
Abstract
High pressure reactivity of phosphorus and hydrogen is relevant to fundamental chemistry, energy conversion and storage, and materials science. Here we report the synthesis of (PH3)2H2, a crystalline van der Waals (vdW) compound (I4cm) made of PH3 and H2 molecules, in a Diamond Anvil Cell by direct catalyst-free high pressure (1.2 GPa) and high temperature (T ≲ 1000 K) chemical reaction of black phosphorus and liquid hydrogen, followed by room T compression above 3.5 GPa. Group 15 elements were previously not known to form H2-containing vdW compounds of their molecular hydrides. The observation of (PH3)2H2, identified by synchrotron X-ray diffraction and vibrational spectroscopy (FTIR, Raman), therefore represents the discovery of a previously missing tile, specifically corresponding to P for pnictogens, in the ability of non-metallic elements to form such compounds. Significant chemical implications encompass reactivity of the elements under extreme conditions, with the observation of the P analogue of the Haber-Bosch reaction for N, fundamental bond theory, and predicted high pressure superconductivity in P-H systems.
Similar content being viewed by others


Introduction
The history of the layered structures of phosphorus has been intimately related to pressure since 19141, when Percy Bridgman, who may be considered the founder of high pressure science, first synthesized black phosphorus (Pblack), whose characteristic crystalline layered structure corresponds to the thermodynamically stable allotrope of the element2. Rhombohedral A7, another layered structure of phosphorus, was later discovered by compression of Pblack above 5 GPa3. Recently, a high pressure study, based on synchrotron X-ray diffraction (XRD) in Diamond Anvil Cell (DAC), has made another mark in the history of the layered structures of phosphorus, providing a clear insight about the mechanism of interlayer bond formation and significantly raising the pressure limit for the existence of the phosphorus layers up to ~30 GPa at room T4,5.
In 2014, the advent of phosphorene6, a 2D corrugated monoatomic layer of phosphorus atoms with extraordinary properties, whose stacking actually builds up the the crystal structure of Pblack, has sharply raised the attention of the scientific community about the layered structures of element 15, with considerable experimental and theoretical efforts nowadays in attempting to stabilize and functionalize the phosphorus layers by the introduction of molecular fragments7 or atoms, like for example H8.
During the last two decades, the generation of pressure in the GPa range has greatly expanded the horizon of chemistry under extreme conditions9. In particular, the combination of pressure with high temperature or electronic photo-excitation has been shown to be a very effective and extremely powerful tool for opening selective reactive paths10,11, activating chemical reactivity in notoriously non interacting systems at ambient conditions12, like here P and H, and for synthesizing new unexpected compounds13,14, thus suggesting the idea of investigating the so far unexplored chemistry of the phosphorus-hydrogen system under high pressure conditions.
Besides H-functionalization and stabilization of the layered structures of phosphorus, the direct chemical reactivity between elemental phosphorus and hydrogen under high pressure conditions is indeed currently of extreme interest for relevant issues essentially related to the chemistry and physics of phosphorus hydrides. Molecular hydrides of non-metallic elements have indeed always attracted the attention of high pressure chemists, physicists and materials scientists for their potential applications as superconducting15 and H-storage materials, due to their high H content and to the ability of forming stoichiometric van der Waals (vdW) compounds in the presence of H216,17. Since the first report of a vdW solid made of He and N218, many H2-containing vdW compounds involving elements from group 14 to group 18, including nobles gases, simple diatomics and molecular hydrides of non-metallic elements, have been experimentally observed at high pressure: CH4(H2)419, CH4H219, (CH4)2H219, SiH4(H2)220, GeH4(H2)221, N2(H2)222, (N2)6(H2)722,23, (O2)3(H2)424, (H2O)6H225, (H2O)H225, (H2S)2H226, (H2Se)2H227, (HI)2H228, Ar(H2)229, Kr(H2)230, Xe(H2)831,32. Nevertheless, among those formed by H2 and by the molecular hydride of a non-metallic element reported so far, none involves any of group 15 elements33. Within this picture, if the elements in the periodic table are to be considered tiles arranged on the basis of their electronic configuration, which determines their properties, then group 15 elements, named pnictogens, represent the missing tiles in this arrangement.
Phosphorus is here of particular relevance. Indeed the recent discovery of high superconducting Tc of 203 K in H2S at high pressure34, in agreement with the Bardeen-Cooper-Schrieffer (BCS) phonon mediated theory of high temperature superconductivity35, has further promoted the search for a similar behavior in the hydrides of neighbor elements in the periodic table. In particular, in the case of phosphorus the report of high pressure superconductivity in phosphine (Tc > 100 K, P > 200 GPa)36, has stimulated several theoretical studies aimed at exploring the structure and stability of PH3 at high pressure and the substantially unknown high pressure behavior of the phosphorus-hydrogen system, with the prediction of superconducting layered structures formed by these two elements above 80 GPa37,38,39.
Finally, the formation, stability and decomposition of PH3 in presence of H2 are relevant astrochemical issues40 related to the composition of giant planets, such as Jupiter and Saturn41,42, and their moons43, where PH3 and H2 have been detected.
Within this picture, in this paper we report a synchrotron XRD and vibrational spectroscopy (FTIR and Raman) study of the high pressure chemistry occurring between black phosphorus and molecular hydrogen at pressure of 1.2–1.5 GPa and temperature ≲1000 K, where phosphorus is in the layered crystalline orthorhombic structure (A17), commonly known as black phosphorus, and H2 is liquid44 (Fig. 1). In these thermodynamic conditions PH3 is directly synthesized from the elements. On further room T compression, between 3.5 and 4.1 GPa, PH3 combined with excess H2 to form the crystalline vdW compound (PH3)2H2, whose identification has remarkable implications. Pressure was statically generated by means of membrane DAC and temperature by laser heating (LH), with Pblack acting at the same time as reactant and laser absorber and H2 as a reactant and pressure transmitting medium.

a Phase diagram of phosphorus (black lines), showing the stability regions of the orthorhombic (A17, Pblack), rhombohedral (A7) and pseudo simple-cubic (p-sc) structures4,5. The melting line of H273 (blue line) and laser heating conditions (red point at P = 1.2 GPa, T ≈ 1000 K) are also displayed. b–g Microphotographs showing the sample aspect after loading (b), before, during and after LH1 (c–e), after LH2 (f), and after LH3 (g) at pressure ranging between 1.2 and 1.5 GPa (LH1, LH2, and LH3, respectively, indicate the first, the second and the third laser heating). The scale bars in the top right corner of b, e correspond to 100 μm.
Results
X-Ray Diffraction
After loading (see “Methods”), the sample of Pblack and H2 was compressed to 1.2 GPa, where three laser irradiations were performed. The irradiations power (<3 W at laser output) and duration (up to ~30 s) were carefully increased up to visually observe the real time chemical transformation of the sample, which occurred in the 900 ± 100 K temperature interval, with only a slight pressure drift from 1.2 to 1.3 GPa, and then to 1.5 GPa, respectively, after the second (LH2) and third (LH3) irradiation (Fig. 1).
