
Abstract
Flavin-mediated photocatalytic oxidations are established in synthetic chemistry. In contrast, their use in reductive chemistry is rare. Deazaflavins with a much lower reduction potential are even better suited for reductive chemistry rendering also deazaflavin semiquinones as strong reductants. However, no direct evidence exists for the involvement of these radical species in reductive processes. Here, we synthesise deazaflavins with different substituents at C5 and demonstrate their photocatalytic activity in the dehalogenation of p-halogenanisoles with best performance under basic conditions. Mechanistic investigations reveal a consecutive photo-induced electron transfer via the semiquinone form of the deazaflavin as part of a triplet-correlated radical pair after electron transfer from a sacrificial electron donor to the triplet state. A second electron transfer from the excited semiquinone to p-halogenanisoles triggers the final product formation. This study provides first evidence that the reductive power of excited deazaflavin semiquinones can be used in photocatalytic reductive chemistry.
Similar content being viewed by others
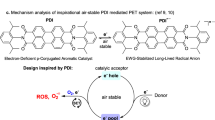


Introduction
Flavins, in the form of flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD), act as cofactors in various types of enzymes1,2. Some organisms also use deazaflavins (dFls; dF0 or dF420, see Fig. 1a) where the N-5 atom of the isoalloxazine ring is replaced by a carbon atom3,4. Despite their structural similarity, their roles in biological systems differ substantially3,5. Flavins (Fls) form semiquinones (Flsqs) (Fig. 1b), which are transiently stable in solution6 and more stable in protein environments7,8. Flsqs are typically involved in one-electron redox processes1,9. Fully reduced flavin (Flred) participates in two-electron reactions or, similarly as Flsq, interacts readily with molecular oxygen (O2), forming reactive oxygen species (ROS) or flavin hydroperoxide (FlOOH)10,11,12,13. FlOOH is the key agent in oxygenations mediated by flavin-dependent monooxygenases14,15. In contrast to Fls, dFls serve exclusively as two-electron carriers, thus behaving rather like nicotinamide adenine dinucleotide (phosphate) (NAD(P)H)3,5. Fully reduced deazaflavins (dFlreds) are considerably stable against oxidation by oxygen, thus avoiding formation of ROS10. This is one reason why dFls have been tested as native coenzyme (FAD or FMN) substitutes16,17,18.

a Structure of flavin and deazaflavin cofactors. b Redox states of flavins and deazaflavins. In contrast to flavins, semiquinone deazaflavins are untypical intermediates in redox chemistry.
Both Fls and dFls absorb visible light enabling their involvement in light-dependent processes19,20,21,22. Fl cofactors in their fully reduced form participate in photo-induced electron transfer (PET) reactions in DNA photolyases repairing pyrimidine dimers formed upon exposure to ultraviolet (UV) light23,24. Fls in their oxidised form are responsible for bacterial bioluminescence25 or perform various functions in photoreceptors7,26,27,28. dFl dF0 in its oxidised form serves as a light-harvesting antenna in photolyases29. Inspired by nature, artificial Fl derivatives as well as flavoenzymes have been applied in photoredox catalysis and photobiocatalysis, mainly in oxidative transformations or in energy transfer processes6,20,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44. Photoenzymatic radical polymerisation employing highly reducing excited FADH*− known from photolyases is rare and a very recent example of a photoreductive process with Fl45. In the dFl series, dFlred has been found to be generated from dFl by PET from ethylenediaminetetraacetic acid used as sacrificial reductant46. This procedure has been utilised in the NAD(P)H-independent regeneration of FADH2 from FAD16,47.
dFl belong to the redox cofactors with the lowest redox potential; the first reduction potential of dFl [E(dFl/dFlsq)] is even more negative than that of Fls by 0.5 V3. Therefore, dFls are better suited for reductive chemistry than Fls. The very negative first reduction potential renders the dFl semiquinone (dFlsq) as a strong reducing agent. Despite this fact, there is no precedence of reductive processes involving this radical species. This might result from the very low stability of the dFlsq free in the solution3,5,48.
Here we demonstrate that the reducing power of excited dFl radicals can be used in the presence of two substrates: (i) a sacrificial electron donor, e.g. diisopropyl(ethyl)amine (DIPEA), allowing the photochemical formation of the radical dFlsq and (ii) a substrate for reduction, p-halogenanisole, undergoing dehalogenation via the excited radical dFlsq*.
Results
Design and synthesis
A series of 5-dFls 1–5 was synthesised with different substituents in position 5 (H-, Me-, iPr-, CF3-, and Ph-) via two different routes depending on the substituent (Fig. 2 and Supplementary Notes 1 and 2). Derivatives 1–4 were obtained following route A starting from 3,4-dimethoxyaniline (6). Reductive amination yielded compound 7, which was subsequently coupled with 6-chloro-3-methylpyrimidine-2,4(1H,3H)-dione to give access to the key intermediate 8. Depending on the reaction conditions for the following step, the corresponding products 1–4 were obtained. As this synthetic method did not allow the synthesis of the phenyl derivative 5, route B was developed. In this three-component microwave-assisted procedure, adapted from Tu et al.49, compound 5 was obtained from a reaction of benzaldehyde with N-butyl-3,4-dimethoxyaniline (7) and N-methylbarbituric acid (9). Following this procedure, the phenyl derivative could be isolated in both the oxidised (5ox) and fully reduced form (5red), which reflects the low sensitivity of the dFlred against O23,50. This observation is further supported by incubation of 5red in darkness for 19 h in non-degassed acetonitrile (ACN) where no considerable formation of 5ox is observed via absorption spectroscopy (Supplementary Note 3). However, light excitation of 5red allows to overcome the activation barrier for re-oxidation by O2 resulting in almost stoichiometric conversion to 5ox (Supplementary Note 3).

Electronic transition energies, E0-0, and first reduction potentials vs. saturated calomel electrode (SCE) are given for all deazaflavins. Full spectra are shown for 5ox and 5red. Data for tetraacetyl riboflavin (TARF) are given for comparison. Further details on synthesis and characterisation are given in Supplementary Notes 1 and 2, respectively.
Photophysical and electrochemical characterisation
Compounds 1–3 have similar UV/visible (Vis) absorption and emission spectra in ACN giving similar electronic transition energies of these dFls with the usual blue-shift of around 40 nm compared to isoalloxazines (Fig. 2 and Supplementary Fig. 1)51. In contrast, derivative 4 with the strong electron withdrawing group -CF3 shows a red-shifted electronic transition by 26 nm in ACN compared to isoalloxazines. The compounds 1–4 are all highly emitting with fluorescence quantum yields ranging from 67% to 85% in ACN. Their emission lifetimes in ACN range between 6.8 and 11.1 ns (Supplementary Note 2). In agreement with literature reports4, cyclo-voltammetry (CV) shows more negative redox potential of the first reduction step of the dFls compared to tetraacetyl riboflavin (TARF) (E(TARF/TARFsq) = −0.79 V, ref. 39) by 0.13–0.52 V (Supplementary Note 4).
