
Abstract
The two-dimensional morphology of molybdenum oxycarbide (2D-Mo2COx) nanosheets dispersed on silica is found vital for imparting high stability and catalytic activity in the dry reforming of methane. Here we report that owing to the maximized metal utilization, the specific activity of 2D-Mo2COx/SiO2 exceeds that of other Mo2C catalysts by ca. 3 orders of magnitude. 2D-Mo2COx is activated by CO2, yielding a surface oxygen coverage that is optimal for its catalytic performance and a Mo oxidation state of ca. +4. According to ab initio calculations, the DRM proceeds on Mo sites of the oxycarbide nanosheet with an oxygen coverage of 0.67 monolayer. Methane activation is the rate-limiting step, while the activation of CO2 and the C–O coupling to form CO are low energy steps. The deactivation of 2D-Mo2COx/SiO2 under DRM conditions can be avoided by tuning the contact time, thereby preventing unfavourable oxygen surface coverages.
Similar content being viewed by others

Introduction
Since the early work by Sinfelt1 and Boudart2 demonstrated that Mo2C and WC exhibit platinum-like catalytic activity in alkane hydrogenolysis and isomerization, a multitude of reports has appeared aiming to exploit molybdenum carbide for industrially-relevant reactions such as methane aromatization3,4, the water-gas shift5,6, and CO2 hydrogenation reactions7,8, to name just a few9. In contrast to middle-to-late transition metal-based catalysts that deactivate by the deposition of coke, sintering or poisoning10, carbide catalysts typically feature a low rate of coking11, are resistant to high temperature sintering as well as to sulfur poisoning12,13. However, the high oxophilicity of carbides fosters their evolution in reactions with oxygenate substrates, leading ultimately to deactivation via the formation of oxide phases; deactivated catalysts require regeneration by re-carburization with CH4/H211. If such deactivation pathways are avoided while the metal utilization of carbides is maximized, for instance by developing catalysts with a high surface-to-bulk ratio and a high dispersion on supports, the replacement of traditional, more expensive middle-to-late transition metal catalysts by carbide-based catalysts will become viable.
The dry reforming of methane, (DRM, CH4 + CO2 ↔ 2CO + 2H2) is an example of a particularly challenging reaction for Mo2C-based catalysts because it combines a CO2-rich feed with high operating temperatures (typically, 800 °C and above)11,14,15,16,17,18,19,20,21. Deactivation of Mo2C in DRM conditions proceeds oxidatively according to Mo2C + 5 CO2 → 2 MoO2 + 6 CO11,17,18 and in order to mitigate it, dry reforming of methane is often conducted at elevated pressures (2–10 bar)11,14,16,18,21. From a mechanistic standpoint, an ill-defined molybdenum oxycarbide phase (MoOxCy) has been reported to play a key role in DRM22, as well as in the water-gas shift23, deoxygenation24,25, and CO2-to-methanol8 conversion processes, suggesting that the partial oxidation of Mo2C is favorable for those reactions. In addition, recent reports on CO2-propane dehydrogenation catalyzed by Mo2C highlighted that control over the oxygen coverage of the surface oxycarbide layer allows tuning the catalytic activity and selectivity26,27. Molybdenum oxycarbide species were also proposed as intermediates during the dehydroaromatization of methane on molybdenum-loaded zeolite ZSM-528. However, establishing the local structure and the nature of the active sites in such molybdenum oxycarbide phases has remained challenging, largely because well-defined oxycarbide catalysts, serving as references, are not available.
In this context, a recently discovered family of two-dimensional atomically-thin early transition metal carbides, nitrides, and carbonitrides called MXenes29 could be exploited as an entry point to supported well-defined 2D carbides and oxycarbides. MXene films are nanocrystalline yet form stable colloidal solutions in protic solvents, and feature metal-terminated surfaces (vide infra) with a lateral size in the submicron scale, which yields highly uniform surface metal sites30. The atomically thin nature of MXenes enables optimal metal utilization provided that these films are highly dispersed on a support. The 2D morphology is also advantageous for mechanistic studies since bulk metal sites in catalysts derived from MXenes, are minimized. Therefore, the characterization methods applied (even when not being inherently surface sensitive) will provide information mostly about the surface sites, which is instructive for structure-activity studies.
In what follows, we describe an approach to yield a supported two-dimensional (2D) model molybdenum oxycarbide material containing well-defined surface sites that are highly active in the dry reforming of methane. We prepare this catalyst from colloidal, delaminated thin films of the molybdenum carbide MXene phase, Mo2CTx, where T stands for OH, O, F surface termination groups. After supporting Mo2CTx on SiO2, these surface groups can be controllably removed (de-functionalized) by reduction in H2, giving 2D-Mo2C/SiO2. The oxidation of 2D-Mo2C/SiO2 by CO2 at 800 °C gives a 2D molybdenum oxycarbide 2D-Mo2COx/SiO2 with a high oxygen surface coverage and an average Mo oxidation state of +5.5. Subjecting this material to DRM conditions reduces the oxygen coverage and forms a catalyst in situ with an average Mo oxidation state of +4. This material is highly active in DRM, in contrast to fully reduced 2D-Mo2C/SiO2 and 2D-Mo2COx/SiO2 material with highest oxygen coverage (confirmed by a Mo oxidation state of ca. +5) which are essentially inactive for the DRM. Our Mo K-edge X-ray absorption near edge structure (XANES) and X-ray photoelectron spectroscopy (XPS) results verify that molybdenum oxycarbide is the active phase in Mo2C-based DRM catalysts31 and density functional theory (DFT) calculations identify that an oxygen coverage of ca. 0.7 monolayer provides a high activity of 2D-Mo2COx/SiO2 in DRM. Reducing or increasing this optimal oxygen coverage decreases the reaction rate. 2D-Mo2COx/SiO2 catalyst with reduced oxygen coverage is also less selective owing to a competing reverse water-gas shift reaction. Hence, supported 2D-Mo2COx catalyst with an initially high coverage of surface oxygen atoms activates first by reduction, that is the oxygen coverage is lowered until a maximum in DRM performance is reached at a Mo oxidation state of ca. +4. Reducing the Mo oxidation state further (i.e., lowering the oxygen coverage below ca. 0.7 monolayers) leads to catalyst deactivation. This observation is in sharp contrast to non-layered Mo2C-based catalysts that readily deactivate by oxidation in a CO2 atmosphere. It is therefore remarkable that deactivated 2D-Mo2COx/SiO2 catalysts do not contain any deposited coke according to temperature-programmed oxidation (TPO) experiments and can be regenerated by oxidation in CO2, fully recovering their highest catalytic activity. Noteworthy, the deactivation of 2D-Mo2COx/SiO2 under DRM conditions can be avoided by optimizing the contact time. Density functional theory calculations corroborate experimental observations and demonstrate that the DRM pathway on an oxycarbidic surface with a submonolayer oxygen coverage is energetically favored compared to the pathway occurring on a fully carbidic surface. The fully oxygen-covered surface is unstable in DRM conditions and reduces in situ its oxygen coverage below 1 oxygen monolayer, also consistent with our experiments.
Results
Synthesis and characterization
Multi-layered Mo2CTx32 (referred to as m-Mo2CTx) was sonicated in ethanol to yield, after centrifugation, a transparent purple colloidal solution of delaminated 2D-Mo2CTx flakes (Fig. 1a and Supplementary Fig. 2)33. A dried aliquot of this solution was analyzed by transmission electron microscopy (TEM) revealing single and a few-layer thin flakes of Mo2CTx, the latter morphology is identified by the characteristic scrolling of edges of the few-layer thin MXene nanosheets (Supplementary Fig. 3)34. The X-ray powder diffraction pattern of the dried delaminated material is typical of a layered Mo2CTx structure (Supplementary Fig. 4)35. The colloidal solution of the Mo2CTx nanoflakes (ca. 0.15 mg mL−1 concentration determined by thermogravimetric analysis) was used for several consecutive incipient wetness impregnations of a SiO2 support (Aerosil 300, calcined at 950 °C, 194 m2 g−1 surface area according to nitrogen physisorption), giving 2D-Mo2CTx/SiO2 that, after drying at 100 °C in air, contained 0.48 wt% Mo by elemental analysis. TEM imaging of 2D-Mo2CTx/SiO2 shows agglomerated amorphous silica grains that are homogeneously covered with thin Mo2CTx nanosheets (Fig. 1b and Supplementary Fig. 5). Consistent with an MXene structure, hexagonally ordered Mo atoms are observed by (high resolution) HR-TEM in 2D-Mo2CTx/SiO2 (Supplementary Fig. 5). The X-ray powder diffraction (XRD) pattern of 2D-Mo2CTx/SiO2 reveals only an amorphous halo corresponding to SiO2 (Supplementary Fig. 6). The absence of crystalline peaks of m-Mo2CTx confirms a high dispersion of individual delaminated Mo2CTx nanosheets on the support.

a Synthesis of 2D-Mo2COx/SiO2 from delaminated Mo2CTx films via incipient wetness impregnation of their colloidal solution onto silica, followed by the reduction of the Tx groups under H2 (giving 2D-Mo2C/SiO2) and the subsequent oxidation in CO2 (giving 2D-Mo2COx/SiO2); b TEM images of 2D-Mo2CTx/SiO2; c in situ reduction of 2D-Mo2CTx/SiO2 under H2 as followed by Mo K-edge XANES revealing an evolution of the pre-edge feature with temperature (shown in the inset as a contour plot); d oxidation state of Mo in reference and synthesized materials determined from the deconvolution of ex situ XPS data of the Mo 3d core levels as well as from e the edge position in XANES spectra.
