
Abstract
Asymmetric hydrogenation (AH) and direct reductive amination (DRA) are both efficient transformations frequently utilized in industry. Here we combine the asymmetric hydrogenation of prochiral olefins and direct reductive amination of aldehydes in one step using hydrogen gas as the common reductant and a rhodium-Segphos complex as the catalyst. With this strategy, the efficiency for the synthesis of the corresponding chiral amino compounds is significantly improved. The practical application of this synthetic approach is demonstrated by the facile synthesis of chiral 3-phenyltetrahydroquinoline and 3-benzylindoline compounds.
Similar content being viewed by others
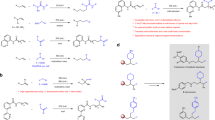
Introduction
Since the manufacture of l-dopa was realized as the first successful industrial-scale asymmetric catalytic process1, asymmetric hydrogenation (AH) has become the main driving force for asymmetric catalysis2,3,4,5,6,7, and the most frequently utilized homogeneous enantioselective catalytic tranformation in large scale8,9,10,11,12,13,14,15,16,17. The striding progress in AH research was evinced by Knowles18 and Noyori19 winning the Nobel prize. AH bears a few outstanding merits: first of all hydrogen gas as the reductant offers 100% atom economy; at the same time, there is a vast library of readily available chiral ligands; more importantly, the catalytic activity is prominent. The catalyst loading can be as low as 0.00002 mol%20. As a result, many important fine chemicals and active pharmaceutical ingredients (APIs) are manufactured via AH of C=C, C=O and C=N bonds, including l-menthol (the most manufactured chiral compound), metolachlor (the best-selling chiral herbicide), dextromethorphan, fluoxetine and sitagliptin2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17.
At the other hand, direct reductive amination (DRA), a one-pot procedure for the construction of C–N bond in which the mixture of carbonyl compound and nitrogen-containing compound is subjected to a reducing agent with H2O as the sole by-product, is a key transformation in organic chemistry21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41. It has been developed into one of the most practical methods for the synthesis of amino pharmaceutical compounds. Common reducing agents include H2, borohydride, formate, silane, isopropyl alcohol and Hantzsch esters. Transition metal-catalyzed DRA using molecular hydrogen as the reducing reagent is highly sustainable in terms of reaction waste control and atom-economy. As both AH and DRA share the same reductant, H2, we propose to combine these two reactions to make the procedure for the synthesis of chiral amino compounds more concise and proficient (Fig. 1). To achieve this purpose, two major challenges have to be addressed: the discovery of a catalytic system which can perform reduction of two different types of bonds (C=N and C=C bonds) and the suppression of side-reactions, namely aldehyde and/or olefin reduction, which might take place before the proposed AH–DRA reaction sequence. We envisioned two strategies that may help to tackle those problems. One is the application of a transition metal which could efficiently coordinate and reduce both the imine bond and the olefin bond. The other is the addition of appropriate additives which facilitate the imine formation and the reduction thereafter, and alleviate the inhibition effect from the amine reactant, imine intermediate and the amine product on the catalyst. Fan and co-workers just disclosed an elegant Ir/Ru-catalyzed AH and DRA of quinoline-2-carbaldehydes with anilines42.

a Asymmetric hydrogenation of olefins. b Direct reductive amination of aldehydes. c This work: the combination of asymmetric hydrogenation and direct reductive amination.
Herein, we describe our efforts toward the integration of AH of olefins and DRA of aldehydes into a one-step cascade reaction. Through the incorporated AH and DRA sequential reactions in one step, the synthetic efficiency of related chiral amino compounds is significantly improved.
Results
Establishment of feasibility
In our previous direct asymmetric reductive amination research, iridium catalyst demonstrated prominent reactivity toward imine reduction43,44,45. So [Ir(cod)Cl]2 precursor along with (R)-Segphos was initially evaluated in the reaction of α,β-unsaturated aldehyde 1a and aniline 2a. Unfortunately only side-product 5 was obtained. Since Brønsted acids have been extensively used in reductive amination to promote the imine formation and imine reduction46,47,48,49,50, 30 mol% p-toluenesulfonic acid (TsOH) was added to the reaction. As expected, TsOH boosted the imine formation and imine reduction to afford 21% product 3a at 64% ee, with 75% intermediate 4 been formed (Table 1, entry 2). When the metal precursor was switched to [Rh(cod)Cl]2, TsOH again exercised positive effects on the reaction (entry 4). At the same time, it suppressed the aldehyde direct reduction. From the results, we can see rhodium was more capable of olefin reduction than iridium. In the brief solvents screening (Table 1, entries 4–8), ethyl acetate, which possesses moderate coordinating ability, provided the highest ee. Next, a few Brønsted acids were examined (Table 1, entries 8–11), among which 4-chlorobenzenesulfonic acid facilitated the catalytic system for better reactivity as well as higher stereoselectivity than other acids. Then the influence of the counterion of the cationic Rh complex was studied51,52. The results demonstrated that the coordinating anions, such as iodide (Table 1, entry 16) and acetate (Table 1, entry 12), deactivated the reaction, while the noncoordinating anion hexafluoroantimonate improved both the reactivity and the selectivity (Table 1, entry 14). Since the coordinating species had influence on the reaction selectivity, we tried some solvents with coordination ability. It turned out the addition of N,N-dimethyllformamide (DMF) to MeOAc (at the ratio of 1:4) enhanced the reaction ee to 98% (Table 1, entry 17). A control experiment, in which aniline 2a was not added, afforded 5 in 39% yield with 61% substrate 1a remaining under the same reaction conditions as in entry 17 (Fig. 2a). The above results demonstrated that the construction of the C–N bond beforehand helped to pave the way for the C=C bond reduction (Fig. 2b).