The phenyl derivative 5 shows deviating characteristics compared to the other dFls. While the electronic transition energies are also blue-shifted compared to isoalloxazines and the redox potential of the first reduction step is also more negative compared to TARF, its emission properties are significantly different. The fluorescence quantum yield reaches only 16% and its excited singlet state lifetime is considerably reduced being only 1.7 ns (Supplementary Note 2). Furthermore, compound 5 is the only derivative that forms a thermally stable fully reduced form 5red, which is only re-oxidised by light as discussed above (Supplementary Note 3).
Photodehalogenation
Owing to the strong reduction potential of dFl derivatives compared to their Fl analogues, we investigated their use in the reductive photoredox catalysis for the dehalogenation of aryl halides, which is considered as a benchmark reaction because of their very negative redox potentials. For instance, with a one-electron reduction potential of −2.75 V52, p-bromoanisole (p-BA) is beyond the limits of most organic photoredox catalysts53,54, or it requires combination of electrocatalysis and photocatalysis for dehalogenation55,56.
We achieved an almost quantitative yield of p-BA dehalogenation using the phenyl derivative 5 (6 mM, 8 mol% relative to substrate), when irradiating at 385 nm in the presence of a sacrificial reducing agent, here we used DIPEA, and caesium carbonate (Cs2CO3) (Table 1). The anisole yield is independent on the oxidation state of the photocatalyst, i.e. 5ox or 5red, since the dFlred 5red is re-oxidised by O2 and light as described above (Supplementary Note 3). Noteworthy, 5ox also showed photo-dehalogenation of p-chloroanisole, whose redox potential is even more negative (Ered = −2.88 V, ref. 52).
We next investigated the influence of the solvent and the excitation wavelength on the total product yield (Supplementary Note 5). Interestingly, when dimethylformamide was used as solvent under otherwise identical conditions the product yield decreased by almost 50%. With respect to the excitation wavelength, the best yield, ca. 80%, was obtained for wavelengths ≤400 nm. When using light of 455 nm, only 50% yield could be obtained. As a control, in the absence of photocatalyst, DIPEA, or irradiation, no reaction occurs. Recording the product build-up in case of 5ox showed that 60% conversion was observed already after 6 h.
Furthermore, we investigated the effect of basicity on the dehalogenation efficiency of p-BA (Table 2). Without any base, only yields between 10% and 30% are observed for all photocatalysts. In the presence of Cs2CO3, under otherwise identical conditions, the yield was increased to >60% for the dFls 3 and 5, while the reactivity of dFls 1 and 4 remained unchanged or increased to only 38% for the methyl derivative 2. For the dFls 1 and 5, we then tested bases with increasing basicity, i.e. NaHCO3, Cs2CO3, and KOtBu. While the activity of 5 was significantly enhanced by Cs2CO3 or KOtBu, the reactivity of 1 only increased in case of KOtBu. These findings already indicate an important acidobasic equilibrium of a potential intermediate species of the photocatalyst that affects the overall performance. Thus a deprotonated form of one key intermediate seems to be beneficial (A more detailed discussion can be found in Supplementary Note 6).
Excited singlet state reaction results in a loss channel
The reactivity of the excited states of the compounds 1 and 5ox with the sacrificial substrate DIPEA was investigated by time-resolved absorption and emission spectroscopy in the absence of any additional base, since the main focus was on the understanding of the underlying photocatalytic reaction mechanism (Further studies on the role of specific deprotonation states is currently ongoing in our laboratories and will be presented elsewhere.). In both cases, the excited state quenching was observed without the formation of any additional transient species (Fig. 3 and Supplementary Note 7). This indicates the formation of a singlet born radical pair that recombines faster than it is formed owing to spin-allowed radical pair recombination (Fig. 3e and Supplementary Note 7). Such a fast recombination reaction was already observed in the Fl photo-oxidation of aromatic alcohols and represents a pure loss channel for photocatalytic applications6. In case of the reaction between DIPEA and the excited singlet of the dFl, the bi-molecular reactions rates for 1 and 5ox under low substrate concentrations (<200 mM DIPEA) are (1.05 ± 0.01) × 1010 M−1 s−1 and (9.0 ± 0.1) × 109 M−1 s−1, respectively (see Stern–Volmer analysis in Supplementary Note 7). These values are smaller by 56% and 47%, respectively, compared to the theoretical diffusion limited bi-molecular reaction limit (see Supplementary Note 7 for a detailed discussion), which might indicate a geometrical preference for reactive encounters allowing for necessary interactions, and thus enabling a high probability for the reaction to proceed.

a–c False colour representation of the time-resolved absorption spectra of 5ox (360 μM) in non-degassed ACN in the absence (a) or presence of either 800 mM DIPEA (b) or 100 mM p-BA (c) exciting at λexc = 450 nm. d Species-associated spectra (SAS) that contribute to the time-resolved absorption signals in a–c with excited singlet state lifetimes as indicated. e Model used to describe the time-resolved absorption data that results in physically reasonable SAS. On this time window, kisc is negligible, and since no further species spectrum is detectable, the assumption 1k'beT ≫ 1k′eT [Ssac] is justified. SE stimulated emission, rad radiative, ic internal conversion, (b)eT (back) electron transfer, (b)isc (back) intersystem crossing, S0 singlet ground state, S1 excited singlet state, T1 triplet state, 1RP singlet born radical pair, Ssac sacrificial electron donor, S substrate.
In the presence of only the actual substrate p-BA (with a concentration of 100 mM, which is sufficiently high for diffusion controlled encounters with the excited singlet state of the photocatalyst), the transient absorption (TA) data of 1 and 5ox resemble those of the photocatalysts alone demonstrating no reaction between p-BA and the corresponding excited states (Fig. 3c and Supplementary Note 7). In addition, in case of the stable fully reduced form, 5red, also no reaction between the excited singlet state and the substrate p-BA is observed (Supplementary Note 7).