Next, we fitted the experimentally acquired Fourier-transformed Mo K-edge extended X-ray absorption fine structure (EXAFS) functions of 2D-Mo2CTx/SiO2 with two coordination spheres, i.e., Mo−C/O/OH and Mo−Mo. The fitting of the first coordination sphere of Mo gives two scattering paths with interatomic distances of 1.74(1) and 2.03(1) Å and coordination numbers (CN) of 1.0(2) and 1.2(3), respectively (Table 1, Supplementary Fig. 12). The shorter scattering path is attributed to molybdenum oxo (Mo=O) surface sites since the fitted distance 1.74(1) Å is in good agreement with the reported Mo=O bond lengths of ca. 1.7 Å found in a molybdenum oxycarbide in a zeolite structure31 as well as in molecular Mo complexes36,37 (determined by EXAFS and X-Ray crystallography, respectively). The second scattering path of the first coordination shell with an interatomic distance of 2.03(1) Å is attributed to the Mo−C and Mo−O bonds where O belongs to a hydroxy surface termination site and C is the interlayer carbidic carbon, consistent with the 2.1 Å Mo−C distance in the parent m-Mo2CTx32. The second Mo coordination sphere is fitted with three Mo–Mo scattering paths with interatomic distances of 2.63(1), 2.92(2), and 3.42(2) Å and with coordination numbers of 0.8(4), 1.4(5), and 1.1(4), respectively (Table 1, Supplementary Fig. 12). The presence of three different Mo–Mo paths indicates a strong distortion of Mo sites in silica-supported Mo2CTx, deviating notably from the Mo coordination environment in parent m-Mo2CTx that features only two Mo–Mo paths at 2.9 Å and 3.2 Å with coordination numbers of 4 and 2, respectively (as determined by EXAFS)32. This deviation is likely owing to (i) the higher fraction of Mo=O sites in 2D-Mo2CTx/SiO2 relative to the m-Mo2CTx, (ii) a disorder of 2D-Mo2CTx/SiO2 caused by the geometrical curvature of the delaminated nanosheets supported on the silica grains and (iii) the interaction with the support. All these factors could contribute to the lower total CNs of Mo in 2D-Mo2CTx/SiO2 as determined by EXAFS.
Subsequently, we explored the transformation of these silica-supported 2D-Mo2CTx sheets in reductive, oxidative, and DRM conditions. Note that, unless specified otherwise, all these treated materials were characterized by avoiding exposure to air, using in situ experimentation, gloveboxes and air-tight transfers for characterization (XPS, fourier-transform infrared (FTIR) spectroscopy, TEM and XANES). Reductive de-functionalization of 2D-Mo2CTx/SiO2 from the passivating Tx surface groups was first studied in H2 flow by in situ XANES (Mo K-edge, Fig. 1c). Interestingly, in contrast to the bulk Mo2CTx32, a well-defined pre-edge feature, typical for molybdenum in a non-centrosymmetric environment38,39, appears in the room temperature XANES spectrum of 2D-Mo2CTx/SiO2 (Supplementary Fig. 13). Under H2 flow and with increasing temperature (in situ XANES experiment), the Mo K-edge in 2D-Mo2CTx/SiO2 shifts to lower energies and this process continues up to ca. 450 °C (from 20015.8 to 20002.0 eV), indicating a gradual reduction of Mo due to the de-functionalization of the Tx groups (Fig. 1c). Simultaneously, the intensity of the characteristic pre-edge feature of 2D-Mo2CTx/SiO2 reduces until its disappearance at ca. 450 °C. Increasing the temperature further to 750 °C induces only a minor shift of the Mo K-edge position compared to 450 °C, that is from 20002.0 to 20000.8 eV, the latter value is very close to the edge position of β-Mo2C (20000.7 eV). This suggests that H2 treatment at 750 °C leads to a Mo terminated carbidic surface predominantly free from oxo, hydroxy and fluoro terminations, supporting the assignment of the aforementioned pre-edge feature to molybdenum sites in 2D-Mo2CTx/SiO2 as opposed to MoO2 or MoO3 (conceivable oxidation products of 2D-Mo2CTx). Noteworthy, the temperature for the de-functionalization of most of the Tx groups in 2D-Mo2CTx/SiO2 (according to XANES) is approximately 450 °C, which is 150 °C lower than that of m-Mo2CTx32, probably owing to the high dispersion of the 2D-Mo2CTx sheets on the silica surface that eliminates gas diffusion limitations in the pores of m-Mo2CTx.
Since the DRM reaction typically requires high temperatures to yield high equilibrium conversions (>700 °C)40, 2D-Mo2CTx/SiO2 was treated in a H2 flow at 800 °C for 1 h to obtain 2D-Mo2C/SiO2. This high temperature, reductive treatment did not affect significantly the original morphology and high dispersion of the silica-supported nanosheets, as confirmed by TEM imaging (Supplementary Fig. 8). That said, in few cases small nanoparticles (ca. 2–3 nm) decorating molybdenum carbide sheets could be discerned in TEM images (Supplementary Fig. 8) possibly due to the partial sintering of 2D-Mo2C sheets to Mo2C nanoparticles at areas where nanosheets overlap. Overall, 2D-Mo2CTx/SiO2 behaves very differently in reductive de-functionalization conditions when compared to m-Mo2CTx that sinters under these conditions forming a porous bulk β-Mo2C phase32. Similarly, a multi-layer m-V2CTx was reported to sinter around 600 °C, which appears to be a general temperature limitation of multi-layer MXenes41. These results underline the importance of the dispersion of the nanosheets on a support to avoid sintering. Next, we prepared silica-supported model 2D molybdenum oxycarbide 2D-Mo2COx/SiO2 by treating 2D-Mo2C/SiO2 in a flow of CO2 (800 °C, 1 h, Fig. 1a). According to TEM, the CO2 treatment of 2D-Mo2C/SiO2 does not alter the morphology or dispersion of the supported nanosheets (Supplementary Fig. 9).
A comparison of the Mo 3d XPS spectra of the prepared silica-supported materials and β-Mo2C and MoO3 references is given in Fig. 1d. The Mo 3d spectrum of 2D-Mo2CTx/SiO2 was fitted with Mo4+, Mo5+, and Mo6+ components with the latter two being the dominating oxidation states (Supplementary Table 2). In contrast, the Mo 3d spectrum of m-Mo2CTx can be fitted using only Mo4+ and Mo5+ components (Supplementary Table 2)32 indicating that Mo sites are more oxidized in the silica-supported delaminated Mo2CTx nanosheets than in m-Mo2CTx. In contrast, the Mo 3d XPS spectrum of 2D-Mo2C/SiO2 is described by mostly carbidic Mo sites in addition to a smaller fraction of Mo4+ and Mo6+ sites (Supplementary Table 2). To understand better the nature of the Mo4+ and Mo6+ sites, observed by XPS of the reduced materials, 2D-Mo2C/SiO2 was analyzed by transmission IR spectroscopy revealing a low intensity band at ca. 2290 cm–1 that is tentatively assigned to surface [≡Si–H] sites (Supplementary Fig. 15). We speculate that the high temperature H2 treatment of 2D-Mo2CTx/SiO2 may give molybdenum hydrides (carbidic or oxycarbidic) that further react by opening the siloxane bridges of silica forming [Mo–Os] and [≡Si–H] bonds (Supplementary Fig. 15, inset). Such reactivity has been observed previously for surface hydrides of tantalum that cleaved siloxane bridges of silica42. Therefore Mo4+ sites in 2D-Mo2C/SiO2 are likely formed due to the grafting43 of the reduced nanosheets onto the silica surface.
Interestingly, the fitting of the 3d XPS spectrum of Mo in 2D-Mo2COx/SiO2 requires 4 components, namely Mo6+, Mo5+, Mo4+ and metallic Mo0 (Supplementary Table 2). Note that Mo0 is not observed in the XPS spectra of 2D-Mo2C/SiO2 and 2D-Mo2CTx/SiO2. We rationalize this result by a partial removal of the carbidic carbon from the 2D-Mo2C layer by CO2 at 800 °C (forming 2 equiv of CO) and attribute the presence of the Mo6+ component in the fit to the oxygen-rich oxycarbide phase of 2D-Mo2COx/SiO2. In contrast, heating multi-layer m-Mo2CTx in a CO2 flow transforms it into a mixture of β-Mo2C and MoO2 already at ca. 550 °C, according to our in situ XRD experiment (Supplementary Fig. 16). This result underlines again that dispersion and isolation of MXene nanosheets on a support is a critical step, prior to high temperature treatments in both reducing and oxidizing conditions, to retain the nanosheet morphology of the (oxy)carbide phase. In a sharp contrast to the stability of 2D-Mo2COx/SiO2 in an atmosphere of pure CO2 at 800 °C, other supported Mo2C-based catalysts yield MoO2 already in CO2-rich DRM feeds, which is the main deactivation pathway of these catalysts11,19.
