2 mL dichloromethane/methanol (v:v = 1:1) and centrifuged at 10,000 rpm for 5 min. Red, plate-like crystals of E-Au13Ag12 and S-Au13Ag12 were obtained via slow diffusion of diethyl ether into the cluster solution over 6 weeks at −10 or 25 °C. The yields are 26.0% for E-Au13Ag12 and 34.9% for S-Au13Ag12 based on the consumption of Ph3PAuCl.
Data availability
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 1888164–1888165. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The datasets generated and/or analyzed during this study are available from the corresponding author upon reasonable request.
References
Petermayer, C. & Dube, H. Indigoid photoswitches: visible light responsive molecular tools. Acc. Chem. Res. 51, 1153–1163 (2018).
Wei, P. et al. Multiple yet controllable photoswitching in a single AIEgen system. J. Am. Chem. Soc. 140, 1966–1975 (2018).
Zhang, B. et al. Thermally-induced reversible structural isomerization in colloidal semiconductor CdS magic-size clusters. Nat. Commun. 9, 2499–2508 (2018).
Fan, X., Wang, J., Wu, K., Zhang, L. & Zhang, J. Isomerism in titanium-oxo clusters: molecular anatase model with atomic structure and improved photocatalytic activity. Angew. Chem. Int. Ed. 58, 1320–1323 (2019).
Wang, H., Zhang, J. & Xie, Z. Reversible photothermal isomerization of carborane-fused azaborole to borirane: synthesis and reactivity of carbene-stabilized carborane-fused borirane. Angew. Chem. Int. Ed. 129, 9326–9329 (2017).
Sun, J. et al. Stimuli-directed dynamic reconfiguration in self-organized helical superstructures enabled by chemical kinetics of chiral molecular motors. Adv. Sci. 5, 1700613–1700621 (2018).
Zhang, J. et al. The proton-controlled synthesis of unprecedented diol functionalized Anderson-type POMs. Chem. Commun. 52, 2378–2381 (2016).
Zhang, J. et al. Unprecedented χ isomers of single-side triolfunctionalized Anderson polyoxometalates and their proton-controlled isomer transformation. Chem. Commun. 51, 9097–9100 (2015).
Konishi, K., Iwasaki, M. & Shichibu, Y. Phosphine-ligated gold clusters with core+exo geometries: unique properties and interactions at the ligand−cluster interface. Acc. Chem. Res. 51, 3125–3133 (2018).
Cook, A. W. & Hayton, T. W. Case studies in nanocluster synthesis and characterization: challenges and opportunities. Acc. Chem. Res. 51, 2456–2464 (2018).
Lei, Z., Wan, X.-K., Yuan, S.-F., Guan, Z.-J. & Wang, Q.-M. Alkynyl approach toward the protection of metal nanoclusters. Acc. Chem. Res. 51, 2465–2474 (2018).
Yao, Q., Chen, T., Yuan, X. & Xie, J. Toward total synthesis of thiolate-protected metal nanoclusters. Acc. Chem. Res. 51, 1338–1348 (2018).
Zheng, K. et al. Motif-mediated Au25(SPh)5(PPh3)10X2 nanorods with conjugated electron delocalization. Nano Res. 12, 501–507 (2019).
Zhang, J. et al. Diphosphine-induced chiral propeller arrangement of gold nanoclusters for singlet oxygen photogeneration. Nano Res. 11, 5787–5798 (2018).
Nieto-Ortega, B. & Bürgi, T. Vibrational properties of thiolate-protected gold nanoclusters. Acc. Chem. Res. 51, 2811–2819 (2018).
Chakraborty, P., Nag, A., Chakraborty, A. & Pradeep, T. Approaching materials with atomic precision using supramolecular cluster assemblies. Acc. Chem. Res. 52, 2–11 (2019).
Zhou, M. et al. Three-orders-of-magnitude variation of carrier lifetimes with crystal phase of gold nanoclusters. Science 364, 279–282 (2019).
Jin, R., Zeng, C., Zhou, M. & Chen, Y. Atomically precise colloidal metal nanoclusters and nanoparticles: fundamentals and opportunities. Chem. Rev. 116, 10346–10413 (2016).