Triplet state reaction forms transiently stable semiquinone
TA spectroscopy in the ns to μs range was used in order to study the triplet state formation of 5ox (Fig. 4a, e as well as further details in Supplementary Notes 2 and 8). In comparison to isoalloxazines, its yield is low being only ca. 11% (Supplementary Table 1). In the presence of the substrate p-BA (50 mM), no reaction is observed with either the excited singlet or triplet state of 5ox (Fig. 4b, e). In contrast, in the presence of the sacrificial electron donor DIPEA (50 mM) electron transfer (eT) from DIPEA to the triplet state occurs forming the semiquinone 5sq (Fig. 4c, f), whose spectrum agrees well with the electrochemically generated spectrum (Fig. 5g and Supplementary Note 9). The yield for eT can be determined from the corresponding decay rates to \(\Phi _{{\mathrm{eT}}} = 1 - k_0k_{\mathrm{q}}^{ - 1} = 87\%\), where k0 is the total decay rate constant of the triplet in the absence and kq in the presence of DIPEA. Considering the concentration of DIPEA (50 mM), the corresponding bi-molecular reaction rate with the triplet state is given to be (8.7 ± 0.1) × 108 M−1 s−1 (keT = ΦeTkq ([DIPEA])−1). However, in the available spectral window of our experiment, we do not observe spectral signatures of the corresponding DIPEA radical cation. The assignment as the semiquinone radical is further proven by recording its electron paramagnetic resonance spectrum under photo-stationary conditions (Supplementary Note 10). In the presence of both p-BA and DIPEA, again, only the triplet reaction with DIPEA is observed, but no indication for a reaction between p-BA and 5sq is found (Fig. 4d, f). In summary, while the excited singlet state reaction with DIPEA results in a singlet born spin correlated radical pair, 1[5sq, DIPEA•+], which recombines faster than it is formed owing to spin allowance, the triplet reaction with DIPEA involves a triplet born radical pair, 3[5sq, DIPEA•+], which is spin forbidden for recombination, thus allowing its accumulation (Fig. 4g).

a–d False colour representation of the time-resolved absorption spectra of 5ox (60 μM) in non-degassed ACN in the absence (a) or presence of either 50 mM p-BA (b), 50 mM DIPEA (c), or both 50 mM p-BA and 50 mM DIPEA (d) exciting at λexc = 430 nm. e Species-associated spectra (SAS) that contribute to the time-resolved absorption signals in a–b, which were generated by applying the model in g to the decay-associated difference spectra (DADS) shown in the inset. f Same as e but for the data in c, d. SE stimulated emission, rad radiative, ic internal conversion, (b)eT (back) electron transfer, (b)isc (back) intersystem crossing, superscript 1/3 singlet or triplet state, RP spin correlated radical pair containing the 5sq (D doublet state), Ssac sacrificial electron donor, S substrate.

a, b, d, e Sequences of UV/Vis absorption spectra after application of −1.2 V to the sample at conditions as indicated. c, f Concentration–time profiles from fitting the species spectra shown in g to the data as indicated. The grey vertical lines in g correspond to the quantum chemically calculated stick spectrum of p-BA•‒ as described in the text. h Summary of all the observed processes. The stick and ball structure of p-BA and p-BA•‒ are the result of quantum chemical calculations as described in the main text and Supplementary Note 11. The significantly reduced Br− dissociation energy in p-BA•− demonstrates the final bromine dissociation upon one electron reduction.
Reactivity of the excited dFlsq
In the next step, we investigated the reactivity of 5sq in its excited state 5sq* towards the dehalogenation of p-BA. For this purpose, we generated the semiquinone form 5sq electrochemically without the need of the light-induced reaction with DIPEA by applying constantly −1.2 V to the sample in ACN and followed its reactivity by UV/Vis absorption over time under the following four conditions: (1) alone in the dark, (2) in the dark and the presence of p-BA (250 mM), (3) alone in the light (λexc = 385 nm), and (4) in the light (λexc = 385 nm) and in the presence of p-BA (250 mM). Alone in the dark, initially the concentration of 5ox depletes completely and 5sq is formed simultaneously (Fig. 5a). On longer delays, the concentration of 5sq reduces by ca. 18% with the simultaneous formation of 5red indicating partial disproportionation of two 5sq molecules into 5ox and 5red19. The former does not recover considerably because the electrode-driven one electron reduction immediately forms 5sq. Using the species spectra obtained from the spectro-electrochemical experiment (Fig. 5g and Supplementary Note 9), the sequence of spectra can be perfectly decomposed in order to obtain the corresponding concentration–time profiles of the contributing species (Fig. 5c). Repeating this experiment in the dark but now in the presence of p-BA, exactly the same behaviour is observed within the experimental error (Fig. 5b, c) demonstrating no reaction between p-BA and either of the dFl redox species in their ground states. This additionally confirms the results of the TA spectroscopy.
However, in the experiments with simultaneous illumination of the sample, significantly different kinetics are observed compared to the situation in the dark. In the absence of p-BA but presence of light, again, a rapid reduction of 5ox is observed (Fig. 5d). However, under these conditions 5sq does not accumulate to the same extent (here only to 75% in the transient maximum) as in the case without illumination. Instead, a significantly enhanced formation of 5red up to 70% in the transient maximum is observed, indicating a light-induced second eT from the electrode to the excited 5sq*. Again, these data can be decomposed well with the known species spectra of the three redox states of 5 (Fig. 5f). In the final experiment with light and p-BA, again, different kinetics are observed compared to the three experiments described before. In this case, 5ox is only converted to ca. 75%. This is accompanied by a significantly reduced accumulation of 5sq and 5red (Fig. 5e), thus indicating a reaction between p-BA and 5sq in its excited state reforming 5ox. Using only the known species-associated spectra (SAS) of the dFl intermediates, this time, the decomposition of the data shows significant deviations. Closer inspection of the arising absorption band at 300 nm reveals an asymmetric shape compared to the expected absorption band of 5red indicating some underlying spectral contributions of a different species than those already known from the dFls. After subtraction of a fitted linear combination of the known spectral features of dFl, an absorption spectrum with a single absorption band peaking at 330 nm remains (magenta spectrum in Fig. 5g). Considering a potential radical anion of p-BA as an intermediate arising after eT from excited 5sq* to p-BA, we calculated the absorption spectrum of p-BA•‒ quantum chemically using a high level of theory, i.e. state-averaged XMCQDPT-CASSCF(12,12), considering also the solvent environment by the polarisable continuum model (PCM) for ACN (see Supplementary Note 11). As seen in Fig. 5g and Supplementary Fig. 12a, the calculated spectrum matches the measured spectrum accurately, so that we assign this spectrum to p-BA•‒. Considering the p-BA•‒ spectrum together with the already determined species spectra for all dFl intermediates now results in a good fit with deviation within the experimental error. Since the fit does not depend on the scaling of the p-BA•‒ spectrum, we scaled the spectrum arbitrarily so that the resulting concentration–time profile stays below all Fl contributions. Furthermore, the quantum chemical calculation revealed a significantly longer bond length between the bromine and the adjacent carbon atom in p-BA•‒ (2.86 Å) than in p-BA (1.92 Å) as well as a significantly reduced dissociation energy being close to thermal energy fluctuations (Supplementary Fig. 12b), which explains well the observed final dissociation of the bromine during the dehalogenation, which is triggered by a single electron reduction step as summarised in Fig. 5h (Further details are given in Supplementary Note 11.).