Next, ex situ Mo K-edge XANES spectra of 2D-Mo2CTx/SiO2, 2D-Mo2C/SiO2 and 2D-Mo2COx/SiO2 were compared (Fig. 1e). Note that the edge position is defined as the first inflection point of the XANES spectra after the pre-edge feature (where the pre-edge corresponds to the forbidden 1s-4d transition)39. In agreement with XPS results, 2D-Mo2CTx/SiO2 features an edge position at 20015.8 eV that is notably higher than in m-Mo2CTx (20010.9 eV) indicating more oxidized Mo sites in supported Mo2CTx nanosheets32. The observed Mo edge energy is consistent with an average oxidation state of 2D-Mo2CTx/SiO2 of ca. +5.5 (Fig. 1e, inset and Supplementary Table 1). The reduction of 2D-Mo2CTx/SiO2 to 2D-Mo2C/SiO2 in H2 leads to the disappearance of the pre-edge feature, as was also observed in the in situ XANES reduction experiment, and shifts the Mo edge positions significantly to lower energies, i.e., from 20015.8 to 20000.8 eV. While the Mo K-edge position confirms the carbidic nature of 2D-Mo2C/SiO2, the white line region of the XANES spectrum of 2D-Mo2C/SiO2 differs from the spectrum of bulk β-Mo2C (Fig. 1e) owing to the different morphologies of these two materials. This observation corroborates the results of TEM analysis and confirms further that sintering to the bulk carbide phase did not occur during the reductive treatment of 2D-Mo2CTx/SiO2. Consistent with XPS analysis, the XANES spectrum of 2D-Mo2COx/SiO2 reveals a significant shift of the Mo K-edge towards higher energies compared to 2D-Mo2C/SiO2 (i.e., 20015.7 vs 20000.7 eV), suggesting an average Mo oxidation state for 2D-Mo2COx/SiO2 between Mo5+ and Mo6+ (Fig. 1e inset, Supplementary Table 1). The XANES spectrum of 2D-Mo2COx/SiO2 displays a characteristic pre-edge feature, similarly to 2D-Mo2CTx/SiO2. We conclude that all available characterization data (XANES, XPS, TEM) are consistent with a predominantly nanosheet morphology and a carbidic and oxycarbidic nature of, respectively, 2D-Mo2C/SiO2 and 2D-Mo2COx/SiO2.
Correlating catalytic activity to the surface oxygen coverage of 2D-Mo2COx
The catalytic activities of 2D-Mo2C/SiO2 and 2D-Mo2COx/SiO2 for the dry reforming of methane (800 °C, 1 bar, space velocity (SV) 1200 L gMo−1 h−1, (weight/volume flow rate) W/F = 3 ms gMo mL−1 contact time, 1:1 CO2:CH4 ratio) are strikingly different (Fig. 2a). While 2D-Mo2C/SiO2 shows negligible DRM activity, 2D-Mo2COx/SiO2 is highly active with methane conversion rates of 0.42 mol(CH4) mol(Mo)−1 s−1 after 10 min on stream at 80% CH4 conversion (i.e., ca. 10% below the thermodynamic equilibrium in those conditions, Fig. 2a)40. Note that the 2D nature of our catalysts enables a very high utilization of Mo such that when normalized by weight of Mo, the initial methane consumption rate of 2D-Mo2COx/SiO2 is ~10–200 times higher than the rates that have been reported for other Mo2C-based catalysts at similar conditions (Supplementary Table 3). The reference catalysts β-Mo2C and m-Mo2CTx exhibit a significantly lower DRM activity in these conditions (ca. 0.0003 and 0.002 mol(CH4) mol(Mo)−1 s−1 after ca. 5 min of time in stream, respectively), and deactivated entirely after 30 min of reaction (Supplementary Fig. 17). The CO chemisorption capacity was determined for in situ reduced 2D-Mo2CTx/SiO2 and the reference β-Mo2C catalysts in order to establish if the superior catalytic performance of 2D-Mo2CTx/SiO2 with respect to β-Mo2C (normalized per mass of Mo) is due to the improved utilization of Mo atoms in the 2D-nanosheet catalyst, or due to a higher activity of the Mo sites in 2D-Mo2CTx/SiO2 owing to their different structure and/or morphology, or a combination of these factors. Our results show that the estimated areal CO capacity in 2D-Mo2CTx/SiO2 and the reference β-Mo2C catalysts are nearly the same (ca. 0.2 and 0.25–0.5 μmol m–2, respectively; see Supplementary Table 4 and “Methods” for details). However, the measured methane conversion rate is ca. 3 orders of magnitude higher for 2D-Mo2CTx/SiO2 relative to the β-Mo2C catalyst. These results provide strong evidence that the improved catalytic activity is due to the different nature of Mo sites in 2D-nanosheet catalyst.

a Catalytic performance of 2D-Mo2C/SiO2 (90–150 min) and 2D-Mo2COx/SiO2 (240–300 min and from 390 min after the CO2 regeneration step) in DRM at a contact time (W/F) of 3 ms gMo mL−1; b correlation of the Mo oxidation state, determined by Mo K-edge XANES, and the catalytic activity of the material; c Mo K-edge XANES spectra of 2D-Mo2COx/SiO2 when exposed to DRM (0.1 ms gMo mL−1 contact time), reductive and oxidative conditions at 730 °C; d stable performance of 2D-Mo2COx/SiO2 at contact times of 1 or 2 ms gMo mL−1.
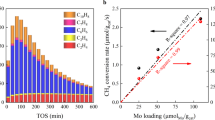
When using a CH4:CO2 feed ratio of 1:1 and a contact time of 3 ms gcat L−1, 2D-Mo2COx/SiO2 continuously deactivates, with the methane consumption rate dropping within 100 min on stream by almost a factor of three to 0.15 mol(CH4) mol(Mo)−1 s−1. At this point, the CH4 conversion has declined from ca. 80 to 30%. However, the initial activity can be fully regenerated by subjecting the catalyst, after 100 min time on stream (TOS), to a flow of pure CO2 at 800 °C for 1 h. While carbide catalysts are generally not known for deactivation by coking10, we evaluated the possible deposition of carbon during the DRM catalytic test in a temperature programmed oxidation (TPO) experiment performed in a thermogravimetric analyzer (TGA) with a simultaneous detection of the off gas by mass spectrometer (MS, Supplementary Fig. 19). Noteworthy, no increase in the intensity of the CO2 peak was observed for the used catalysts after 10 and 100 min of reaction, 2D-Mo2COx/SiO2−TOS10 and 2D-Mo2COx/SiO2−TOS100 (note: these materials remained light gray before and after catalytic testing). Consistent with this, 2D-Mo2COx/SiO2−TOS10 and 2D-Mo2COx/SiO2−TOS100 did not lose any weight during the TPO experiment. Thus, TPO data provide strong evidence that no detectable carbon deposition occurred on 2D-Mo2COx/SiO2 under the experimental conditions of the DRM tests.
While the activation of metallic DRM catalysts by CO2 treatment has been reported before44, in particular through the removal of deposited carbon by gasification45, the reactivation of carbide-based DRM catalysts by CO2 is, to the best of our knowledge, unprecedented and remarkable given that Mo2C-based catalysts for DRM are typically oxidized by CO2 yielding inactive MoO211,15. Note that the reference materials could not be reactivated in CO2, which is explained by the formation of MoO2 layer covering the active phase (Supplementary Fig. 18). We hypothesized that the deactivation of 2D-Mo2COx/SiO2 with time on stream (Fig. 2a) occurred due to a continuous depletion of the oxygen coverage of the CO2-activated molybdenum oxycarbide surface. Therefore, to relate the decrease of the catalytic performance of our Mo oxycarbide catalyst to the oxidation state of Mo, 2D-Mo2COx/SiO2 that has been exposed for 10 or 100 min to DRM conditions was analyzed by XPS and XANES. The deconvolution of the Mo 3d spectrum of 2D-Mo2COx/SiO2−TOS10 reveals Mo6+, Mo5+, Mo4+, and carbidic Mo states with Mo4+ being the main component at 41% (Supplementary Table 2). Note that metallic Mo is absent in the fit of 2D-Mo2COx/SiO2−TOS10 in contrast to 2D-Mo2COx/SiO2 discussed above. This might be due to the facile carburization of Mo0 with time on stream or the removal of volatile Mo carbonyl species. The deconvolution of the Mo 3d spectrum of 2D-Mo2COx/SiO2−TOS100 reveals the same oxidation states as those found in 2D-Mo2COx/SiO2−TOS10 (Mo6+, Mo5+, Mo4+, and carbidic states, Supplementary Fig. 23). However, carbidic Mo is the main component in 2D-Mo2COx/SiO2−TOS100 with a fitted fraction of 45% (Supplementary Table 2). This comparison of 2D-Mo2COx/SiO2 after 10 or 100 min of TOS clearly points at a reduced surface oxygen coverage and therefore a higher fraction of carbidic Mo in the less active catalyst, namely 2D-Mo2COx/SiO2−TOS100. Noteworthy, similarly to the as prepared 2D-Mo2CTx/SiO2, the XRD pattern of 2D-Mo2COx/SiO2-TOS100 features no Bragg peaks, thereby further confirming that no sintering to crystalline molybdenum oxides or carbides had occurred after this time (Supplementary Fig. 6).