Each irradiation produced an increasing transformation of Pblack, as confirmed by the intensity decrease of the Pblack peaks in the diffraction patterns acquired before and after laser heating. After LH3 the absence of diffraction peaks indicated the complete consumption of Pblack, suggesting the formation of an amorphous or liquid product in the laser heated area (Fig. 2).

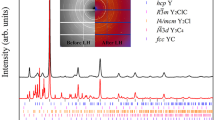
Integrated panoramic XRD patterns and corresponding detector images of the sample, acquired at room T before laser heating (LH) at 1.2 GPa (black trace, bottom image), after the third laser heating (LH3) at 1.5 GPa (red trace, middle image), and after compression at 4.1 GPa (blue trace, top image). The blue ticks and labels respectively refer to the refined peak positions of the crystalline product at this pressure (Supplementary Note 1) and to the corresponding h,k,l indexing. The tetragonal I4cm structure of P atoms as obtained from single-crystal data at 5.5 GPa is also shown in the top left corner of the Figure (see the “X-Ray Diffraction” section).
The sample was then compressed at room T and between 3.5 and 4.1 GPa the sudden appearance of single diffraction spots in the detector image, observed up to the highest explored pressure (5.5 GPa), unambiguously marked the formation of a crystalline product (Fig. 2). A careful XRD mapping of the sample over a grid with 10 μm spacing was performed to identify the regions where single spot diffraction could be observed. These regions were further explored by higher resolution mapping (4 μm spacing grid) and single-crystal patterns acquired on selected points of the laser heated area.
As H2 is liquid below 5.7 GPa (Fig. 1) and H atoms are weak X-ray scatterers, only information about the P atoms could be evinced from our single-crystal data, whose structure solution at 5.5 GPa indicates a tetragonal structure (a = 7.6075(6) Å, b = 7.6075(6) Å, c = 6.3303(13) Å, α = 90∘, β = 90∘, γ = 90∘, V = 366.36(9) Å3, Z = 8) belonging to space group I4cm (n. 108, \({C}_{{\rm{4v}}}^{10}\)), where the shortest distance between P atoms is 3.608 Å (Fig. 2 and Supplementary Note 1) and which does not correspond to any known structure of phosphorus. An equivalent solution corresponding to centrosymmetric space group I4/mcm (n. 140, D\({\,}_{4h}^{18}\)) was initially considered (see “Discussion”).
The first reaction product that we thought about was of course PH3. Unfortunately, even if PH3 is expected to solidify at higher pressure with respect to NH3 (1.0 GPa) due to the smaller electric dipole moment and to the absence of H-bonding45,46, the solidification pressure of PH3 at room T is unknown.
Furthermore, the high pressure structure of solid PH3 is also unknown and only ambient pressure low T XRD data by Natta and Casazza dating back to 193047 are available in the literature, indicating that PH3 crystallizes into a compact packing face centered cubic structure, possibly belonging to space group \({T}_{{\rm{h}}}^{2}\) (Pn\(\bar{3}\), P2/n\(\bar{3}\), n. 201) or \({O}_{{\rm{h}}}^{4}\) (Pn\(\bar{3}\)m, P42/n\(\bar{3}\) 2/m, n. 224), none of which is compatible with our single-crystal data (Supplementary Note 2).
Even considering the P positions in the I4cm and I4/mcm structures obtained from the single-crystal data as occupied by PH3 molecules, some inconsistencies emerge with a compact packing structure. Indeed, assuming orientationally disordered spherical shaped PH3 molecules in contact with each other, deriving the molecular volume using as molecular radius half of the shortest distance between P atoms ((4/3)π(3.608/2)3 = 24.592 Å3, in agreement with literature48,49), and considering 8 molecules per unit cell, then a filled volume of 196.736 Å3 out of the 366.36 Å3 unit cell volume obtained from the single-crystal data can be estimated, corresponding to a 0.537 filling ratio, which is significantly lower than the 0.74 ratio expected for a close packing structure.
This occurrence, indicating the presence of free volume, which can not be accounted for by a compact packing of PH3, provided the first hints suggesting the presence of interactions between PH3 molecules and a different composition of our reaction product, as indeed confirmed by the spectroscopic data.
Fourier Transform InfraRed absorption spectroscopy
The Fourier Transform InfraRed (FTIR) spectra acquired after LH3 at 6.7 GPa and at different pressures during decompression are shown in Fig. 3 with the relevant band frequencies listed in Table 1. At 6.7 GPa, infrared absorption maxima are observed at 983, ~1100 (out of scale) 2358, 3346, 3466, 4121, 4250, 4625, and 4814 cm−1.

a Room T FTIR absorption spectra of the sample acquired at 6.7 GPa after LH3 and at different pressure values during decompression to ambient conditions. The two bands at 3346 and 4121 cm−1 labeled in blue disappear on releasing pressure to 3.1 GPa. The spectra have been vertically translated for clarity and the values on the absorbance scale are intended for relative comparison. The break on the wavenumber axis excludes the spectral range corresponding to the Ia diamond saturating absorptions, which partially covers the ν4 absorption of PH3. The absorption features at ~2640 cm−1 (6.7 GPa) and ~2900 cm−1 (appearing on releasing pressure to 3.1 GPa) are assigned respectively to the diamond from the ambient pressure reference and to traces of oil on the optics of the interferometer. b Significant spectral regions of type-1 (red, lower trace) and type-2 (blue, upper trace) Raman spectra acquired on different selected spots of the mapping grid across the sample at 6.8 GPa and room T after LH. With respect to type-1 spectrum the type-2 one clearly shows the simultaneous presence of PH3 and of the extra band at 4123 cm−1 in the H2 stretching region at lower frequency compared to pure H2.
The last three bands can be assigned to infrared active absorptions of H2 (4250 (Q1(1), 4625 (Q1(1)+S0(0)), and 4814 (Q1(1)+S0(1)) cm−1) in agreement with literature44.
Even if the infrared bands of solid PH3 have been measured only at low T50,51 and those of liquid PH3 only at low temperature and modest high pressure (up to 35 atm)52, the bands observed at 983, ~1100, 2358, and 3466 cm−1 can be confidently assigned to the fundamental and combination vibrational modes of PH3, as indicated in Table 1.
Finally, the two remaining bands at 3346 and 4121 cm−1 can not be assigned either to PH3 or H2 (Supplementary Note 3).
On decompression from 6.7 to 3.1 GPa, below the crystallization threshold of the reaction product, the two extra bands at 3346 cm−1 and 4121 cm−1 disappear. All the other bands of PH3 exhibit a high-frequency shift, as typically observed in H-bonded systems when releasing pressure, and the bands of H2 a low-frequency shift, as expected. On further decompression to ambient conditions, both PH3 and H2 bands shift to lower frequency, until disappearing with the opening of the cell.
No bands could be detected in the FTIR spectra after completely releasing the membrane pressure and opening/closing the cell under glove box in an inert atmosphere.
Raman spectroscopy
A detailed Raman mapping, consisting of a 130 × 130 μm2 mesh with 10 μm grid spacing, was performed to gain insight about the reaction products and their spatial distribution within the sample at each pressure point during decompression, covering the entire frequency range between 40 and 4700 cm−1. The analysis of the Raman spectra acquired across the sample at 6.8 GPa revealed the presence of two limit spectra, referred to in the following respectively as type-1 and type-2 Raman spectra, and a variety of combinations of them, with significant bands in three spectral regions: 200–1250 cm−1, 2000–2600 cm−1, and 4000–4300 cm−1 (Fig. 3 and Supplementary Fig. 5).