Analogous experiments were conducted with the non-substituted dFl 1. However, in contrast to the experiments with 5, the semi-reduced form could not be observed on the time scale of the measurements. The failure of detecting the semi-reduced form 1sq indicates that its lifetime is significantly shorter compared to that of 5sq. This agrees with the observation of a significantly lower yield in the dehalogenation using 1 compared to 5, since less 1sq may be accumulated.
Photocatalytic reaction mechanism based on consecutive PET (conPET)
In summary, our results can be compiled to the photocatalytic reaction mechanism depicted in Fig. 6. With respect to the redox potential neither 5red (E(5sq/5red) = −1.6 V) nor 5sq (E(5ox/5sq) = −1.41 V) can reduce p-BA (Ered = −2.75 V) in their ground states. The participation of the excited fully reduced form 5red* is not observed most likely due to very efficient intrinsic deactivation of the excitation energy as seen in fast deactivation kinetics on a ps time scale (Supplementary Note 7 and Supplementary Fig. 8). In the presence of a sacrificial electron donor DIPEA, we observe different reactivities for either the excited singlet or triplet state of 5ox. In the excited singlet state reaction, a singlet born spin correlated radical pair, 1[5sq, DIPEA•+], is formed, which recombines faster than it is formed. Accordingly, this reaction represents a pure loss channel. In contrast, eT to the triplet state results in a triplet born radical pair, 3[5sq, DIPEA•+], that is spin forbidden for recombination, thus allowing the accumulation of the considerably stable 5sq. As a consequence, 5sq can be excited by a second photon (E*(5ox/5sq) = −3.3 V), which enables a conPET57 from 5sq* to the substrate, p-BA, regenerating 5ox in its ground state and closing the photocatalytic cycle. The dissociation energy for the bond between the bromine and the adjacent carbon in p-BA•‒ is significantly reduced leading to thermally driven dissociation into Br– and the aryl radical. Finally, anisole is formed via hydrogen abstraction from either a DIPEA radical cation or a solvent molecule57. Owing to the lack of the observation of any protonated 5sq form (see Supplementary Note 6), the radical anion of 5sq is the active species.

A sacrificial electron donor, e.g. DIPEA, reacts either with the excited singlet or triplet state of the deazaflavin. In the excited singlet state reaction, a singlet born spin correlated radical pair, 1[5sq, \({\mathrm{S}}_{{\mathrm{sac}}}^ {•+}\)], is formed after eT. This radical pair recombines faster than it is formed. In contrast, eT to the triplet state results in a triplet born radical pair, 3[5sq, \({\mathrm{S}}_{{\mathrm{sac}}}^ {•+}\)], allowing the accumulation of the considerably stable 5sq. 5sq can be excited by a second photon enabling a consecutive photo-induced electron transfer from 5sq* to the substrate, p-BA, regenerating 5ox in its ground state and closing the photocatalytic cycle. Finally, anisole is formed via hydrogen abstraction from either a \({\mathrm{S}}_{{\mathrm{sac}}}^ {•+}\) or a solvent molecule.
Discussion
We have described the detailed photocatalytic mechanism of a reductive dehalogenation of p-halogenanisole by dFls. The key point is the conPET via the anionic semiquinone form of the dFl. In doing so, we have revealed that dFls are valuable photocatalysts for reductive chemistry. In summary, after photoexcitation of dFl followed by intersystem crossing a sacrificial electron donor, at a moderate concentration, will enable a first eT to the triplet state resulting in a considerably stable anionic semiquinone. Additional excitation of the anionic semiquinone provides sufficient energy for a second eT process from the semiquinone to compounds with highly negative redox potentials, such as p-halogenanisoles. Finally, downstream reactions resulting in product formation occur spontaneously.
The action spectrum of the conPET reaction comprises the absorption of two different coloured photons by the two key photocatalytic forms of dFl, where one is formed from the other having a distinct lifetime in the ns regime. Therefore, a concrete timed illumination of the photocatalytic system with short and intense pulses at the appropriate excitation wavelengths, which are temporally optimised to the lifetime of the key intermediates, should allow further improvement on the efficiency. This aspect is of general importance for all conPET-type reactions and should be addressed in future work.
To our knowledge, this study shows for the first time that the reductive power of excited dFlsq can be used in photocatalytic reductive chemistry. Moreover, because of its very negative potential, this species seems to belong to the most powerful reductants53,55,56,58,59,60,61. Therefore, this study expands the photocatalytic toolbox for new chemical transformations and should serve as a guide for engineering photocatalysts of the next generation.
Methods
Materials
Starting materials and reagents were purchased from commercial suppliers (Sigma Aldrich, Alfa Aesar, Acros, Fluka, VWR, or Fluorochem) and were used without further purification. Solvents were used as p.a. grade or dried and distilled according to literature known procedures.
Nuclear magnetic resonance (NMR) spectroscopy
NMR spectra were recorded at room temperature using a Bruker Avance 300 (300 MHz for 1H, 75 MHz for 13C), a Bruker Avance 400 (400 MHz for 1H, 101 MHz for 13C), an Agilent 400-MR DDR2 (400 MHz for 1H and 101 MHz for 13C), or a Varian Mercury Plus 300 (300 MHz for 1H, 75 MHz for 13C) with internal solvent signal as reference. All chemical shifts are reported in δ-scale as parts per million (multiplicity, coupling constant J, number of protons) relative to the solvent residual peaks as the internal standard (IS). NMR spectra are available in Supplementary Figs. 13–22.
Mass spectrometry
The mass spectrometric measurements were performed at the Central Analytical Laboratory of the University of Regensburg or at the Central Analytical Laboratory of UCT, Prague on a Finnigan MAT 95, ThermoQuest Finnigan TSQ 7000, Finnigan MATSSQ 710A, Agilent Q-TOF 6540 UHD, or a LTQ Orbitrap Velos (Thermo Fisher Scientific).
Gas chromatography (GC)
GC measurements were performed on a GC 7890 from Agilent Technologies. Data acquisition and evaluation was done with Agilent ChemStation Rev.C.01.04. A capillary column HP-5MS/30 m × 0.25 mm/0.25 μM film and helium as carrier gas (flow rate of 1 mL/min) were used. The injector temperature (split injection: 40:1 split) was 280 °C, detection temperature 300 °C (FID). The GC oven temperature programme was adjusted as follows: the initial temperature of 40 °C was kept for 3 min and was increased at a rate of 15 °C/min until the injection temperature of 280 °C was reached. After 5 min, the temperature was further increased at a rate of 25 °C/min until the final temperature of 300 °C was reached and kept for 5 min. Using 4-methylanisole as an IS, we obtained the GC calibration given in Supplementary Fig. 23 for the quantitative analysis.