XANES data agree well with the XPS results as the catalyst with the highest activity (2D-Mo2COx/SiO2−TOS10) features a Mo K-edge position at 20011.5 eV, which corresponds to an average oxidation state of Mo of +4 (Supplementary Fig. 14 and Fig. 2b). Note that the pre-edge feature in 2D-Mo2COx/SiO2 is well-defined, but poorly resolved in 2D-Mo2COx/SiO2−TOS10, suggesting a change in the geometry of the Mo sites due to the partial reduction during DRM conditions (Supplementary Fig. 14). 2D-Mo2COx/SiO2−TOS100 displays a Mo K-edge position at 20009.0 eV, corresponding to an average Mo oxidation state of ca. +3. This means that 2D-Mo2COx/SiO2 does not produce any measurable amount of coke when operated under a contact time of 3 ms gMo mL−1 and deactivates by reduction, lowering the oxygen coverage with TOS compared to the as-CO2-treated 2D-Mo2COx/SiO2 material. Subjecting 2D-Mo2COx/SiO2−TOS100 to a CO2 atmosphere (800 °C, 1 h) recovers the fully-oxidized 2D-Mo2COx/SiO2 characterized by a Mo oxidation state of ca. +5.5, that is the same as in the as-prepared 2D-Mo2COx/SiO2 material (Supplementary Fig. 14). Likewise, the identical XANES spectra of 2D-Mo2COx/SiO2 prior to DRM testing and after a CO2-based regeneration step are also in line with a fully recovered oxygen coverage after regeneration (Fig. 2a and Supplementary Fig. 14). TEM images of the regenerated material reveal that the 2D-Mo2COx nanosheets have a similar morphology to the initial 2D-Mo2CTx nanosheets as the hexagonal ordering of the Mo atoms and the characteristic scrolling of the nanosheets at the edges are still observed (Supplementary Fig. 10). Overall, these results highlight the remarkable structural stability of silica-supported 2D-Mo2COx nanosheets under catalytic DRM environments as well as in an oxidative CO2 atmosphere at 800 °C.
To further interrogate the relationship between the oxygen coverage of 2D-Mo2COx/SiO2 and the catalytic activity in DRM, an in situ XANES experiment was performed (BM31, ESRF)46 in a capillary cell reactor wherein 2D-Mo2CTx/SiO2 was first treated under H2 (750 °C, 0.5 h) to prepare 2D-Mo2C/SiO2 followed by exposing the material to DRM conditions at 730 °C (CH4: CO2 = 1: 1, SV = 3 × 104 L gMo−1 h−1, contact time of 0.1 ms gMo mL−1). In those DRM conditions, an immediate oxidation of 2D-Mo2C/SiO2 to 2D-Mo2COx/SiO2 occurred as indicated by a change of the Mo oxidation state from ca. +0.2 to +5 (Mo edge positions of 20000.7 and 20014.4 eV, respectively), consistent with a high oxygen coverage of the material (Supplementary Fig. 23). Monitoring the off-gas composition using a compact gas chromatograph (GC)47 shows no catalytic activity at those high oxygen coverages of 2D-Mo2COx. Hence, no reduction of 2D-Mo2COx with TOS occurs at low contact times of the synchrotron experiment. The oxygen coverage of 2D-Mo2COx/SiO2 is high under those DRM conditions as a change to a CO2 flow does not affect the XANES spectra significantly (Fig. 2c and Supplementary Fig. 24). Switching to a N2 atmosphere and co-feeding ca. 5% of H2 instantly reduces Mo to the carbidic state, yielding XANES spectra similar to that of 2D-Mo2C/SiO2 (Fig. 2c). However, introducing the DRM feed re-forms immediately 2D-Mo2COx/SiO2 with a Mo oxidation state of ca. +5, inactive for the DRM. The major difference between the laboratory (Fig. 2a) and synchrotron experiments lies in the space velocity that is roughly 25 times higher in the synchrotron experiment. We conclude that while the ex situ XANES data associate the highest DRM activity of 2D-Mo2COx/SiO2 with an average Mo oxidation state of +4 and subsequent deactivation with TOS with the reduction of Mo, in situ Mo K-edge DRM experiment revealed that the oxycarbidic surface with a high oxygen coverage and an average oxidation state of +5 is inactive for DRM. The latter observation is consistent with the reduction of the average Mo oxidation state from +5.5 to +4 in 2D-Mo2COx/SiO2 within 10 min TOS in a laboratory DRM catalytic test (W/F = 3 ms gMo mL−1). Note that no characteristic MoO2 features were observed with both ex and in situ XANES spectra for the catalyst after DRM catalytic tests, indicating that 2D-Mo2COx/SiO2 does not oxidize to bulk Mo oxides in the presence of both CO2 and H2O (the latter, owing to the parallel reverse water-gas shift (RWGS, CO2 + H2 ↔ CO + H2O) reaction).
Next, we varied the contact time of the laboratory DRM experiment and found that for a contact time between 1 and 2 ms gMo mL−1, the performance of 2D-Mo2COx/SiO2 in DRM is stable over 20 h TOS (Fig. 2d). Note that the measured ratio of CO:H2 of ca. 2 and 3 for, respectively, 2 and 1 ms gMo mL−1, indicates that the RWGS reaction takes place as a side reaction under these conditions. Noteworthy, the contact times associated with a stable catalytic performance are intermediate between the contact times used in the synchrotron experiment and laboratory experiments in which catalyst deactivation occurred (ca. 0.1 and 3 ms gMo mL−1, respectively). This suggests that at short contact times, dissociation of CO2 proceeds fast and yields a too high oxygen coverage of the Mo2COx surface, while at high contact times CH4 and/or DRM products (CO and H2) deplete the surface coverage of Mo2COx beyond the optimal value, leading to catalyst deactivation. Note that 2D-Mo2COx/SiO2 exhibits a high stability over 20 h on stream at W/F = 1–2 ms gMo mL−1 despite the presence of steam produced by the competing RWGS reaction. However, while at these conditions the selectivity and catalytic activity are stable, both values are lower than the initial (viz. TOS = 10 min) activity and selectivity of 2D-Mo2COx/SiO2 at W/F = 3 ms gMo mL−1 (Fig. 2a, d). Our search for a stable catalytic performance with increased activity and selectivity as compared to conditions of W/F = 1–2 ms gMo mL−1 uncovered that performing the catalytic test at a higher pressure (i.e., 8 bar instead of 1 bar) and keeping W/F = 3 ms gMo mL−1 yields a CO:H2 ratio close to 1.5 and only a slightly lower methane conversion rate as compared to that at 1 bar (0.42 vs 0.34 mol(CH4) mol(Mo)−1 s−1) (Supplementary Fig. 22). Thus, oxygen coverage higher than optimal (i.e., corresponding to an Mo oxidation state of +4) not only decreases the methane conversion rates, but also compromizes the selectivity due to the competing RWGS reaction.
Finally, aiming to induce major structural changes in 2D-Mo2COx/SiO2, we subjected the material to three subsequent DRM-catalysis-CO2-regeneration cycles with a total TOS of ca. 30 h using a contact time of 2 ms gMo mL−1 for the DRM step (Supplementary Fig. 20). Note, that the resulting material collected after the DRM step (labeled 2D-Mo2COx/SiO2−spent) was only partially deactivated, featuring a methane conversion rate of 0.08 mol(CH4) mol(Mo)−1 s−1. Similarly to 2D-Mo2COx/SiO2−TOS10, deconvolution of the Mo 3d spectrum of the spent catalyst reveals Mo6+, Mo5+, Mo4+, and carbidic Mo components, with Mo4+ being the main component at 62% (Supplementary Fig. 23 and Supplementary Table 2). TEM analysis of 2D-Mo2COx/SiO2−spent (exposed to air) reveals the presence of 2D nanosheets with a hexagonal ordering (Supplementary Fig. 11) and overlapping signals of Mo, C, and O in the EDX maps, confirming the oxycarbidic nature of the catalyst. Additionally, areas containing a bulk crystalline phase of MoO2 with a distorted rutile structure were revealed by FT HR-TEM imaging (Supplementary Fig. 11), in agreement with the high fraction of Mo4+ identified in the XPS fittings of this sample and its reduced activity relative to the most active 2D-Mo2COx/SiO2−TOS10 catalyst. In agreement with TEM results, the XRD analysis of 2D-Mo2COx/SiO2-spent revealed small but discernable peaks corresponding to MoO2. That said, supported nanosheets remain the dominant morphology of the spent catalyst, underlining its remarkable stability.