Zhuang, S. et al. Fcc versus non-fcc structural isomerism of gold nanoparticles with kernel atom packing dependent photoluminescence. Angew. Chem. Int. Ed. 58, 4510–4514 (2019).
Teo, B. K. & Zhang, H. Molecular machines: molecular structure of [(p-Tol3P)10Au13Ag12Cl8](PF6)-a cluster with a biicosahedral rotorlike metal core and an unusual arrangement of bridging ligands. Angew. Chem. Int. Ed. 31, 445–447 (1992).
Teo, B. K., Shi, X. B. & Zhang, H. Cluster of clusters. Structure of a novel cluster [(Ph3P)10Au13Ag12Br8](SbF6) containing an exact staggered-eclipsed-staggered metal configuration: evidence of icosahedral units as building blocks. J. Am. Chem. Soc. 113, 4329–4331 (1991).
Teo, B. K., Shi, X. B. & Zhang, H. Cluster rotamerism of a 25-metal-atom cluster [(Ph3P)10Au13Ag12Br8]+ monocation: a molecular rotary unit. J. Chem. Soc. Chem. Commun., 1195–1196 (1992).
Teo, B. K. & Zhang, H. Cluster of clusters. Structure of a new 25-metal-atom cluster [(p-Tol3P)10Au13Ag12Cl7](SbF6)2 containing a nearly staggered-eclipsed-staggered metal configuration and five doubly bridging ligands. Inorg. Chem. 30, 3115–3116 (1991).
Tian, S. et al. Structural isomerism in gold nanoparticles revealed by X-ray crystallography. Nat. Commun. 6, 8667–8672 (2015).
Chen, Y. et al. Isomerism in Au28(SR)20 nanocluster and stable structures. J. Am. Chem. Soc. 138, 1482–1485 (2016).
Song, Y. et al. How a single electron affects the properties of the “non-superatom” Au25 nanoclusters. Chem. Mater. 28, 2609–2617 (2016).
Liao, L. et al. Quantitatively monitoring the size-focusing of Au nanoclusters and revealing what promotes the size transformation from Au44(TBBT)28 to Au36(TBBT)24. Anal. Chem. 88, 11297–11301 (2016).
Molard, Y. Clustomesogens: liquid crystalline hybrid nanomaterials containing functional metal nanoclusters. Acc. Chem. Res. 49, 1514–1523 (2016).
Liu, C., Abroshan, H., Yan, C., Li, G. & Haruta, M. One-Pot synthesis of Au11(PPh2Py)7Br3 for the highly chemoselective hydrogenation of nitrobenzaldehyde. ACS Catal. 6, 92–99 (2016).
Acknowledgements
G.L. acknowledges financial support from the National Natural Science Foundation of China (No. 21701168) and Liaoning Revitalization Talents Program (XLYC1807121).
Author information
Authors and Affiliations
Contributions
G.L., C.W., H.A., and R.J. conceived the work. Z.Q., J.Z., S.L., and G.L. performed the experiments. G.L., H.A., and R.J. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Yuichi Negishi and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qin, Z., Zhang, J., Wan, C. et al. Atomically precise nanoclusters with reversible isomeric transformation for rotary nanomotors. Nat Commun 11, 6019 (2020). https://doi.org/10.1038/s41467-020-19789-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-19789-4
This article is cited by
-
Impact of the metal core on the electrochemiluminescence of a pair of atomically precise Au20 nanocluster isomers
Communications Chemistry (2023)
-
Atomically precise copper dopants in metal clusters boost up stability, fluorescence, and photocatalytic activity
Communications Chemistry (2023)
-
Key factors for connecting silver-based icosahedral superatoms by vertex sharing
Communications Chemistry (2023)
-
Tailoring optical and photocatalytic properties by single-Ag-atom exchange in Au13Ag12(PPh3)10Cl8 nanoclusters
Nano Research (2022)
-
Three-stage alloying of [Ag44(p-MBA)30]4− cluster with [Au2(p-NTP)2Cl]−
Nano Research (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