Cyclo-voltammetry
CV measurements were performed with the three-electrode potentiostat galvanostat (PGSTAT302N, Metrohm Autolab). The control of the measurement instrument, the acquisition, and processing of the CV data were performed with the software Metrohm Autolab NOVA 1.10.4. Electrochemical studies were carried out under argon atmosphere in ACN containing 0.1 M tetra-N-butylammonium hexafluorophosphate using ferrocene/ferrocenium (Fc/Fc+) as an internal reference. A glassy carbon electrode (working electrode), platinum wire counter electrode, and Ag quasi-reference electrode were employed.
Spectro-electrochemistry
Measurements were performed in an Ottle Cell (Optically transparent thin-layer electro-chemical cell), pathlength = 0.02 cm, working electrode: Pt minigrid, counter electrode: Pt minigrid, pseudo reference electrode: Ag wire. Samples were prepared by degassing a solution of 5ox in ACN (3 mM) via bubbling of Argon for several minutes. The substrate, p-BA, was added via a Hamilton syringe resulting in a solution of 250 mM. A constant electric field of −1.2 V was applied to the cell, and UV/Vis absorption spectra were recorded every 5 s (using an Agilent 8453 spectrometer). Irradiation was done via a handheld LED (λmax = 385 nm) for a time of ca. 2 s in between each spectrum. Spectro-electrochemical data are given in Fig. 5 and Supplementary Note 9.
General irradiation conditions
Samples were irradiated at 254 nm (UV handlamp from Herolab GmbH Laborgeräte, 254 nm, 8 W), at 365 nm (UV handlamp from Herolab GmbH Laborgeräte, 365 nm, 8 W), at 370 nm (Opulent Starboard, Luminus SST-10-UV-A130, λmax = 370 nm, Ityp = 500 mA, Imax = 1.0 A, Φtyp = 875 mW), at 385 nm (Opulent Starboard, Luminus SST-10-UV-A130, λmax = 385 nm, Ityp = 500 mA, Imax = 1.5 A, Φtyp = 1015 mW), or at 455 nm (CREE XLamp Me-C LED, λmax = 455 nm, Imax = 700 mA). The corresponding emission spectra of each LED are given in Supplementary Fig. 24 or in the corresponding figures containing sequences of UV/Vis absorption spectra.
Stationary UV/Vis absorption and emission spectroscopy
UV/Vis absorption (Cary 60 or Cary 100 UV-Vis spectrophotometer; Agilent) and UV/Vis emission spectra (Horiba FluoroMax-4 spectrofluorometer) were recorded at room temperature. Fluorescence quantum yields were determined with a Hamamatsu C9920-02 system equipped with a Spectralon integrating sphere. The quantum yield accuracy is <10% according to the manufacturer.
Stepwise illumination and recording of absorption spectra
Stationary UV/Vis absorption spectra were recorded using an Agilent Cary 50 UV/Vis spectrophotometer in a sample cell of 10 × 10 mm that can be flanged to the freeze pump and thaw apparatus and closed after degassing. A sample of 5red was illuminated using an UV handlamp (UV handlamp from Herolab GmbH Laborgeräte, 254 nm, 365 nm, 8 W) providing either 254 or 365 nm. Continuous light was delivered onto the sample that was reproducibly positioned in ca. 2 cm distance without any further optics allowing for direct comparison of data sets recorded under different sample preparation conditions. After each illumination period, an absorption spectrum was recorded directly afterwards. The sample volume of 3 mL was continuously stirred during the illumination periods providing a homogeneously illuminated sample.
Time-resolved UV/Vis emission spectroscopy
A self-constructed time correlated single photon counting set-up62 was used to record emission decay data at single detection wavelength. A quartz cuvette with four optical windows of the dimension 2 mm × 10 mm was used. The sample was excited along the 2 mm pathlength, and the emission was recorded orthogonally to this. The optical density of the sample was set to ca. 0.1 at the excitation wavelength over 2 mm pathlength.
Ns to ms time-resolved UV/Vis absorption spectroscopy
The streak camera set-up described first in refs. 62,63 was adapted as follows. The third harmonic of a Nd:YAG laser (10 Hz, Surelite II, Continuum) pumping an Optical Parametric Oscillator (OPO, Continuum) tuned to 450 nm (10 mJ, ca. 10 ns) was used for sample excitation. As a probe light, a pulsed 150 W Xe-flash lamp (Applied Photophysics) was used that was focussed three times via toric mirror optics: (i) before probe shutter, (ii) into sample, and (iii) into spectrograph. The entire white light probe pulse was analysed by a combination of a spectrograph (200is, Bruker) and a streak camera (C7700, Hamamatsu Photonics). The use of mechanical shutters enabled the recording of a sequence of three individual data sets: (i) an image (DFL) with both flash lamp and laser, (ii) an image (D0) without any incoming light, and (iii) an image (DF) only with the flash lamp. One hundred such sequences were recorded, and corresponding data sets were averaged. Then the TA was calculated as:
A 10-mL sample was stepwise cycled by a peristaltic pump (ecoline, ISMATEC) through a flow cell with a pathlength of 2 mm for pump and 10 mm for probe beams (dimensions: 2 mm × 10 mm × 30 mm, Starna) ensuring a total replacement of the sample prior to each individual measurement. No photocatalyst degradation was observed under the used conditions.