Theoretical modeling of DRM pathways on 2D-Mo2COx/SiO2
DFT calculations provided further insight into the role of oxygen coverage for DRM activity and the energetically preferred structure of 2D-Mo2C nanosheets after CO2 treatment and under DRM conditions. 2D-Mo2C was modeled by three atomic layers, with two Mo layers exposed to reactants and a carbon layer in between these Mo layers (Fig. 3). The stability of the orthorhombic Mo2C and Mo2COx phases has been previously evaluated by DFT calculations48,49. We derived 2D-Mo2C surface from the crystal structure of Mo2Ga2C, slicing it along the 001 direction, since it is the most convenient approach to obtain a 2D system with the desired stoichiometry (Supplementary Fig. 29). The computational model used has Mo sites surrounded by three carbon atoms (Mo–C distance of 2.09 Å), with the inner layer of carbon atoms separating the Mo atoms of the top and bottom surfaces by 2.88 Å. The coordination number of the Mo surface atoms is 6; the Mo–Mo distances at the surface are 3.03 Å. Note that EXAFS fitting of 2D-Mo2CTx/SiO2, as discussed above, revealed that Mo is coordinated with O and with C/O at a distance of ca. 1.74–2.03 Å, which is close to the 2.09 Å Mo–C distance of the theoretical model (Supplementary Table 3). The EXAFS analysis revealed Mo–Mo distances in the range of 2.63–3.42 Å (Table 1); this splitting of distances, as compared with the single Mo–Mo shell of the ideal model, is an indication of lattice strain in the Mo2CTx sheets. We note that our idealized model does not contain lattice strain or curvature of the experimental 2D-Mo2COx/SiO2 nanosheets, and the model does not include the interaction of 2D-Mo2COx with dehydroxylated SiO2 (i.e., covalent grafting observed by IR spectroscopy). Additionally, the average Mo oxidation state at one monolayer (ML) oxygen coverage of our model is lower than the experimentally determined (by XANES) Mo oxidation state for 2D-Mo2COx/SiO2 (ca. +5.5, Fig. 1e and Supplementary Table 5). Thus, it is likely that 2D-Mo2COx/SiO2 contains more oxygen atoms than 1 O ML. That being said, our idealized model is able to rationalize the experimental trends and it provides insights on the oxygen coverage (and the oxidation state of Mo), which has a significant impact on the DRM catalytic activity. Our evaluation of the thermodynamic stability of the different oxygen coverages of the 2D-Mo2C slab at 800 °C under CO2 reveals that a molybdenum oxycarbide phase with one oxygen monolayer (1 O ML) adsorbed on each of the Mo layers, i.e., 2D-Mo2CO2, is preferred thermodynamically (see the Supplementary computational details for further details). 2D-Mo2CO2 is formed from 2D-Mo2C and CO2 that react yielding one oxygen atom chemisorbed on the carbide surface with a concomitant release of one gas-phase CO molecule. To reach this full oxygen surface coverage, 18 CO2 molecules react per unit cell (9 CO2 molecules per each side of the slab) in a highly exergonic reaction releasing Gibbs energy of −928 kJ mol−1 per unit cell (ca. −52 kJ mol−1 per CO2 molecule). The reaction of an additional CO2 molecule yields an overall Gibbs free energy of −903 kJ mol−1, therefore, this step is unfavorable and the most stable configuration corresponds to 1 O ML coverage (i.e. one oxygen atom per Mo atom on the 2D-Mo2C surface), which is in agreement with the observed stability of 2D-Mo2COx/SiO2 in a CO2 atmosphere at 800 °C. Under DRM conditions, the reaction of 2D-Mo2C 1 O ML with one methane molecule has favorable energetics for the formation of one CO and two H2 gas-phase molecules, reducing thereby the surface oxygen coverage. Specifically, decreasing the oxygen coverage of 2D-Mo2COx from a full coverage of 1 O ML to 0.89, 0.78, and 0.67 O ML proceeds with reaction energetics of −30, −18 and 0 kJ mol−1, that is changing from exergonic to isoergonic energetics. This data confirm that the full coverage of the surface by 1 O ML is thermodynamically unstable under DRM conditions, as observed experimentally.

Top view of the a clean 2D-Mo2C, b 2D-Mo2C-1 O ML and c 2D-Mo2C-0.67 O ML surfaces. d Gibbs free energy profile of the dry reforming of methane catalyzed by the 2D-Mo2C-0.67 O ML surface. Gibbs energies are given with respect to initial reactants, CH4 and CO2, at 800 °C and under 1 bar pressure. For brevity, the reaction profile is presented without H–H coupling steps. The full profile is shown in Supplementary Fig. 25. TS stands for transition state.
In the following, we selected 2D-Mo2C-0.67 O ML as a starting point to calculate the energy profile of the DRM reaction pathways since the oxygen coverage of this structure is consistent with the optimal oxygen coverage observed experimentally (characterized by an average Mo oxidation state of +4). This partial 0.67 O ML oxygen coverage allows for free reactive Mo sites on the surface. The energetic penalty to form 2D-Mo2C-0.67 O ML from 2D-Mo2CO2 is merely +30 kJ mol−1 higher than forming the most stable oxygen coverage under the DRM conditions, i.e., 2D-Mo2C-0.88 O ML. The adsorption of CO2 on 2D-Mo2C-0.67 O ML is endergonic by 143 kJ mol−1, leading to a bent CO2 on the Mo sites of the oxycarbide surface. Dissociation of this bent CO2 to adsorbed CO* and O* products requires 173 kJ mol−1 of Gibbs free energy, and is overall exergonic by 12 kJ mol−1 (Fig. 3). This Gibbs energy barrier is even lower than the one obtained for a Ni(111) surface at 650 °C (209 kJ mol−1)50. In terms of required electronic energy, CO2 cleavage of the adsorbed, bent CO2 intermediate has a very low energy barrier of +31 kJ mol−1. This bent CO2 adsorbate was evaluated previously on several metal carbides51 and was suggested to enhance the reactivity of CO2 on interfaces between a metal and a metal carbide52 or a metal oxide53,54,55. DFT calculations on the clean, Mo-terminated surface of δ-Mo2C(001) showed that CO2 activation on molybdenum carbides is also associated with low activation barriers56. In turn, the activation of CH4 via C–H bond cleavage requires a higher Gibbs activation energy of 242 kJ mol−1 and the reaction occurs on Mo sites of the oxycarbide surface. The calculated energy barrier is comparable to the ones computed for Ni(111), Pd(111), and Pt(111) surfaces50. Conversely, CH4 activation on oxygen sites to form *OCH3 and *OH is endergonic by 105 kJ mol−1, which is by 45 kJ mol−1 less favorable than CH4 activation on the Mo sites of the same surface.
Subsequent CH3 and CH2 activation steps to form CH2* and CH* species are significantly less energy demanding than the initial C–H activation of CH4, with respective Gibbs energy barriers of 63 and 38 kJ mol−1. However, the cleavage of the C–H bond in CH* to form adsorbed C and H has a high Gibbs energy barrier of 117 kJ mol−1. Note that each successive C–H activation of methane generates an adsorbed hydrogen that can be released as an H2 in a strongly entropically favored process, releasing 59 kcal mol−1 at 800 °C per each desorbed H2 molecule (Fig. 3).
Besides the activation of CO2 and CH4, another key step in the dry reforming of methane is the oxidation of either CH* or C* species on the oxycarbide surface by adsorbed oxygen (O* or structural oxygen)57 ultimately producing adsorbed CO. In the case of 2D-Mo2COx, and in contrast to metallic surfaces50, the preferred mechanism is the direct oxidation of adsorbed C* by adsorbed O* to form CO*. This pathway has a Gibbs energy barrier of 175 kJ mol−1, and it is endergonic by only 22 kJ mol−1.The alternative pathway involving the coupling of CH* and O* to form adsorbed CHO*, followed by its decomposition to co-adsorbed CO* and H*, is less favorable (Supplementary Fig. 26). Finally, desorption of CO is exergonic by 20 kJ mol−1 per CO molecule. Overall, the calculated energy barriers of the proposed mechanism are feasible under the DRM conditions studied here (800 °C, CH4: CO2 = 1:1) and are thermodynamically exergonic by 32 kJ mol−1. The most energy demanding step is the initial methane activation step, which is associated with a significantly higher barrier than the direct CO2 activation or the C–O coupling step forming CO. In addition, the relative rates for the formation and consumption of intermediate oxygen and carbon surface species, and, therefore their ratio on the 2D-oxycarbide surface, has to be similar in order to obtain a good catalytic performance. Our theoretical calculations suggest that the ratio of C* to O* is another possible factor (besides the oxidation state of Mo atoms on the surface) that can contribute to the significantly higher catalytic activity of the 2D-Mo2C-0.67 O ML system in comparison to the clean 2D-Mo2C surface.
We also evaluated the energy profile of the DRM reaction on a clean 2D-Mo2C surface and found that the activation of CO2 occurs on this surface easier than on 2D-Mo2C-0.67 O ML. The produced CO* and O* surface species are significantly more stabilized on 2D-Mo2C compared to 2D-Mo2C-0.67 O ML, featuring a lower Gibbs energy barrier for CO2 activation of 109 kJ mol−1 (compare to 173 kJ mol−1 for the 2D-Mo2C-0.67 O ML surface). The resulting adsorbed CO* and O* species are located at −99 kJ mol−1 for the clean 2D-Mo2C surface, which is significantly lower than for the 2D-Mo2C-0.67 O ML surface (−12 kJ mol−1, Supplementary Fig. 27). That said, the free energy barrier for CH4 activation is very similar for both surfaces (238 vs. 243 kJ mol−1). Another difference with implications for the catalytic activity is the substantial stabilization of CH* and C* species on the 2D-Mo2C surface with respect to the initial reactants. The high adsorption energy of C* and O* species lead to a significantly higher energy barrier for the C–O coupling step (+219 kJ mol−1) on 2D-Mo2C, in contrast to the respective barrier on the 2D-Mo2C-0.67 O ML surface (+175 kJ mol−1). This overstabilization of CH* and C* and O* intermediates on a clean 2D-Mo2C surface explains its lower activity for DRM. In contrast, the 2D-Mo2C-0.67 O ML surface does not bind intermediates too strongly. Figure 4 summarizes our computational studies, which agree well with the experimental results discussed above.

The most active catalyst has a partial oxygen surface coverage, corresponding to Mo(+4) oxidation state. The C–H bond activation of methane is proposed as RDS, i.e., rate determining step, based on DFT calculations.