Type-1 Raman spectrum (Fig. 3, lower panel, red trace) exhibits broad bands at 388, 449, 613, 833, 1064, 2198, 2520, and 4212 cm−1, whereas in type-2 Raman spectrum additional bands are detected at 984, 1114, 2352, 4123 cm−1. The two broad features observed in all the spectra at 388 and 449 cm−1 can be assigned to amorphous Pred (Supplementary Note 5). The weak band at 2198 cm−1, which has significantly lower frequency compared to phosphine (PH3 2321 cm−1)52, or to higher phosphane homologues like diphosphane (P2H4 2283 cm−1)53 and triphosphane (P3H5 2267 cm−1 at 193 K)53, is compatible with the typical vibrational frequency of P–H bonds and further confirms the occurrence of chemical reactivity between P and H2. The sharp band at 4212 cm−1 identifies the characteristic stretching vibration of H2 and those at 613, 833, and 1064 cm−1 the corresponding pressure broadened S0(1), S0(2), and S0(3) rotational bands (S0(0) overlaps with the band of Pred at 388 cm−1)54.
Type-2 Raman spectrum exhibits additional bands at 984, 1114, and 2352 cm−1, which can be readily assigned respectively to the ν2, ν4 and ν1: ν3 fundamental modes of PH351, consistently with the IR spectra (Fig. 3, lower panel, blue trace). However, differently from the IR spectra, no combination bands of PH3 are detected in the Raman spectra. The Raman band at 4123 cm−1, observed only in type-2 Raman spectra together with the presence of PH3, almost exactly matches the corresponding IR absorption band at 4121 cm−1 (6.7 GPa) and can not be assigned either to P, H2 or PH3.
As in the case of the IR spectra, during decompression to 3.1 GPa, below the crystallization pressure of the reaction product observed by XRD, the extra band at 4123 cm−1 disappears, while the bands of PH3 exhibit a high-frequency shift (Supplementary Fig. 5) and those of H2 a low-frequency shift. Both PH3 and H2 bands frequencies all undergo a low-frequency shift on further decompression and disappear after opening the cell.
At 1.95 GPa three sharp bands, unambiguously identified as the characteristic \({A}_{{\rm{g}}}^{(1)}\), Eg and \({A}_{{\rm{g}}}^{(2)}\) signatures of Pblack, respectively appear at 373, 443, and 467 cm−1, remaining observable on decompression down to ambient pressure (Supplementary Fig. 5). Even if the detection of these three peaks could suggest an incomplete transformation of Pblack and its missed observation, the Raman spectra acquired at 0.2 GPa and at ambient pressure on the recovered sample show characteristic bands at ~385 cm−1, which is not present in any of the Pred forms nor in Pblack (Supplementary Fig. 7), and at 2245 cm−1 in the P–H stretching region, which closely resemble the Raman spectra reported by Yuan and coauthors for the recovered products of the decomposition of PH3 quenched from 25 GPa to 31 GPa55 (Supplementary Fig. 8), thus suggesting a laser-induced decomposition of PH3 during the acquisition of the Raman spectra, after releasing pressure below the crystallization threshold of the reaction product.
Interestingly, the P–H stretching band observed at 2200 cm−1 at 6.8 GPa exhibits a high-frequency shift to ~2226 cm−1 on releasing pressure to 3.1 GPa, which further increases to 2245 cm−1 on releasing pressure to ambient conditions (Supplementary Fig. 5), providing evidence of the presence of H-bonding in the recovered solid product. Unfortunately, no additional insight could be gained on the recovered solid material responsible for type-1 Raman spectrum, which appears to consists of a hydrogenated (H-functionalized) mixture of amorphous Pred and crystalline Pblack.
Discussion
Our data provide clear experimental evidence for direct high-pressure and high-temperature chemical reactivity between elemental Pblack and H2. The resulting formation of PH3 according to the following chemical equation
represents the so far unreported catalyst-free phosphorus analogue of the nitrogen-based Haber-Bosch reaction for the synthesis of NH3 (Supplementary Note 9).
Compressing PH3 in excess H2 at room T, between 3.5 and 4.1 GPa, the XRD data indicate the crystallization of a reaction product. As type-1 Raman spectrum was assigned to an amorphous solid product, we related the X-ray diffraction pattern of our crystalline product to type-2 Raman spectra, in which PH3 is observed, and considered the P positions of the corresponding structure to be occupied by PH3 molecules. The presence of PH3 in type-2 Raman spectra is always associated to the detection of an extra band in the H2 stretching region at lower frequency compared to pure H2, which disappears on releasing pressure below the crystallization threshold of the crystalline reaction product. A similar behavior is observed in the IR spectra, where an extra band is detected at 4121 cm−1 (6.7 GPa), almost exactly coinciding with the frequency of the extra Raman band at 4123 cm−1 (6.8 GPa).
Whereas the sharp higher frequency band perfectly matches with the literature data about H2 molecules in crystalline phase I (4250 cm−1 in IR and 4212 cm−1 in Raman at 6.8 GPa)56, the unassigned and unexpected broader extra band at lower frequency (4121 cm−1 at 6.7 GPa in IR and 4123 cm−1 at 6.8 GPa in Raman), indicates the presence of a second type of H2 molecules, which experience a significant weakening of the bond force constant (5.15%, average value between 4.30% Raman and 6.00% IR weakening), likely due to a different local force field, as indeed consistently attested by their larger bandwidth in comparison to pure H2.
A possible interpretation for this occurrence is the formation of a van der Waals crystalline compound made of PH3 and H2 molecules with (PH3)2H2 stoichiometry and a tetragonal Al2Cu-like structure belonging to I4cm space group26, where PH3 and H2 respectively occupy 8c (Cs) and 4a (C4) Wyckoff sites (Fig. 4). In this structure four molecules of PH3 are located on a plane parallel to the [a, b] direction at 0.5z and occupy the positions around a 4-fold rotation axis (C4) along the c direction. Four additional molecules occupy the positions generated by a rotation along C4 and a translation along +0.5z, giving rise to alternatively rotated layers of PH3 molecules.

Crystal structure of the (PH3)2H2 vdW compound at 5.5 GPa and room T (a) with views along the c (b), a (c), and b (d) crystal axes. The orange spheres represent the PH3 molecules, whereas the white spheres represent the H2 molecules. The size of the spheres has been drawn by estimating their radius as the sum of the P–H bond length (1.421 Å)2 and H van der Waals radius (1.20 Å)49 in the case of PH3 (2.62 Å) and as the sum of the H van der Waals radius (1.20 Å)49 and half of the H–H distance (0.742 Å)74 in the case of H2 (1.571 Å).
This interpretation of our data is in agreement with the I4cm tetragonal structure of P atoms obtained from the single-crystal data and also accounts for the existence of free volume in the unit cell, in the case PH3 only would be present.