Sub-ps and sub-ns pump/supercontinuum probe spectroscopy
Ultrafast broadband TA experiments were carried out using UV/Vis pump–supercontinuum probe spectroscopy at 1 kHz repetition frequency. In short, a Ti-sapphire amplifier system (Coherent Libra) was used to generate 800 nm with 1.2 mJ pulses at 1 kHz. The output was split into three parts of which only two were used: (1) Ca. 50% of the 800 nm pulses were used to pump a collinear Optical Parametric Amplifier (OPA, TOPAS-800-fs, Light Conversion) tuned to pump pulses centred at ca. 450 nm (ca. 100 fs, ca. 400 nJ at the sample position) for sample excitation. (2) Ca. 10% were used to pump a non-colinear OPA (in-house build) tuned to pulses centred at ca. 530 nm (ca. 100 fs, ca. 5 μJ at the CaF2 position) for generation of supercontinuum white light probe pulses by focussing into a moving CaF2 disc of 1-mm thickness giving a probe spectrum ranging from 310 to 700 nm. Pump pulses were delayed via a motorised delay line equipped with an open corner cube mirror up to 2 ns. Two complementary high-speed spectrographs (Entwicklungsbüro EB Stresing) for signal and reference recording were used. The pump and probe pulses were focussed colinearly into the sample to spot sizes of ca. 80 and 60 μm full width at half maximum, respectively. For longer delays reaching out from ns to μs time ranges, a similar spectrometer was used in which the pump laser was electronically delayed relative to the probe laser. A detailed description can be found in ref. 64. The relative polarisations between the pump and probe were set by a half-wave plate in the pump beam path to magic angle (54.71°) for observations of pure population changes or to either parallel (0°) or orthogonal (90°) for observation of the anisotropy. The averaged pre-t0 laser scatter signal was subtracted from the data and the ca. 1 ps chirp of the white light was corrected for prior to data analysis using the coherent artefact as an indicator for time zero at each wavelength. Throughout the probe range, the spectral resolution was better than 4 nm and the temporal resolution was better than 150 fs. Ten individual scans with averaging 100 spectra per time point were typically recorded. The time axis—within total 500 points—was linear between −1 and 2 ps and logarithmic from 2 ps to the maximum time delay ensuring that the dynamics on every time scale will have equal weighting in the fitting analysis. In the sub-ps TA set-up, 10 mL of the sample were cycled through an in-house build cell with a pathlength of 100 μm for pump and probe beams. In sub-ns TA set-up, 10 mL of the sample were cycled through a flow cell (Starna) with a pathlength of 2 mm for pump and probe beams. In all cases, all scans resulted in reproducible data sets. In addition, the integrity of the sample was checked by recording stationary absorption spectra before and after each measurement. No photocatalyst degradation was observed under the used conditions. The shown data correspond to one representative measurement. No smoothing or filtering procedures were applied to the data.
TA data analysis and modelling
Singular value decomposition (SVD)-based rank analysis and global fitting were performed using an in-house written programme described previously62,63. In brief, the linear least squares problem
is solved, where ∆A is the time-resolved absorption data matrix, F is the matrix containing the analytical functions accounting for the temporal changes in the data, i.e. exponential decays (convoluted with the instrument response, typically a Gaussian function), and B is the matrix with the to be determined spectra. Further optimisation of χ2 is achieved by optimising the rate constants in F by a nonlinear least squares algorithm. As a result of such fits, the so-called decay-associated difference spectra (in matrix B) and their associated optimised rate constants are obtained. These are the unique result of the global fit, and this treatment does not require any model for the kinetics involved in the transient processes. The number of exponentials in the global fit is determined by the SVD-based rank analysis, which is described elsewhere65. The model that relates the actual species kinetics to the elementary function is applied afterwards resulting in SAS. The shape of the SAS in terms of identity with well-known spectra or following physical laws decides about the appropriateness of the model. This step does not change the χ2 value found in the global fit, and therefore, this procedure has the advantage that all interpretation is performed with the same quality of fit.
As an alternative analysis, known species spectra, taken either from literature or recorded in this work, were taken in order to decompose the recorded time-resolved data matrix using the transpose of the data matrix and using the basis spectra instead of analytical functions. The resulting concentration–time profiles inform about the appropriateness of the basis spectra and the physical reasonability, i.e. total sum of species being constant to 1.
Quantum chemical calculations
Quantum chemical calculations on all molecular moieties were performed using either the Firefly QC package66, which is partially based on the GAMESS (US)67 source code, or the Orca package68,69. All ground state structures were optimised on the level of restricted open shell density functional theory (ROHF-DFT) using the B3LYP functional and the aug-cc-pDVZ basis set. Complete active space self-consistency field (CASSCF) theory was used in order to calculate the static correlation energy. The highest 12 contributing π and n electrons were included into the CAS, distributed over 12 molecular orbitals (MO), and energy averaging over 10 states with equal weights, i.e. CASSCF(12,12)10, was performed. In order to calculate the dynamic correlation energy, extended multi-configuration quasi-degenerate perturbation theory (XMCQDPT) was used on top of the CASSCF optimised MOs70. In all cases, an intruder state avoidance denominator shift of 0.02 was used. The absorption spectrum for 5sq was calculated via unrestricted open shell time-dependent DFT (TD-DFT) with the aug-cc-pDVZ basis set. Solvent effects were taken into account with the conductor-like PCM for the TD-DFT calculation and with the PCM for the XMCQDPT-CASSCF calculations. Relaxed potential energy surfaces along the C-Br bond in p-BA and p-BA•− were performed on the DFT (B3LYP) level of theory with the aug-cc-pVDZ basis set.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and Supplementary Information, as well as from the authors upon reasonable request.
References
Walsh, C. T. & Wencewicz, T. A. Flavoenzymes: versatile catalysts in biosynthetic pathways. Nat. Prod. Rep. 30, 175–200 (2013).
Joosten, V. & Van Berkel, W. J. H. Flavoenzymes. Curr. Opin. Chem. Biol. 11, 195–202 (2007).
Greening, C. et al. Physiology, biochemistry, and applications of F420 and FO-dependent redox reactions. Microbiol. Mol. Biol. Rev. 80, 451–493 (2016).
Walsh, C. Naturally occurring 5-deazaflavin coenzymes: biological redox roles. Acc. Chem. Res. 19, 216–221 (1986).
Hemmerich, P., Massey, V. & Fenner, H. Flavin and 5-deazaflavin: a chemical evaluation of ‘modified’ flavoproteins with respect to the mechanisms of redox biocatalysis. FEBS Lett. 84, 5–21 (1977).
Megerle, U. et al. Unraveling the flavin-catalyzed photooxidation of benzylic alcohol with transient absorption spectroscopy from sub-pico- to microseconds. Phys. Chem. Chem. Phys. 13, 8869–8880 (2011).
Kutta, R. J., Archipowa, N. & Scrutton, N. S. The sacrificial inactivation of the blue-light photosensor cryptochrome from Drosophila melanogaster. Phys. Chem. Chem. Phys. 20, 28767–28776 (2018).
Kutta, R. J., Archipowa, N., Johannissen, L. O., Jones, A. R. & Scrutton, N. S. Vertebrate cryptochromes are vestigial flavoproteins. Sci. Rep. 7, 44906 (2017).
Massey, V. The chemical and biological versatility of riboflavin. Biochem. Soc. Trans. 28, 283–296 (2000).
Holtmann, D. & Hollmann, F. The oxygen dilemma: a severe challenge for the application of monooxygenases? ChemBioChem 17, 1391–1398 (2016).
Insińska-Rak, M. & Sikorski, M. Riboflavin interactions with oxygen—a survey from the photochemical perspective. Chem. Eur. J. 20, 15280–15291 (2014).