To conclude, we have shown that supporting and dispersing delaminated 2D-Mo2CTx nanosheets on silica prevents to a large extent their thermal sintering to bulk Mo2C and MoO2 phases, paving the way for the application of 2D Mo-carbides and oxycarbides for high temperature heterogeneous catalysis. XANES and XPS analysis indicate that oxycarbidic 2D-Mo2COx with an average Mo oxidation state of +4 is the active phase for DRM and oxidizing or reducing this state reduces the methane consumption rate. DFT calculations suggest that the rate-limiting step in the DRM on 2D-Mo2COx is CH4 cleavage and that the oxygen coverage resulting from the initial CO2 activation steps reduces the binding energy of C* and O* species and lowers, in turn, the energy barriers of the C–O coupling step. Importantly, when deactivation by reduction of the oxygen coverage has occurred, the catalyst can be reactivated to its initial activity by re-oxidation in a pure stream of CO2. Noteworthy, the nanosheet morphology of 2D-Mo2COx is maintained during catalysis and regeneration. In addition, we identified hydrodynamic conditions that ensured a stable performance over 20 h time on stream, owing to an in situ regeneration of the optimal oxygen coverage in those conditions. Overall, the results obtained indicate that supported 2D-Mo2COx is a very active and robust catalyst for high temperature catalytic applications, exhibiting a remarkable stability in oxidative environments.
Methods
Materials
Multi-layered Mo2CTx material (m-Mo2CTx) was synthesized following a reported method32. 2D-Mo2CTx nanosheets were delaminated from m-Mo2CTx (50 mg) in ethanol (10 mL) using a pulse sonication (1.5 h, 85 W, 20 kHz) (Fisher Scientific, FB120). Resulting suspension was centrifuged (7000 rpm, 7 min) and the supernatant solution containing colloidal delaminated Mo2CTx nanosheets (d-Mo2CTx) was further used for the incipient wetness impregnation (IWI) onto SiO2 support (150–300 μm particle size fraction of Aerosil 300 that had been calcined at 950 °C, 194 m2 g−1 surface area by nitrogen physisorption). The IWI procedure using ethanol colloidal solution of d-Mo2CTx was repeated multiple times and after drying (100 °C, 12 h) yielded 2D-Mo2CTx/SiO2 material with Mo loading of 0.48 wt.% (batch 1) and 0.35 wt.% (batch 2) according to elemental analysis (ICP-AES). 2D-Mo2CTx/SiO2 was subsequently reduced in hydrogen (20 vol % H2/N2, 800 °C, 1.5 h, 50 mL min−1, 10 °C min−1) and oxidized in CO2 (100% CO2 10 mL min−1, 800 °C, 1.5 h) to give 2D-Mo2C/SiO2 and 2D-Mo2COx/SiO2 materials, respectively. To avoid surface modification of the activated materials, 2D-Mo2C/SiO2, 2D-Mo2COx/SiO2, and all activated catalysts were transferred into a nitrogen-filled glovebox (H2O and O2 levels <0.5 ppm) and handled without exposure to air.
Characterization
Ex situ XRD data were examined within the 2θ range of 5–90° (step size and scan time per step were 0.0167° and 3 s, respectively). XRD data were collected on a PANalytical Empyrean X-ray diffractometer equipped with a Bragg–Brentano HD mirror and operated at 45 kV and 40 mA (Cu Kα radiation, λ = 1.5418 nm). TGA was performed on Mettler Toledo TGA/DSC 3 instrument. The concentration of 2D-Mo2CTx flakes in the colloidal solutions was determined by drying a 750 μL aliquot in a sapphire crucible (900 μL) at 80 °C for 1 h (5 °C min−1). TPO was performed on Mettler Toledo TGA/DSC 1 instrument. Mo material (20 mg) was placed in an alumina crucible (70 μL) and heated to 800 °C under air flow (10 °C min−1, 125 mL min−1). The H2O and CO2 content in the outlet gas was followed by a mass spectrometer MKS Cirrus 3. Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analyses were performed in Remagen, Germany, by the Mikroanalytisches Labor Pascher. XPS data were acquired using a Sigma II instrument of Thermo Fisher Scientific, equipped with an UHV chamber (non-monochromatic 200 W Al Kα source, a hemispherical analyzer, and a seven channel electron multiplier, with the analyzer-to-source and the emission angles of 50° of 0°, respectively). The survey and narrow scans used a pass energy of 50 eV and 25 eV, respectively, and C 1s peak of adventitious carbon was set at 284.8 eV to compensate for charge induced shifts. We used a home-build air-tight cell to transfer reduced or used specimens between the glovebox and the XPS instrument58. CasaXPS Version 2.3.19PR1.0 software was used to analyze XPS data. The background was subtracted according to Shirley59, and the application of the atomic sensitivity factors (ASF) of Scofield allowed to estimate the atomic composition60. HAADF STEM images were recorded using FEI Talos F200X with the accelerating voltage of 200 kV. All samples were prepared by shaking the holey carbon TEM grid in the vial with the dry specimen powder. 2D-Mo2C/SiO2 and 2D-Mo2COx/SiO2 were deposited on TEM grids in the glove box and transferred to the microscope without ambient exposure by using the TEM vacuum transfer holder (Fischione, 2560). FTIR Spectroscopy was conducted using a 200 mL glass reactor with IR-transparent CaF2 windows. A thin pellet of the sample was pressed in a glovebox and loaded to the reactor using a glass sample holder. A spectrum was recorded in an argon atmosphere in a transmission mode on a Nicolet 6700 FTIR spectrophotometer using 64 scans at a resolution of 2 cm−1. CO chemisorption was performed using an AutoChem system (Micromeritics) with a thermal conductivity detector (TCD). Approximately 100 mg of the material was loaded in a U-shape reactor and reduced at 800 °C under 5% H2/Ar for 1.5 h (10 °C min−1, 50 mL min−1). The sample was then cooled down under a He atmosphere to 450 °C and degassed for 2 h. The first CO adsorption isotherm was carried out at 50 °C. The reactor was then purged with He for 30 min to remove weakly adsorbed species prior to performing the second CO isotherm, also at 50 °C. The amount of strongly adsorbed CO was determined as a difference between the first and the second adsorption isotherms, which provided the CO adsorption capacity. The surface area of the 2D-Mo2C nanosheets in 2D-Mo2C/SiO2 was estimated based on the crystal structure of Mo2CTx32. Considering the hexagonal ordering of the Mo atoms with a Mo–Mo distance of 2.8629 Å gave an area estimate of 7.098 Å2 per Mo atom. Based on the Mo content in 2D-Mo2CTx/SiO2, determined by ICP, and the mass of the 2D-Mo2CTx/SiO2 specimen used for CO chemisorption, the amount of strongly chemisorbed CO was normalized per the surface area of the two-dimensional molybdenum nanosheets (Supplementary Table 4).
Experiments using X-ray absorption spectroscopy (XAS) were performed at the Swiss-Norwegian Beamlines (SNBL, BM31)46 at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). A double-crystal Si (111) monochromator with continuous scanning in transmission mode (unfocused beam, 1–2 ∙ 109 ph s−1 mm−1) was used to collect XAS spectra at the Mo K-edge. Edge position of the Mo foil set at 20000 eV served for calibration of the XAS data. A quartz capillary reactor (outer diameter 1.5 mm, wall thickness 0.1 mm) was used to perform the in situ DRM experiment (Supplementary Fig. 1)46. 2D-Mo2CTx/SiO2 (ca. 2 mg), placed between two quartz wool plugs in a quart capillary reactor, was reduced in 20 vol% H2 in N2 (10 mL min−1 total flow rate, 50–750 °C the temperature range, ca. 9 °C min−1, 20 min holding time). The oxidation was performed in pure CO2 (10 mL min−1, 730 °C, 2 h). DRM catalytic experiments were performed at 730 °C using a mixture of CH4 and CO2 of varied ratio diluted in N2 (5 mL min−1 total flow rate, SV ca. 30000 L gMo−1 h−1). The composition of the outlet gas was followed by a portable GC (Global Analyser Solutions) with TCD and FID detectors. Ex situ XAS data were collected from pellets with an optimized amount of specimen mixed with cellulose. All reduced materials or used catalysts derived from 2D-Mo2C/SiO2 were handled in a glovebox and analyzed in an air-tight sealed bags. XAS data were processed using the Athena software, and EXAFS data were fitted using the Artemis software (Demeter 0.9.25 software package)61. The edge position for Mo and Mo2C was defined as the position of the first maximum of the first derivative curve. The second maximum of the first derivative curve was chosen as the edge position for oxide and oxycarbide materials.
Catalytic testing
The catalytic testing of freshly-prepared materials in conditions of the dry reforming of methane was carried out in a fixed-bed quartz reactor at atmospheric pressure. In a typical experiment, 100 mg of freshly prepared 2D-Mo2CTx/SiO2 material was used. Prior to the activity tests, 2D-Mo2CTx/SiO2 was in situ reduced in 20 vol % H2/N2 (800 °C, 1.5 h, 50 mL min−1, 10 °C min−1) and then oxidized in pure CO2 (800 °C, 1.5 h, isothermal). The activity test was then performed at 800 °C with a variable total flow rate of the feed (10–50 ml min−1, SV = 1200–6000 L gMo–1 h–1, 45% CH4, 45% CO2, and 10% N2). The composition of the off-gas was analyzed via a GC (PerkinElmer Clarus 580) equipped with a thermal conductivity TCD detector. The carbon balance during the catalytic tests was generally close to 100 %, maximal deviations noted were ±10%, observed when switching gases.