Furthermore, in this structure the H2 molecules are encaged within square antiprismatic voids delimited by eight PH3 molecules (four on one layer and four on the adjacent layer) and occupy a single type of crystal site (C4), corresponding to 4a Wyckoff positions, whose occupancy, according to group theory, is consistent with the appearance of one infrared and Raman active crystal component for the H2 stretching vibration (Supplementary Note 6 and Supplementary Fig. 10).
In addition, the occupation by PH3 molecules of 8c Wyckoff sites (Cs), is consistent with the splitting of the (ν1 + ν4);(ν3 + ν4) combination band. The extra band observed in the IR spectra at 3346 cm−1 at 6.8 GPa, disappearing on decompression to 3.1 GPa, can be thus assigned to PH3 molecules forming the (PH3)2H2 crystal structure (Supplementary Note 6 and Supplementary Fig. 10).
A density of 1.269 g cm−3 can be calculated at 5.5 GPa from the refinement of the single-crystal data, with 2.89% in weight of H2 and total 11.5% in weight of H (H2 + H in PH3).
The molecular nature of the reaction product is further confirmed by the bulk modulus B = 6.7 ± 0.8 GPa derived from the 2nd order Birch-Murnaghan equation of state in the investigated pressure range, which is in absolute agreement with analogous systems26, and by the pressure evolution of the nearly constant c/a axial ratio, which indicates an almost isotropic compression within the applied pressure range (Supplementary Note 7).
Pressure has greatly extended the number of known hydrides synthesized under high-density conditions15,33. Among non metallic elements, H2-containing hydrides have been reported so far in literature for elements ranging from group 14 to group 18 of the periodic table, and include van der Waals hydrides of noble gases, simple diatomic molecules, and covalent molecular hydrides (Supplementary Fig. 16).
If among these hydrides we only consider those involving elements which are able to form covalent molecular hydrides (Supplementary Fig. 17) and particularly focus on those which have been reported to adopt a I4cm (I4/mcm) crystal structure with X2H2 composition, where X represents the corresponding molecular hydride (Supplementary Fig. 18), then we observe that this structure has been experimentally reported in the case of: carbon, with methane (CH4)19, for group 14; sulphur, with H2S26, and selenium, with H2Se27, for group 16; and iodine, with HI57, for group 17. Interestingly, to the best of our knowledge no such structure has been reported so far for any of the elements of group 15.
In particular (CH4)2H2, (H2S)2H2, (H2Se)2H2 and (HI)2H2 all reportedly exhibit the same I4/mcm structure, with the H2 and X molecules respectively occupying 4a and 8h Wyckoff positions, whereas (PH3)2H2 exhibits a I4cm structure with the H2 and PH3 molecules occupying the 4a and 8c Wyckoff positions. The I4cm and I4/mcm structures are closely related and only differ for the presence of an inversion center in I4/mcm, with identical lattice parameters and atomic positions. Noticeably, even if the occupation of 8h Wyckoff positions of C2v site symmetry by CH4, H2S, H2Se and HI, respectively in (CH4)2H2, (H2S)2H2, (H2Se)2H2 and (HI)2H2 does not rise any symmetry issue (such as in the case of PH3 (C3v) occupying a C2v site in the I4/mcm structure), no infrared spectra for any of these compounds have been acquired in the H2 stretching region, where, according to the analysis of the Davydov components activity using group theory arguments, the appearance of an extra band would unambiguously support the formation of a structure belonging to the I4cm rather than to the I4/mcm space group (Supplementary Note 6). Indeed, the possibility of the I4cm lower symmetry structure has been proposed also for (H2S)2H226, whereas the I4/mcm structures of (H2Se)2H2 and (HI)2H2 were assigned according to similarity with (H2S)2H2, thus suggesting all these structures to belong to I4cm rather than I4/mcm space group. Furthermore, the consistency of the IR and Raman optical activity with the application of group theory to the crystal symmetry, indicates that the PH3 molecules are not randomly oriented and that their orientations reflect the symmetry and periodicity of the intermolecular potential originating from their symmetry. This apparently contrasts with the orientational disorder reported for CH4, H2S, H2Se, and HI, respectively, in (CH4)2H2, (H2S)2H2, (H2Se)2H2, and (HI)2H2, which has been speculated from the behavior of the pure hydrides, without any conclusive evidence to support it like IR absorption spectra in the H2 stretching region (Supplementary Note 10)58,59.
The identification of (PH3)2H2 thus represents the discovery of the missing tile for group 15, specifically corresponding to phosphorus, in the puzzle of the periodic properties of non-metallic elements, which are able to form van der Waals molecular compounds containing their covalent hydrides and H2 molecules (Fig. 5).

Non-metal region of the Periodic Table showing the elements (yellow and orange) able to form crystalline X2H2 vdW compounds (X = CH419, PH3, H2S26, H2Se27, HI57) having the I4cm (I4/mcm) structure, with P (X = PH3), highlighted in orange, representing the so far missing tile for group 15 elements reported in this study (see text).
A further chemical insight can be gained from the Raman data. All these five X2H2 isostructural compounds feature the extra Raman band in the H2 stretching region, due to the vibration of the H2 molecules inside their structure, in addition to the signal of the surrounding pure H2, which is always present as excess reactant from the synthesis. These frequencies are listed in the Table in Fig. 6, together with the corresponding frequency shift with respect to pure H2. An interesting feature emerging from this comparison is that, at similar high-pressure conditions, the frequency shift of the extra band with respect to pure H2 is always negative, except in the case of methane. According to the valence shell electron pair repulsion (VSEPR) theory, and to the fulfillment of the octet rule for the outer electronic shell45, the main difference between methane and the other hydrides is that methane does not possess an electron lone pair on carbon, whereas PH3, H2S, H2Se and HI all have at least one electron lone pair on the hydride forming element (Fig. 6).

Upper panel. Table reporting the Raman frequency (cm−1) of the H–H stretching vibration of H2 molecules in different isostructural X2H2 crystals (\({\nu }_{{H}_{2}}\)(X2H2) with X = CH419, PH3, H2S26, H2Se27, and HI57) and their frequency shift (Δν) with respect to bulk H2 (\({\nu }_{{H}_{2}}\)(H2)) in comparable pressure conditions. Ref. 19 directly provides the frequency shift value for CH4. Lower panel. The molecular structures of CH4, PH3, H2S, H2Se, and HI, drawn according to the VSEPR theory and fulfilling the octet rule for the electron outer shell, are shown to highlight the absence of lone pairs on C in CH4, comparing to the other hydride forming elements.
The presence of lone pairs is typically associated to the ability of forming H-bonding, as indeed observed for all these systems, and of behaving as an electron donor Lewis base. H-bonding between the corresponding hydride molecules, evidenced by a negative frequency shift with increasing pressure of the internal stretching modes involving H atoms, has been indeed reported for (H2S)2H2, (H2Se)2H2, and (HI)2H2.
The high-frequency shift, observed both in the infrared and Raman spectra acquired on releasing pressure from 6.8 to 3.1 GPa across the melting threshold of the crystalline product (Supplementary Fig. 6), suggests also PH3 to behave like the analogous X2H2 isostructural van der Waals compounds, exhibiting H3P ⋯ H-PH2 H-bonding interactions, which disappear on decompression from 6.8 to 3.1 GPa after the decomposition of (PH3)2H2, as attested by the decrease of the vibrational frequencies of PH3 on further decompression.