Gelalcha, F. G. Heterocyclic hydroperoxides in selective oxidations. Chem. Rev. 107, 3338–3361 (2007).
Gadda, G. Oxygen activation in flavoprotein oxidases: the importance of being positive. Biochemistry 51, 2662–2669 (2012).
Huijbers, M. M. E., Montersino, S., Westphal, A. H., Tischler, D. & van Berkel, W. J. H. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 544, 2–17 (2014).
Torres Pazmiño, D. E., Winkler, M., Glieder, A. & Fraaije, M. W. Monooxygenases as biocatalysts: classification, mechanistic aspects and biotechnological applications. J. Biotechnol. 146, 9–24 (2010).
Zilly, F. E., Taglieber, A., Schulz, F., Hollmann, F. & Reetz, M. T. Deazaflavins as mediators in light-driven cytochrome P450 catalyzed hydroxylations. Chem. Commun. 46, 7152–7154 (2009).
Su, Q., Boucher, P. A. & Rokita, S. E. Conversion of a dehalogenase into a nitroreductase by swapping its flavin cofactor with a 5-deazaflavin analogue. Angew. Chem. Int. Ed. 56, 10862–10866 (2017).
Taglieber, A., Schulz, F., Hollmann, F., Rusek, M. & Reetz, M. T. Light-driven biocatalytic oxidation and reduction reactions: scope and limitations. ChemBioChem 9, 565–572 (2008).
Duchstein, H.-J., Fenner, H., Hemmerich, P. & Knappe, W.-R. (Photo)chemistry of 5-deazaflavin. Eur. J. Biochem. 95, 167–181 (1979).
Schmermund, L. et al. Photo-biocatalysis: biotransformations in the presence of light. ACS Catal. 9, 4115–4144 (2019).
Iwata, T. et al. Hydrogen bonding environments in the photocycle process around the flavin chromophore of the AppA-BLUF domain. J. Am. Chem. Soc. 140, 11982–11991 (2018).
Silva, E. & Edwards, A. M. (eds) Flavins: Photochemistry and Photobiology (RSC Publishing, 2006).
Sancar, A. Mechanisms of DNA repair by photolyase and excision nuclease (Nobel Lecture). Angew. Chem. Int. Ed. 55, 8502–8527 (2016).
Sancar, A. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 103, 2203–2238 (2003).
Lee, J., Müller, F. & Visser, A. J. W. G. The sensitized bioluminescence mechanism of bacterial luciferase. Photochem. Photobiol. 95, 679–704 (2019).
Conrad, K. S., Manahan, C. C. & Crane, B. R. Photochemistry of flavoprotein light sensors. Nat. Chem. Biol. 10, 801 (2014).
Zollitsch, T. M. et al. Magnetically sensitive radical photochemistry of non-natural flavoproteins. J. Am. Chem. Soc. 140, 8705–8713 (2018).
Cashmore, A. R., Jarillo, J. A., Wu, Y.-J. & Liu, D. Cryptochromes: blue light receptors for plants and animals. Science 284, 760–765 (1999).
Kiontke, S., Gnau, P., Haselsberger, R., Batschauer, A. & Essen, L.-O. Structural and evolutionary aspects of antenna chromophore usage by class II photolyases. J. Biol. Chem. 289, 19659–19669 (2014).
Lee, S. H., Choi, D. S., Kuk, S. K. & Park, C. B. Photobiocatalysis: activating redox enzymes by direct or indirect transfer of photoinduced electrons. Angew. Chem. Int. Ed. 57, 7958–7985 (2018).
König, B., Kümmel, S., Svobodová, E. & Cibulka, R. Flavin photocatalysis. Phys. Sci. Rev. 3, 1–17 (2018).
Metternich, J. B., Mudd, R. J. & Gilmour, R. Science of Synthesis: Photocatalysis in Organic Synthesis, Vol. 1 (Thieme, 2018).
Zelenka, J., Cibulka, R. & Roithová, J. Flavinium catalysed photooxidation: detection and characterization of elusive peroxyflavinium intermediates. Angew. Chem. Int. Ed. 58, 15412–15420 (2019).
Ramirez, N. P., König, B. & Gonzalez-Gomez, J. C. Decarboxylative cyanation of aliphatic carboxylic acids via visible-light flavin photocatalysis. Org. Lett. 21, 1368–1373 (2019).
März, M. et al. Azodicarboxylate-free esterification with triphenylphosphine mediated by flavin and visible light: method development and stereoselectivity control. Org. Biomol. Chem. 16, 6809–6817 (2018).
Mühldorf, B. & Wolf, R. C-H photooxygenation of alkyl benzenes catalyzed by riboflavin tetraacetate and a non-heme iron catalyst. Angew. Chem. Int. Ed. 55, 427–430 (2016).
Hering, T., Mühldorf, B., Wolf, R. & König, B. Halogenase-inspired oxidative chlorination using flavin photocatalysis. Angew. Chem. Int. Ed. 55, 5342–5345 (2016).
Hartman, T. & Cibulka, R. Photocatalytic systems with flavinium salts: from photolyase models to synthetic tool for cyclobutane ring opening. Org. Lett. 18, 3710–3713 (2016).
Mühldorf, B. & Wolf, R. Photocatalytic benzylic C-H bond oxidation with a flavin scandium complex. Chem. Commun. 51, 8425–8428 (2015).
Mojr, V. et al. Tailoring flavins for visible light photocatalysis: organocatalytic [2+2] cycloadditions mediated by a flavin derivative and visible light. Chem. Commun. 51, 12036–12039 (2015).
Feldmeier, C., Bartling, H., Magerl, K. & Gschwind, R. M. LED-illuminated NMR studies of flavin-catalyzed photooxidations reveal solvent control of the electron-transfer mechanism. Angew. Chem. Int. Ed. 54, 1347–1351 (2015).
Metternich, J. B. & Gilmour, R. A bio-inspired, catalytic E → Z isomerization of activated olefins. J. Am. Chem. Soc. 137, 11254–11257 (2015).
Zelenka, J. et al. Combining flavin photocatalysis and organocatalysis: metal-free aerobic oxidation of unactivated benzylic substrates. Org. Lett. 21, 114–119 (2019).
Zhang, W., Carpenter, K. L. & Lin, S. Electrochemistry broadens the scope of flavin photocatalysis: photoelectrocatalytic oxidation of unactivated alcohols. Angew. Chem. Int. Ed. 59, 409–417 (2020).
Zhou, F., Li, R., Wang, X., Du, S. & An, Z. Non-natural photoenzymatic controlled radical polymerization inspired by DNA photolyase. Angew. Chem. Int. Ed. 58, 9479–9484 (2019).