We note that catalytic results reported were obtained with freshly-prepared 2D-Mo2CTx/SiO2. When as prepared 2D-Mo2CTx/SiO2 was left for a prolonged time (1 month–1.5 years) in air, a complete or partial fragmentation of 2D-Mo2CTx nanosheets into nanoparticles had occurred according to TEM characterization (Supplementary Fig. 7). The fully fragmented material is inactive in DRM, which emphasizes the importance of 2D nanosheets morphology for the high catalytic activity.
Computational details
The Vienna Ab Initio Simulation Package (VASP) was utilized to conduct the periodic DFT calculations62,63,64. The projector-augmented-wave (PAW) method was used, where pseudopotentials describe interactions between valence electrons and ion cores while the electronic wave functions are expanded as a discrete plane wave (PW) basis set65. All calculations utilized a PW energy cutoff of 500 eV. The generalized gradient approximation (GGA), including dispersion corrections by the BEEF-vdW density functional, were used to treat electron exchange and correlation66.
The sampling of the Brillouin zone was performed using Monkhorst–Pack grids67 with K-point values equal to 3 × 3 × 1 for the Mo2C surface. Electronic occupancies were determined according to a Gaussian Smearing (with a smearing value of 0.05 eV). Self-consistent field (SCF) calculations of the electronic structure were considered converged once the electronic energy change between two consecutive steps was below 10−5 eV. All geometries were optimized fully, i.e., until the forces acting on each atom were converged below 10−4 eV Å−1. Dipole corrections were applied in the z-direction and placed in the center in the unit cell. The energy of isolated molecules was determined by a Γ-point calculation such that each species was placed in a box with dimensions 15 × 15.5 × 16 Å. The climbing image nudge elastic band (CI-NEB) method allowed locating transition states (TS) using eight intermediate images68. A frequency analysis confirmed further that identified TS structures corresponded to saddle points. Normal vibration modes of adsorbed species were calculated by diagonalization of the Hessian matrix, obtained using a central finite difference approximation with displacements equal to 0.015 Å in the direction of each Cartesian coordinate. All Mo and C atoms from the carbide were kept fixed during the frequency calculations of the adsorbed species. All the reported energy values in the main text correspond to Gibbs Energies at 800 °C. The theoretical model of the (001) facet of 2D-Mo2C was constructed as presented in Supplementary Fig. 29. The experimental bulk structure of Mo2Ga2C was taken as a starting point. Then, the Ga atoms (in green) were removed from this structure to generate the bulk Mo2C structure. Next, an ideal 2D-facet model for Mo2C was obtained by selecting the inner fragment of the cell in the 001 direction (Supplementary Fig. 29). This was followed by the full geometry optimization of the resulting structure. The Mo2C (001) surface slab were taken with dimensions equal to 3 × 3, using two Mo layers on the top and the bottom of the slab and one middle carbon layer. 15 Å of vacuum were added in the direction perpendicular to the surface.
Data availability
The data supporting the findings of this study are available from the corresponding authors upon reasonable request.
References
Sinfelt, J. H. & Yates, D. J. Effect of carbiding on the hydrogenolysis activity of molybdenum. Nat. Phys. Sci. 229, 27–28 (1971).
Levy, R. B. & Boudart, M. Platinum-like behavior of tungsten carbide in surface catalysis. Science 181, 547–549 (1973).
Solymosi, F., Cserényi, J., Szöke, A., Bánsági, T. & Oszkó, A. Aromatization of methane over supported and unsupported Mo-based catalysts. J. Catal. 165, 150–161 (1997).
Siaj, M., Oudghiri-Hassani, H., Maltais, C. & McBreen, P. H. Thermally stable alkylidene groups on the surface of β-Mo2C: relevance to methane aromatization and olefin−metathesis catalysis. J. Phys. Chem. C. 111, 1725–1732 (2007).
Patt, J., Moon, D. J., Phillips, C. & Thompson, L. Molybdenum carbide catalysts for water-gas shift. Catal. Lett. 65, 193–195 (2000).
Moon, D. J. & Ryu, J. W. Molybdenum carbide water-gas shift catalyst for fuel cell-powered vehicles applications. Catal. Lett. 92, 17–24 (2004).
Porosoff, M. D., Yang, X., Boscoboinik, J. A. & Chen, J. G. Molybdenum carbide as alternative catalysts to precious metals for highly selective reduction of CO2 to CO. Angew. Chem., Int. Ed. 53, 6705–6709 (2014).
Posada-Pérez, S. et al. The conversion of CO2 to methanol on orthorhombic β-Mo2C and Cu/β-Mo2C catalysts: mechanism for admetal induced change in the selectivity and activity. Catal. Sci. Technol. 6, 6766–6777 (2016).
Hwu, H. H. & Chen, J. G. Surface chemistry of transition metal carbides. Chem. Rev. 105, 185–212 (2005).
Chorkendorff, I. & Niemantsverdriet, J. W. Concepts of Modern Catalysis and Kinetics (Wiley, 2003).
Claridge, J. B. et al. New catalysts for the conversion of methane to synthesis gas: molybdenum and tungsten carbide. J. Catal. 180, 85–100 (1998).
Schaidle, J. A., Lausche, A. C. & Thompson, L. T. Effects of sulfur on Mo2C and Pt/Mo2C catalysts: Water gas shift reaction. J. Catal. 272, 235–245 (2010).
Da Costa, P. et al. Tetralin hydrogenation catalyzed by Mo2C/Al2O3 and WC/Al2O3 in the presence of H2S. Catal. Today 65, 195–200 (2001).
York, A. P. E., Claridge, J. B., Brungs, A. J., Tsang, S. C. & Green, M. L. H. Molybdenum and tungsten carbides as catalysts for the conversion of methane to synthesis gas using stoichiometric feedstocks. Chem. Commun. 1, 39–40 (1997).
Brungs, A. J., York, A. P. E., Claridge, J. B., Marquez-Alvarez, C. & Green, M. L. H. Dry reforming of methane to synthesis gas over supported molybdenum carbide catalysts. Catal. Lett. 70, 117–122 (2000).
Xiao, T., Wang, H., Da, J., Coleman, K. S. & Green, M. L. H. Study of the preparation and catalytic performance of molybdenum carbide catalysts prepared with C2H2/H2 carburizing mixture. J. Catal. 211, 183–191 (2002).
Xiao, T.-c, Hanif, A., York, A. P. E., Nishizaka, Y. & Green, M. L. H. Study on the mechanism of partial oxidation of methane to synthesis gas over molybdenum carbide catalyst. Phys. Chem. Chem. Phys. 4, 4549–4554 (2002).
LaMont, D. C., Gilligan, A. J., Darujati, A. R. S., Chellappa, A. S. & Thomson, W. J. The effect of Mo2C synthesis and pretreatment on catalytic stability in oxidative reforming environments. Appl. Catal., A 255, 239–253 (2003).
LaMont, D. C. & Thomson, W. J. Dry reforming kinetics over a bulk molybdenum carbide catalyst. Chem. Eng. Sci. 60, 3553–3559 (2005).
Liang, P. et al. Simple synthesis of ultrasmall β-Mo2C and α-MoC1−x nanoparticles and new insights into their catalytic mechanisms for dry reforming of methane. Catal. Sci. Technol. 7, 3312–3324 (2017).
Kurlov, A. et al. Molybdenum carbide and oxycarbide from carbon-supported MoO3 nanosheets: phase evolution and DRM catalytic activity assessed by TEM and in situ XANES/XRD methods. Nanoscale 12, 13086–13094 (2020).
Wan, W., Tackett, B. M. & Chen, J. G. Reactions of water and C1 molecules on carbide and metal-modified carbide surfaces. Chem. Soc. Rev. 46, 1807–1823 (2017).
Liu, P. & Rodriguez, J. A. Water-gas-shift reaction on molybdenum carbide surfaces: essential role of the oxycarbide. J. Phys. Chem. B 110, 19418–19425 (2006).
Chen, C.-J. & Bhan, A. Mo2C Modification by CO2, H2O, and O2: effects of oxygen content and oxygen source on rates and selectivity of m-cresol hydrodeoxygenation. ACS Catal. 7, 1113–1122 (2017).
Schaidle, J. A. et al. Experimental and computational investigation of acetic acid deoxygenation over oxophilic molybdenum carbide: surface chemistry and active site identity. ACS Catal. 6, 1181–1197 (2016).
Sullivan, M. M. & Bhan, A. Effects of oxygen coverage on rates and selectivity of propane-CO2 reactions on molybdenum carbide. J. Catal. 357, 195–205 (2018).
Sullivan, M. M., Chen, C.-J. & Bhan, A. Catalytic deoxygenation on transition metal carbide catalysts. Catal. Sci. Technol. 6, 602–616 (2016).
Lezcano-Gonzalez, I. et al. Molybdenum speciation and its impact on catalytic activity during methane dehydroaromatization in zeolite ZSM-5 as revealed by operando X-ray methods. Angew. Chem. Int. Ed. 55, 5215–5219 (2016).
Anasori, B., Lukatskaya, M. R. & Gogotsi, Y. 2D metal carbides and nitrides (MXenes) for energy storage. Nat. Rev. Mater. 2, 16098 (2017).