The presence of a H-bonding between PH3 molecules has noticeable chemical relevance as PH3, in contrast to NH3, is known for not forming H-bonding at ambient conditions45,46, due to the small electronegativity difference of phosphorus with respect to hydrogen and to the consequent smaller electric dipole moment60. Furthermore, the existence of such interaction, together with the presence of H2 molecules, is in agreement with the larger volume of the crystalline cell of (PH3)2H2 compared to what expected in pure PH3.
The softening of the stretching vibration in the H2 molecules forming the (PH3)2H2 crystal clearly indicates the presence of chemical interaction between H2 and PH3. In the case of the isolated molecules such interaction has been described by ab initio computational methods61 in terms of two possible contributions: 1) the electron lone pair of P can act as a Lewis base and the σ* anti-bonding molecular orbital of H2 as a Lewis acid (n → σ*); 2) the σ bonding molecular orbital electrons of H2 act as Lewis base and the first anti-bonding molecular orbital of PH3 as a Lewis acid (σ → σ*(H-PH2)).
The first interaction is essentially a HOMO-LUMO orbital overlap interaction involving the highest occupied molecular orbital (HOMO) of PH3 (2a1 symmetry), which hosts the electron lone pair and has a prevalent non-bonding character, and the unoccupied σ* anti-bonding molecular orbital of H2, technically the lowest unoccupied molecular orbital (LUMO), whereas the second one corresponds to the opposite situation, where the HOMO σ bond electron density of H2 interacts with the LUMO orbital of PH3 (3a1) (Fig. 7). Energetically, the first interaction is larger when the 2a1 HOMO of PH3 and the σ* of H2 have maximum overlap, with the electron lone pair and the molecular axis of H2 aligned, but is present, even to a smaller extent, also in other interaction configurations, whereas the second one requires the σ electron density of H2 to interact with the 3a1 LUMO of PH3 in a configuration where the electron lone pair is perpendicular to H2 molecular axis.

a Qualitative molecular orbital diagram for pyramidal AH3 (where A = N, P, Sb, Bi) obtained from the symmmetry adapted linear combination of atomic orbitals of H3 (C3v) and A fragments (adapted from Figures 4–9 in ref. 75). The orbitals of each fragment are aligned vertically, in order of increasing energy from bottom to top, under the corresponding column of panel a, with H3 on the left side, AH3 in the middle and A on the right side. The 2a1 highest occupied molecular orbital (HOMO), where the electron lone pair is located, is highlighted by a red circle. b Molecular orbital diagram for H2, showing the completely filled σ bonding molecular orbital and the empty σ* anti-bonding lowest unoccupied molecular orbital (LUMO) highlighted by a red circle. c Isolobal orbital overlap interaction between the 2a1 molecular orbital of AH3 and the σ* anti-bonding molecular orbital of H2.
In solid state, as group 15 elements are concerned, the first kind of interaction has been recently reported to be responsible for the softening of the H2 stretching vibration in the As4O6 ⋅ 2H2 crystal by electron density transfer from the As electron lone pair to the σ* anti-bonding orbital of H262. Gúnka et al. achieved this result by adopting the ICOHP (integrated projected crystal orbital Hamilton population) and ICOOP (integrated projected crystal orbital overlap population) computational methods, which, based on the crystal orbital overlap population (COOP) approach originally developed by R. Hoffmann63, indeed relate the local molecular orbitals to the band structure of crystals through the projection decomposition of the electron density of states, allowing to gain insight about the frontier orbitals that control structure and reactivity in extended systems.
Accordingly, a qualitative interpretation for the softening of the stretching vibration of the H2 molecules in the (PH3)2H2 crystal, certainly deserving appropriate theoretical investigation for effective electronic band structure calculation, is here proposed in terms of isolobal frontier molecular orbital overlap interaction between the HOMO of PH3, hosting the electron lone pair, and the σ* anti-bonding LUMO of H2 (Fig. 7). Even if the orientation of PH3 and H2 molecules is not known, considering that H2 is expected to undergo hindered rotations at this pressure, it is indeed likely that the 2a1 HOMO of some of the PH3 molecules building up the cavities, where the H2 molecules are hosted, and the σ* anti-bonding orbital of H2 dynamically adopt the correct orientation for an effective overlap. The σ* anti-bonding orbital of H2 is normally not occupied, which makes the H2 molecule stable. The electron density transfer from the lone pair of PH3 to the σ* anti-bonding orbital of H2 may decrease the bonding electron density of the H2 molecule, thus causing a reduction of the force constant (~5.15%) and finally a frequency decrease of the H2 stretching mode, according to the harmonic oscillator frequency equation (ν = \(\frac{1}{2\pi }\sqrt{\frac{k}{\mu }}\)). The existence of the σ(H2) → σ*(H-PH2) interaction may further contribute to this effect. As a result, the H2 molecules within the crystal structures of (PH3)2H2 exhibit a lower vibrational frequency compared to bulk solid H2.
Finally, the observation of (PH3)2H2 is also somehow relevant for superconductivity in PH3. PH3 has been experimentally reported to become metallic at 40 GPa and superconducting at 207 GPa with a Tc of 103 K, but with no structural characterization so far36. Since then quite a lot of theoretical efforts have been made to account for such observation. At the moment, theory and experiments seem to agree about the instability of pure PH3 at high pressure, whose decomposition proceeds through the release of H2. Experimentally, a couple of recent papers have reported the decomposition of PH355,64, with the initial formation of diphosphane followed by decomposition into elemental phosphorus and H2. However, no further convincing characterization was proposed, suggesting that, like in the case of H2S, other species may be responsible for the superconductivity observed by Drozdov et al.36. Theoretically, besides predicting the decomposition of PH3, different studies have calculated the stabilization of PH2 phases above 80 GPa, which, from a stoichiometric point of view, is consistent with the release of H237,38. Furthermore, a key role of molecular H2 in stabilizing the high-pressure superconducting phases of phosphorus hydrides has been recently proposed39. Interestingly, even if the pressure range is here much lower, our data show that PH3 and H2 form a crystalline vdW compound, in which molecular H2 is indeed involved, possibly stabilizing PH3, or other related species, even at higher pressure.
To summarize, the results of this study have multiple chemically relevant implications. First of all, using LH in DAC, we have successfully induced direct reactivity between Pblack and H2 at 1.2 GPa and temperature lower than 1000 K, without the use of any catalyst or precursor. To our knowledge this is the first report about a direct chemical reaction between Pblack, the thermodynamically stable allotrope of P, and H2 at high pressure and high temperature to form PH3, somehow mimicking and representing the P analogue of the Haber-Bosch process for the synthesis of NH3 from N2 and H2.
Secondly, at room T and pressure between 3.5 and 4.1 GPa PH3 combines with excess H2 to form the crystalline (PH3)2H2 van der Waals compound, whose observation consistently fills a gap existing for pnictogens in the periodic properties of non-metallic elements able to form crystalline vdW compounds made of the corresponding hydride and of molecular hydrogen. The identification of (PH3)2H2 represents the so far missing tile of P in this puzzle and confirms a general trend in the formation of H2-containing vdW compounds with X2H2 stoichiometry (X = molecular hydride) and I4cm (I4/mcm) structure.