Link, P. A. J., van der Plas, H. C. & Müller, F. Photoreduction of 5-deazaflavins [1,3,9-triaza-anthracene-2(3H),4(10H)-diones]. J. Chem. Soc. Chem. Commun. 17, 1385–1387 (1986).
van Schie, M. M. C. H. et al. Deazaflavins as photocatalysts for the direct reductive regeneration of flavoenzymes. Mol. Catal. 452, 277–283 (2018).
Goldberg, M., Pecht, I., Kramer, H. E. A., Traber, R. & Hemmerich, P. Structure and properties of 5-deazaflavin radicals as compared to natural flavosemiquinones. Biochim. Biophys. Acta 673, 570–593 (1981).
Shi, F. et al. A facile and efficient synthesis of novel pyrimido[5,4-b][4,7]phenanthroline-9,11(7H,8H,10H,12H)-dione derivativesviamicrowave-assisted multicomponent reactions. J. Heterocycl. Chem. 46, 563–566 (2009).
Khalafi-Nezhad, A., Sarikhani, S., Shahidzadeh, E. S. & Panahi, F. l-Proline-promoted three-component reaction of anilines, aldehydes and barbituric acids/malononitrile: regioselective synthesis of 5-arylpyrimido[4,5-b]quinoline-diones and 2-amino-4-arylquinoline-3-carbonitriles in water. Green Chem. 14, 2876–2884 (2012).
Edmondson, D. E., Barman, B. & Tollin, G. Importance of the N-5 position in flavine coenzymes. Properties of free and protein-bound 5-deaza analogs. Biochemistry 11, 1133–1138 (1972).
Neumeier, M. et al. Dichromatic photocatalytic substitutions of aryl halides with a small organic dye. Chem. Eur. J. 24, 105–108 (2018).
Bardagi, J. I., Ghosh, I., Schmalzbauer, M., Ghosh, T. & König, B. Anthraquinones as photoredox catalysts for the reductive activation of aryl halides. Eur. J. Org. Chem. 1, 34–40 (2018).
Ghosh, I. & König, B. Chromoselctive photocatalysis: controlled bond activation through light-color regulation of redox potentials. Angew. Chem. Int. Ed. 55, 7676–7679 (2016).
Kim, H., Kim, H., Lambert, T. H. & Lin, S. Reductive electrophotocatalysis: merging electricity and light to achieve extreme reduction potentials. J. Am. Chem. Soc. 142, 2087–2092 (2020).
Cowper, N. G. W., Chernowsky, C. P., Williams, O. P. & Wickens, Z. K. Potent reductants via electron-primed photoredox catalysis: unlocking aryl chlorides for radical coupling. J. Am. Chem. Soc. 142, 2093–2099 (2020).
Ghosh, I., Ghosh, T., Bardagi, J. I. & König, B. Reduction of aryl halides by consecutive visible light-induced electron transfer processes. Science 346, 725–728 (2014).
Meyer, A. U., Slanina, T., Heckel, A. & König, B. Lanthanide ions coupled with photoinduced electron transfer generate strong reduction potentials from visible light. Chem. Eur. J. 23, 7900–7904 (2017).
Kerzig, C., Guo, X. & Wenger, O. S. Unexpected hydrated electron source for preparative visible-light driven photoredox catalysis. J. Am. Chem. Soc. 141, 2122–2127 (2019).
MacKenzie, I. A. et al. Discovery and characterization of an acridine radical photoreductant. Nature 580, 76–80 (2020).
Cibulka, R. Strong chemical reducing agents produced by light. Nature 580, 31–32 (2020).
Kutta, R. J. Blitzlichtphotolyse - Untersuchung zu LOV-Domänen und Photochromen Systemen. Dissertation, Naturwissenschaftliche Fakultät IV -Chemie und Pharmazie- der Universität Regensburg (2012).
Kutta, R. J., Langenbacher, T., Kensy, U. & Dick, B. Setup and performance of a streak camera apparatus for transient absorption measurements in the ns to ms range. Appl. Phys. B 111, 203–216 (2013).
Baudisch, B. Time Resolved Broadband Spectroscopy from UV to NIR. Dissertation, Ludwig-Maximilians-Universität München (2018).
Lanzl, K., Sanden-Flohe, M. V., Kutta, R. J. & Dick, B. Photoreaction of mutated LOV photoreceptor domains from Chlamydomonas reinhardtii with aliphatic mercaptans: implications for the mechanism of wild type LOV. Phys. Chem. Chem. Phys. 12, 6594–6604 (2010).
Granovsky, A. A. Firefly version 8. http://classic.chem.msu.su/gran/firefly/index.html (1997).
Schmidt, M. W. et al. General atomic and molecular electronic structure system. J. Comput. Chem. 14, 1347–1363 (1993).
Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2, 73–78 (2012).
Neese, F. Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 8, e1327 (2018).
Granovsky, A. A. Extended multi-configuration quasi-degenerate perturbation theory: the new approach to multi-state multi-reference perturbation theory. J. Chem. Phys. 134, 214113 (2011).
Acknowledgements
This project was supported by the Czech Science Foundation (project No 19-09064S), the Deutsche Forschungsgemeinschaft (KO 1537/18-1) and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement 741623). R.J.K. thanks the Deutsche Forschungsgemeinschaft for a Research Fellow stipend (KU 3190/1-1, KU 3190/2-1) supporting this project. The authors are grateful to Professor Eberhard Riedle (BioMolekulare Optik, Ludwig-Maximillians-Universität München) for use of the ns to μs transient absorption apparatus.
Author information
Authors and Affiliations
Contributions
R.J.K., R.C., and A.G. wrote the paper with input from all of the authors. A.G. and T.N. performed the entire synthesis of all used compounds and conducted all photocatalytic dehalogenation reactions. A.G. did the electrochemical and EPR characterisation. R.J.K. together with A.G. and T.N. designed the spectro-electrochemistry experiment, which A.G. performed. R.J.K. together with A.G. and T.N. collected the stationary spectroscopic data. R.J.K. performed all time-resolved spectroscopic experiments and conducted the computational studies. Experimental data were analysed by R.J.K. together with A.G., computational data were analysed by R.J.K., and results were discussed by all authors. The project was conceived by R.C., R.J.K., and B.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Graml, A., Neveselý, T., Jan Kutta, R. et al. Deazaflavin reductive photocatalysis involves excited semiquinone radicals. Nat Commun 11, 3174 (2020). https://doi.org/10.1038/s41467-020-16909-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-16909-y
This article is cited by
-
5-Deazaalloxazine as photosensitizer of singlet oxygen and potential redox-sensitive agent
Photochemical & Photobiological Sciences (2023)
-
Tuning riboflavin derivatives for photodynamic inactivation of pathogens
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