Li, Z. & Wu, Y. 2D early transition metal carbides (MXenes) for catalysis. Small 15, e1804736 (2019).
Zheng, Y. et al. Molybdenum oxide, oxycarbide and carbide: controlling the dynamic composition, size and catalytic activity of zeolite-supported nanostructures. J. Phys. Chem. C. 123, 22281–22292 (2019).
Deeva, E. B. et al. In situ XANES/XRD study of the structural stability of two-dimensional molybdenum carbide Mo2CTx: implications for the catalytic activity in the water–gas shift reaction. Chem. Mater. 31, 4505–4513 (2019).
Halim, J. et al. Synthesis and characterization of 2D molybdenum carbide (MXene). Adv. Funct. Mater. 26, 3118–3127 (2016).
Shah, S. A. et al. Template-free 3D titanium carbide (Ti3C2Tx) MXene particles crumpled by capillary forces. Chem. Commun. 53, 400–403 (2017).
Wang, H.-W., Naguib, M., Page, K., Wesolowski, D. J. & Gogotsi, Y. Resolving the Structure of Ti3C2Tx MXenes through multilevel structural modeling of the atomic pair distribution function. Chem. Mater. 28, 349–359 (2016).
Graham, P. M., Buschhaus, M. S. A., Baillie, R. A., Semproni, S. P. & Legzdins, P. Hydroperoxide-Initiated Intramolecular Insertions of NO into metal−carbon bonds. Organometallics 29, 5068–5072 (2010).
Varjas, C. J., Powell, D. R. & Thomson, R. K. Rapid access to an oxido-alkylidene complex of Mo(VI). Organometallics 34, 4806–4809 (2015).
Takenaka, S., Tanaka, T., Funabiki, T. & Yoshida, S. Structures of molybdenum species in silica-supported molybdenum oxide and alkali-ion-modified silica-supported molybdenum oxide. J. Phys. Chem. B 102, 2960–2969 (1998).
Beale, A. M. & Sankar, G. In situ study of the formation of crystalline bismuth molybdate materials under hydrothermal conditions. Chem. Mater. 15, 146–153 (2003).
Pakhare, D. & Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 43, 7813–7837 (2014).
Thakur, R. et al. Insights into the thermal and chemical stability of multilayered V2CTx MXene. Nanoscale 11, 10716–10726 (2019).
Saggio, G. et al. Synthesis, characterization, and reactivity of the highly unsaturated silica-supported trisiloxy tantalum: (⋮SiO)3Ta(III). Organometallics 21, 5167–5171 (2002).
Coperet, C. et al. Surface organometallic and coordination chemistry toward single-site heterogeneous catalysts: strategies, methods, structures, and activities. Chem. Rev. 116, 323–421 (2016).
Steib, M., Lou, Y., Jentys, A. & Lercher, J. A. Enhanced activity in methane dry reforming by carbon dioxide induced metal-oxide interface restructuring of nickel/zirconia. ChemCatChem 9, 3809–3813 (2017).
Theofanidis, S. A., Batchu, R., Galvita, V. V., Poelman, H. & Marin, G. B. Carbon gasification from Fe–Ni catalysts after methane dry reforming. Appl. Catal., B 185, 42–55 (2016).
van Beek, W., Safonova, O. V., Wiker, G. & Emerich, H. SNBL, a dedicated beamline for combined in situ X-ray diffraction, X-ray absorption and Raman scattering experiments. Phase Transit 84, 726–732 (2011).
Tsoukalou, A. et al. The structural evolution and dynamics of an In2O3 Catalyst for CO2 hydrogenation to methanol: an operando XAS-XRD and in situ TEM study. J. Am. Chem. Soc. 141, 13497–13505 (2019).
Medford, A. J., Vojvodic, A., Studt, F., Abild-Pedersen, F. & Nørskov, J. K. Elementary steps of syngas reactions on Mo2C(001): adsorption thermochemistry and bond dissociation. J. Catal. 290, 108–117 (2012).
Likith, S. R. J. et al. Thermodynamic stability of molybdenum oxycarbides formed from orthorhombic Mo2C in oxygen-rich environments. J. Phys. Chem. C. 122, 1223–1233 (2018).
Foppa, L., Silaghi, M.-C., Larmier, K. & Comas-Vives, A. Intrinsic reactivity of Ni, Pd and Pt surfaces in dry reforming and competitive reactions: Insights from first principles calculations and microkinetic modeling simulations. J. Catal. 343, 196–207 (2016).
Kunkel, C., Viñes, F. & Illas, F. Transition metal carbides as novel materials for CO2 capture, storage, and activation. Energy Environ. Sci. 9, 141–144 (2016).
Asara, G. G., Ricart, J. M., Rodriguez, J. A. & Illas, F. Exploring the activity of a novel Au/TiC(001) model catalyst towards CO and CO2 hydrogenation. Surf. Sci. 640, 141–149 (2015).
Silaghi, M.-C., Comas-Vives, A. & Copéret, C. CO2 Activation on Ni/γ–Al2O3 catalysts by first-principles calculations: from ideal surfaces to supported nanoparticles. ACS Catal. 6, 4501–4505 (2016).
Foppa, L. et al. Contrasting the role of Ni/Al2O3 interfaces in water-gas shift and dry reforming of methane. J. Am. Chem. Soc. 139, 17128–17139 (2017).
Lam, E. et al. CO2 Hydrogenation on Cu/Al2O3: role of the metal/support interface in driving activity and selectivity of a bifunctional catalyst. Angew. Chem., Int. Ed. 58, 13989–13996 (2019).
Posada-Perez, S. et al. The bending machine: CO2 activation and hydrogenation on δ-MoC(001) and β-Mo2C(001) surfaces. Phys. Chem. Chem. Phys. 16, 14912–14921 (2014).
Damaskinos, C. M., Vasiliades, M. A. & Efstathiou, A. M. The effect of Ti4+ dopant in the 5 wt% Ni/Ce1-xTixO2-δ catalyst on the carbon pathways of dry reforming of methane studied by various transient and isotopic techniques. Appl. Catal., A 579, 116–129 (2019).
Cossu, G., Rossi, A., Arcifa, A. & Spencer, N. D. Development and application of a cost-effective transfer cell for X-ray photoelectron spectroscopy. Incontro di Spettroscopia Analitica ISA Cagliari (2018).
Shirley, D. A. High-resolution X-ray photoemission spectrum of the valence bands of gold. Phys. Rev. B 5, 4709–4714 (1972).
Scofield, J. H. Hartree-slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron Spectros. Relat. Phenom. 8, 129–137 (1976).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 12, 537–541 (2005).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Wellendorff, J. et al. Density functionals for surface science: exchange-correlation model development with Bayesian error estimation. Phys. Rev. B 85, 235149 (2012).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Acknowledgements
The authors thank ScopeM, the Laboratory of Surface Science and Technology, the Laboratory of Surface and Interfacial Chemistry (all from ETH Zürich) for the use of their electron microscopy, XPS, and high vacuum and facilities, respectively. The European Synchrotron Radiation Facility (ESRF), through proposal CH5229, is acknowledged for provision of beamtime. Dr. Wouter van Beek and Dr. Dragos Stoian are acknowledged for the support during the XAS experiments. Dr. Agnieszka Kierzkowska, Dr. Elena Willinger and Dr. Davood Hosseini (all ETH Zürich) are acknowledged for collecting TEM data. Dr. Hui Zhou (ETH Zürich) is acknowledged for performing CO chemisorption experiments. This project has received funding from the Swiss Office of Energy (BFE; SI/500881-01), ETH Zurich (ETH-40 17-2), Spanish “Ministerio de Ciencia, Innovación y Universidades” (PGC2018-100818-A-I00; “Proyecto I+D Generación del Conocimiento”), and Spanish MEC and the European Social Fund (RyC-2016-19930; “Ramon y Cajal” Fellowship to A.C.-V.).
Author information
Authors and Affiliations
Contributions
D.L. and A.F. conceived the research project. A.K., A.F., and C.R.M. planned the experimental work. A.K. and E.B.D. performed catalytic tests, synthesized materials, and analyzed data. D.L., A.K., and A.T. performed TEM, XAS, and XPS experiments, respectively. P.M.A. supervised XAS experiments. A.C.-V. designed and performed DFT calculations. A.K. and A.F. wrote the original and final draft of the paper with contributions from all authors. A.F. and C.R.M. supervised the experimental research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kurlov, A., Deeva, E.B., Abdala, P.M. et al. Exploiting two-dimensional morphology of molybdenum oxycarbide to enable efficient catalytic dry reforming of methane. Nat Commun 11, 4920 (2020). https://doi.org/10.1038/s41467-020-18721-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-18721-0
This article is cited by
-
Synergistic promotions between CO2 capture and in-situ conversion on Ni-CaO composite catalyst
Nature Communications (2023)
-
Role of support bio-templating in Ni/Al2O3 catalysts for hydrogen production via dry reforming of methane
Scientific Reports (2023)
-
One-step direct conversion of methane to methanol with water in non-thermal plasma
Communications Chemistry (2022)
-
Optimization of Non-thermal Plasma-Assisted Catalytic Oxidation for Methane Emissions Abatement as an Exhaust Aftertreatment Technology
Plasma Chemistry and Plasma Processing (2022)
-
Two-dimensional molybdenum carbide 2D-Mo2C as a superior catalyst for CO2 hydrogenation
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