The formation of unexpected chemical compounds made of components apparently non-interacting at ambient conditions, such as P and H, is extremely important for their relevant implications, which include H2 storage, the chemistry occurring in extraterrestrial environments of giant planets such as Jupiter, Saturn, and their moons, where PH3 and H2 are present40,41,43, and the identification of astrochemical processes leading to the synthesis of phosphine, which is a critical issue for the detection of the presence of life in harsh extraterrestrial environments of rocky planets, as inferred by the recent observation of anomalous high levels of phosphine in the cloud decks of Venus atmosphere65.
Third, as advancement in fundamental bond theory is concerned, the relevant observation of H-bonding in PH3, which in contrast to NH3 is not reported to exist at ambient pressure, and the existence of a molecular orbital interaction between the electron lone pair in PH3 and the antibonding molecular orbital of H2, provide remarkable insights to understand the effects underlying the predicted stabilization of the P–H systems under high-pressure conditions.
Finally, the synthesis of (PH3)2H2 effectively provides confirmatory experimental evidence for the key role played by H2 molecules in stabilizing the PH system at high pressure, as suggested by recent calculations predicting the presence of H2 units in superconducting PH structures at high pressure.
Once again the history of phosphorus has intertwined with pressure, whose role in enhancing similarities and consistencies in the periodic properties of elements apparently exhibiting particular behavior at ambient conditions5,66,67 is here further highlighted.
Methods
Synthesis of Pblack
Pure crystalline Pblack was synthesized from red phosphorus according to reference68. All the reactants used for the synthesis of Pblack were purchased from Sigma-Aldrich with the following purity: red phosphorus (>99.99%), tin (>99.999%), gold (>99.99%), and SnI4 (99.999%). The purity of the synthesized Pblack crystals was checked by X-ray powder diffraction, Raman spectroscopy, EDX analysis and ICP-MS measurements, the latter giving a purity of 99.999+%. The crystals of Pblack were fragmented by means of a metallic tip for obtaining smaller 20–40 μm chips to be loaded into the DAC.
Sample preparation and the experimental conditions
Pressure was generated by means of membrane Diamond Anvil Cells (DAC) equipped with Ia type standard cut 16-sided beveled anvils having 600 μm culets. Re gaskets 200 μm thick were indented to 80 μm thickness and laser-drilled to obtain a 300 μm sample chamber. Before using them for the sideways containment of the samples, a Au ring was applied to prevent unintended catalytic effect and H2 diffusion and reactivity with Re. For this purpose the 300 μm diameter gasket hole was filled with Au powder, compressed with ~20 bar of He in the membrane until the powder appeared reflective and then laser-drilled again to obtain a 250 μm diameter hole. A small crystal of Pblack was placed in the sample chamber by means of a metallic tip and the remaining volume was filled with fluid H2 using standard gas-loading technique. Au and a ruby chip were used to measure the pressure, whereas the temperature was measured by the fit of the black body thermal radiation emission of the sample during laser heating. High temperature was generated by means of Nd:YAG laser source (λ = 1064 nm) focused on the Pblack crystal (≈30 μm beam spot size diameter), which acted both as reactant and laser absorber, thus avoiding any other source of contamination. No evidence for the formation of Au69 or Re70 hydrides was observed.
X-ray diffraction data acquisition and analysis
XRD experiments were carried out at the ESRF-ID27 beamline using a monochromatic synchrotron radiation beam (λ = 0.3738 Å) focused to ~5 μm to select different areas of the heterogeneous sample. The diffracted radiation was revealed by a MAR CCD165 detector, located approximately at 187 mm from the sample. The setup was calibrated against a CeO2 powder standard and Dioptas software was used to integrate the 2D area images to 1D patterns.
A single-crystal data set was collected at 5.5 GPa. Diffraction intensities were acquired in an ω-oscillation scan mode over the range ±30∘ with a frame width of 0.5∘ and an exposure time of 2 s per single frame. The instrument model was calibrated at the beginning of the beam time by performing a full data collection of an enstatite single-crystal placed in a dummy DAC. The diffraction images were then imported into a CrysAlisPro suite (Supplementary Note 1) and processed accordingly. After determining the unit cell the intensities were reduced applying corrections for Lorentz and polarization effects, and also a multiscan absorption correction at the final step. Reciprocal lattice layers were reconstructed using the unwarp procedure (Supplementary Fig. 1). Careful inspection of the unwarped images did not reveal any twinning, satellite reflections or diffuse scattering, in contrast to the crystal of (H2Se)2H2, where diffuse scattering streaks were observed27. The crystal structure was subsequently solved by direct methods and then refined on F2 by full-matrix least-squares procedures using the SHELXL package (Supplementary Note 1). In addition, at the same pressure and at two other pressure points (4.1 and 4.5 GPa), panoramic oscillation images (ϕ = ±5∘, acquisition time 30 s) of oligocrystalline conglomerate were recorded and used to perform Le Bail fits with JANA 2006 software after azimuthal integration (Supplementary Figs. 2–4).
Spectroscopic data acquisition and analysis
The Raman spectra were acquired at LENS with 1.5 cm−1 spectral resolution using the 647.1 nm emission wavelengths of a Kr ion laser. The details of the Raman setup are described elsewhere71. Raman spectra were acquired performing a 14 × 14 mapping over a 10 μm spaced grid using a single 300 groove/mm grating, which allowed to cover the 200–3300 cm−1 frequency region with 4 cm−1 spectral resolution. The most significant spots of the mapping were further inspected at higher resolution (1.5 cm−1) with different grating configuration down to 23.5 cm−1 (triple grating subtractive configuration 900–900–1800 groove/mm) and up to 4700 cm−1 (single grating configuration 900 groove/mm). No photochemical effect was observed at the employed laser power (1.5 mW) for pressure higher than 1.9 GPa, whereas at this pressure the formation of Pblack could indicate the decomposition of PH355.
The FTIR spectra were acquired with 1 cm−1 spectral resolution, using a Bruker IFS-120HR interferometer, suitably modified for the acquisition of infrared absorption spectra at high pressure in DAC72.
The frequency and intensity of the FTIR and Raman bands were obtained by fitting procedure using Voigt line shapes after baseline subtraction. Fityk software was used for this purpose.
Data availability
The X-ray crystallographic coordinates for the structure of the (PH3)2 vdW compound reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number 2034375. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/datarequest/cif. All other data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Bridgman, P. W. Two new modifications of phosphorus. J. Am. Chem. Soc. 36, 1344–1363 (1914).
Corbridge, D. Phosphorus: Chemistry, Biochemistry and Technology, Sixth Edition. (Taylor & Francis, 2013).
Jamieson, J. C. Crystal structures adopted by black phosphorus at high pressures. Science 139, 1291–1292 (1963).
Scelta, D. et al. Interlayer bond formation in black phosphorus at high pressure. Angew. Chem. Int. Ed. 56, 14135–14140 (2017).
Scelta, D. et al. The p-sc structure in phosphorus: bringing order to the high pressure phases of group 15 elements. Chem. Commun. 54, 10554–10557 (2018).
Peruzzini, M. et al. A perspective on recent advances in phosphorene functionalization and its applications in devices. Eur. J. Inorg. Chem. 2019, 1476–1494 (2019).
Ryder, C. R. et al. Covalent functionalization and passivation of exfoliated black phosphorus via aryl diazonium chemistry. Nat. Chem. 8, 597–602 (2016).
Bódi, D. & Höltzl, T. Thermal stability and flexibility of hydrogen terminated phosphorene nanoflakes. J. Phys. Chem. C. 122, 8535–8542 (2018).
Mao, H.-K., Chen, X.-J., Ding, Y., Li, B. & Wang, L. Solids, liquids, and gases under high pressure. Rev. Mod. Phys. 90, 015007 (2018).
Citroni, M., Ceppatelli, M., Bini, R. & Schettino, V. Laser-induced selectivity for dimerization versus polymerization of butadiene under pressure. Science 295, 2058–2060 (2002).
Chelazzi, D., Ceppatelli, M., Santoro, M., Bini, R. & Schettino, V. High-pressure synthesis of crystalline polyethylene using optical catalysis. Nat. Mater. 3, 470–475 (2004).
Dong, X. et al. A stable compound of helium and sodium at high pressure. Nat. Chem. 9, 440–445 (2017).
Dewaele, A. et al. Synthesis and stability of xenon oxides Xe2 O5 and Xe3 O2 under pressure. Nat. Chem. 8, 784–790 (2016).
Howie, R. T., Turnbull, R., Binns, J., Frost, M., Dalladay-Simpson, P. & Gregoryanz, E. Formation of xenon-nitrogen compounds at high pressure. Sci. Rep. 6, 34896 (2016).
Wang, H., Li, X., Gao, G., Li, Y. & Ma, Y. Hydrogen-rich superconductors at high pressures. Wiley Interdiscip. Rev. Comput. Mol. Sci. 8, e1330 (2018).
Mao, W. L. & Mao, H.-k. Hydrogen storage in molecular compounds. Proc. Natl Acad. Sci. USA 101, 708–710 (2004).
Struzhkin, V. V., Militzer, B., Mao, W. L., Mao, H.-K. & Hemley, R. J. Hydrogen storage in molecular clathrates. Chem. Rev. 107, 4133–4151 (2007).
Vos, W. L. et al. A high-pressure van der Waals compound in solid nitrogen-helium mixtures. Nature 358, 46–48 (1992).
Somayazulu, M. S., Finger, L. W., Hemley, R. J. & Mao, H. K. High-pressure compounds in methane-hydrogen mixtures. Science 271, 1400–1402 (1996).
Strobel, T. A., Somayazulu, M. & Hemley, R. J. Novel pressure-induced interactions in silane-hydrogen. Phys. Rev. Lett. 103, 065701 (2009).
Strobel, T. A., Chen, X.-J., Somayazulu, M. & Hemley, R. J. Vibrational dynamics, intermolecular interactions, and compound formation in GeH4 -H2 under pressure. J. Chem. Phys. 133, 164512 (2010).
Laniel, D., Svitlyk, V., Weck, G. & Loubeyre, P. Pressure-induced chemical reactions in the N2 (H2)2 compound: from the N2 and H2 species to ammonia and back down into hydrazine. Phys. Chem. Chem. Phys. 20, 4050–4057 (2018).
Ciezak, J. A., Jenkins, T. A. & Hemley, R. J. Optical and Raman microspectroscopy of nitrogen and hydrogen mixtures at high pressure. AIP Conf. Proc. 1195, 1291–1294 (2009).
Loubeyre, P. & LeToullec, R. Stability of O2/H2 mixtures at high pressure. Nature 378, 44–46 (1995).
Vos, W. L., Finger, L. W., Hemley, R. J. & Mao, H.-K. Novel H2 -H2 O clathrates at high pressures. Phys. Rev. Lett. 71, 3150–3153 (1993).
Strobel, T. A., Ganesh, P., Somayazulu, M., Kent, P. R. C. & Hemley, R. J. Novel cooperative interactions and structural ordering in H2S–H2. Phys. Rev. Lett. 107, 255503 (2011)
Pace, E. J. et al. Synthesis and stability of hydrogen selenide compounds at high pressure. J. Chem. Phys. 147, 184303 (2017).
Binns, J. et al. Synthesis and stability of hydrogen iodide at high pressures. Phys. Rev. B 96, 144105 (2017).
Ulivi, L., Bini, R., Loubeyre, P., LeToullec, R. & Jodl, H. J. Spectroscopic studies of the Ar(H2)2 compound crystal at high pressure and low temperatures. Phys. Rev. B 60, 6502–6512 (1999).
Kleppe, A. K., Amboage, M. & Jephcoat, A. P. New high-pressure van der Waals compound Kr(H2)4 discovered in the krypton-hydrogen binary system. Sci. Rep. 4, 4989 (2014).
Somayazulu, M. et al. Pressure-induced bonding and compound formation in xenon-hydrogen solids. Nat. Chem. 2, 50–53 (2010).
Somayazulu, M., Dera, P., Smith, J. & Hemley, R. J. Structure and stability of solid Xe(H2)n. J. Chem. Phys. 142, 104503 (2015).
Bi, T., Zarifi, N., Terpstra, T. & Zurek, E. Reference Module in Chemistry, Molecular Sciences and Chemical Engineering. (Elsevier, 2019)
Drozdov, A. P., Eremets, M. I., Troyan, I. A., Ksenofontov, V. & Shylin, S. I. Conventional superconductivity at 203 kelvin at high pressures in the sulfur hydride system. Nature 525, 73–83 (2015).
Bardeen, J., Cooper, L. N. & Schrieffer, J. R. Microscopic theory of superconductivity. Phys. Rev. 106, 162–164 (1957).
Drozdov, A. P. & Troyan, M. I. E. Superconductivity above 100 K in PH3 at high pressures. Preprint at https://arxiv.org/abs/1508.06224.
Flores-Livas, J. A. et al. Superconductivity in metastable phases of phosphorus-hydride compounds under high pressure. Phys. Rev. B 93, 020508 (2016).
Shamp, A. et al. Decomposition products of phosphine under pressure: PH2 stable and superconducting? J. Am. Chem. Soc. 138, 1884–1892 (2016).
Bi, T., Miller, D. P., Shamp, A. & Zurek, E. Superconducting phases of phosphorus hydride under pressure: stabilization by mobile molecular hydrogen. Angew. Chem. Int. Ed. 56, 10192–10195 (2017).
Morton, R. J. & Kaiser, R. I. Kinetics of suprathermal hydrogen atom reactions with saturated hydrides in planetary and satellite atmospheres. Planet. Space Sci. 51, 365–373 (2003).
Lewis, J. S. Physics and Chemistry of the Solar System. in International Geophysics, Vol. 87. (Academic Press, 2004)
Fletcher, L., Orton, G., Teanby, N. & Irwin, P. Phosphine on Jupiter and Saturn from Cassini/CIRS. Icarus 202, 543–564 (2009).
Pasek, M. A., Mousis, O. & Lunine, J. I. Phosphorus chemistry on Titan. Icarus 212, 751–761 (2011).
Mao, H. K. & Bell, P. M. Observations of hydrogen at room temperature (25∘ C) and high pressure (to 500 Kilobars). Science 203, 1004–1006 (1979).
Cotton, F. A., Wilkinson, G., Murillo, C. A. & Bochmann, M. Advanced inorganic chemistry, 6th edn. (John Wiley & Sons, 1999).
Atkins, P., Overton, T., Rourke, J., Weller, M. & Armstrong, F. Shriver and Atkins’ Inorganic Chemistry, 5th edn. (W. H. Freeman, 2010).
Natta, G. & Casazza, E. La struttura dell’idrogeno fosforato (PH3) e dell’idrogeno arsenicale (AsH3). Gazz. Chim. Ital. 60, 851–859 (1930)
Stevenson, D. P. The structure of phosphine and related hydrides. J. Chem. Phys. 8, 285–287 (1940).
Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 68, 441–451 (1964).
Hardin, A. H. & Harvey, K. B. Infrared absorption of solid phosphine. Can. J. Chem. 42, 84–89 (1964).
Francia, M. D. & Nixon, E. R. Infrared study of solid phosphine. J. Chem. Phys. 58, 1061–1065 (1973).
Devyatykh, G. G., Sennikov, P. G. & Nabiev, S. S. Vibrational spectra of volatile inorganic hydrides in the liquid state. Russ. Chem. Bull. 48, 623–639 (1999).
Baudler, M. & Glinka, K. Open-chain polyphosphorus hydrides (Phosphines). Chem. Rev. 94, 1273–1297 (1994).
Ceppatelli, M., Bini, R. & Schettino, V. High-pressure photodissociation of water as a tool for hydrogen synthesis and fundamental chemistry. Proc. Natl Acad. Sci. USA 106, 11454–11459 (2009).
Yuan, Y. et al. Stoichiometric evolutions of PH3 under high pressure: implication for high-Tc superconducting hydrides. Natl Sci. Rev. 6, 524–531 (2019).
Mao, H., Xu, J. & Bell, P. Pressure-Induced Infrared Spectra of Hydrogen to 542 kbar. Year book, Vol. 82, 366–372 (Carnegie Institution of Washington, 1983)
Binns, J., Dalladay-Simpson, P., Wang, M., Ackland, G. J., Gregoryanz, E. & Howie, R. T. Formation of H2 -rich iodine-hydrogen compounds at high pressure. Phys. Rev. B 97, 024111 (2018).
Pace, E. J. et al. Properties and phase diagram of (H2S)2H2. Phys. Rev. B, 101, 174511 (2020).
Snider, E. et al. Room-temperature superconductivity in a carbonaceous sulfur hydride. Nature 586, 373–377 (2020).
Hansen, A. S., Du, L. & Kjaergaard, H. G. Positively charged phosphorus as a hydrogen bond acceptor. J. Phys. Chem. Lett. 5, 4225–4231 (2014).
Grabowski, S. J., Alkorta, I. & Elguero, J. Complexes between dihydrogen and amine, phosphine, and arsine derivatives. hydrogen bond versus pnictogen interaction. J. Phys. Chem. A 117, 3243–3251 (2013).
Guńka, P. A., Hapka, M., Hanfland, M., Chalasiński, G. & Zachara, J. Toward heterolytic bond dissociation of dihydrogen: the study of hydrogen in arsenolite under high pressure. J. Phys. Chem. C. 123, 16868–16872 (2019).
Hoffmann, R. How chemistry and physics meet in the solid state. Angew. Chem. Int. Ed. 26, 846–878 (1987).
Liu, M. et al. Unravelling decomposition products of phosphine under high pressure. J. Raman Spectrosc. 49, 721–727 (2018).
Greaves, J. S. et al. Phosphine gas in the cloud decks of Venus. Nat. Astron. https://doi.org/10.1038/s41550-020-1174-4 (2020).
Lundegaard, L. F., Weck, G., McMahon, M. I., Desgreniers, S. & Loubeyre, P. Observation of an O8 molecular lattice in the ϵ phase of solid oxygen. Nature 443, 201–204 (2006).
Santoro, M. et al. Amorphous silica-like carbon dioxide. Nature 441, 857–860 (2006).
Nilges, T., Kersting, M. & Pfeifer, T. A fast low-pressure transport route to large black phosphorus single crystals. J. Solid State Chem. 181, 1707–1711 (2008).
Donnerer, C., Scheler, T. & Gregoryanz, E. High-pressure synthesis of noble metal hydrides. J. Chem. Phys. 138, 134507 (2013).
Atou, T. & Badding, J. In situ diffraction study of the formation of rhenium hydride at high pressure. J. Solid State Chem. 118, 299–302 (1995).
Ceppatelli, M., Gorelli, F. A., Haines, J., Santoro, M. & Bini, R. Probing high-pressure reactions in heterogeneous materials by raman spectroscopy. Z. Krist. Cryst. Mater. 229, 83–91 (2014).
Bini, R., Ballerini, R., Pratesi, G. & Jodl, H. J. Experimental setup for Fourier transform infrared spectroscopy studies in condensed matter at high pressure and low temperatures. Rev. Sci. Instrum. 68, 3154–3160 (1997).
Bonev, S. A., Schwegler, E., Ogitsu, T. & Galli, G. A quantum fluid of metallic hydrogen suggested by first-principles calculations. Nature 431, 669–672 (2004).
Batsanov, S. S. Van der Waals radii of hydrogen in gas-phase and condensed molecules. Struct. Chem. 10, 395–400 (1999).
Albright, A. T., Burdett, J. K. & Whangbo, M.-H. Orbital Interactions in Chemistry, 2nd Edn, 834 (Wiley, 2013).
Acknowledgements
Thanks are expressed to EC through the European Research Council (ERC) for funding the project PHOSFUN “Phosphorene functionalization: a new platform for advanced multifunctional materials” (Grant Agreement No. 670173) through an ERC Advanced Grant. This study was supported by the Deep Carbon Observatory (DCO) initiative under the project Physics and Chemistry of Carbon at Extreme Conditions, by the project “GreenPhos - alta pressione”, by the Italian Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR), by Ente Cassa di Risparmio di Firenze under the project Firenze Hydrolab2.0 and by Fondazione Cassa di Risparmio di Firenze under the project HP-PHOTOCHEM. The authors acknowledge the European Synchrotron Radiation Facility (ESRF) for provision of synchrotron radiation facilities and thank G. Garbarino, M. Mezouar and J. Jacobs for assistance in using beamline ID27. The authors would like to thank Prof. Roberto Righini for useful discussions.
Author information
Authors and Affiliations
Contributions
M.C., D.S., and R.B. performed the experiments, analyzed all the data and discussed the results. K.D. analyzed the XRD data and discussed the results. M.S.R and M.P discussed the results. M.S.R. synthesized black phosphorus. G.G., J.J., and M.M. assisted at ESRF-ID27. M.C. conceived the experiment and wrote the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ceppatelli, M., Scelta, D., Serrano-Ruiz, M. et al. High pressure synthesis of phosphine from the elements and the discovery of the missing (PH3)2H2 tile. Nat Commun 11, 6125 (2020). https://doi.org/10.1038/s41467-020-19745-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-19745-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