a The reduction of 1 without the addition of aniline 2a. b The reduction of 1 with the addition of aniline 2a.
Examination of substrate scope
With the optimal conditions using the rhodium/Segphos catalyst and the additive set in hand, we first explored the scope of the α,β-unsaturated aldehydes in the AH and DRA reactions with aniline 2a (Table 2). In most cases, the aldehyde substrates could be transformed into the corresponding chiral amino compounds in excellent enantioselectivity and high yields. Regarding the substituted aromatic groups connected to the chiral center (R2) of the products, when the substituents were on para- or meta-positions, the corresponding products were obtained with more than 95% ee and 90% yields, regardless of their electron-donating (3b–3e, 3h–3j) or electron-withdrawing (3f–3g, 3k) properties; but when the substituents were on ortho-positions (3l–3n) or as 1-naphthyl group (3p), the reaction required higher catalyst loading and/or reaction temperature, probably due to the greater steric hindrance. The additive set and catalytic system also worked well for heteroaromatic substrate 1q and aliphatic group substituted 1r. It is worth mentioning that protic groups –OH (3e) and –NHAc (3i), and reducible group –CN (3k) were well-tolerated in the reactions. As for the various –R1 substrates, the results were similar to that of the –R2 substituted ones. As for the limitations of the reaction, the catalytic system did not work well when R1 and R2 were both alkyl groups (3zb), or the α-substituent was switched to β-position (3zc). Alkyl amine was also not suitable for this method (3zd). The reactions of 2-phenylbut-2-enal (1ze) with 2a did not lead to the desired products. As for 3,4-diphenylbut-3-en-2-one (1zf), most of the starting material 1zf remained untouched.
Then the scope of anilines was explored using the same catalytic system under the optimized conditions. The results are summarized in Table 3. All selected anilines, with substituents at para- (3ab–3ad), meta- (3ae–3ag) and ortho- (3ah–3aj) positions, or having electronic-withdrawing or electronic-donating groups, reductively coupled with the 2,3-diphenylacrylaldehyde substrate 1a smoothly to afford the desired products in excellent ees and yields. Notably the protic –OH group (2i) again tolerated in this transformation. In addition, sterically hindered anilines, with ortho-substituents (2 h, 2j) and 1-naphthyl group (2k), were all suitable nitrogen sources for the successful convertion of 1a.
Practical applications
To further showcase the utility of this AH and DRA conbination strategy, we next made efforts on the transformations of these N-(2,3-diarylpropyl)aniline products 3. Tetrahydroquinolines are prevailing natural alkaloids and artificially synthesized molecules, which have found frequent applications in pharmaceutical and agrochemical industry53,54,55,56. The AH of readily available quinolines is the most convenient and straightforward access to these compounds6,57. But the 3-substituted tetrahydroquinolines could not be prepared through this route, since under the typical acidic AH reaction conditions, the 1,4-dihydroquinoline intermediates would undergo isomerization to form 1,2-dihydroquinoline, in which it is hard to control the stereoselectivity6,58. Using our methodology, product 3y from the 3-(2-chlorophenyl)-2-phenylacrylaldehyde 1y substrate could easily been converted into the chiral 3-phenyl-tetrahydroquinoline product 6 via the Buchwald–Hartwig cross-coupling reaction (Fig. 3a), in which the ee value was not affected59,60,61. Similarly, product 3n from the 2-(2-chlorophenyl)-3-phenylacrylaldehyde 1n substrate could be cyclized to form the 3-benzylindoline alkaloid 7 (Fig. 3b).

a The transformation of 3y for the synthesis of 3-phenyl-tetrahydroquinoline 6. b The transformation of 3n for the synthesis of 2-benzyl-indoline 7.
Discussion
In summary, we have successfully integrated in one-pot two efficient reactions, AH and DRA, which share the common reductant, namely hydrogen gas. Catalyzed by the rhodium-Segphos complex, the DRA of aldehydes and the AH of prochiral olefins took place sequencially to afford the chiral amino compounds. The rhodium catalyst precursor was capable of performing the reduction of two different types of bonds. The addition of 30 mol% of 4-chlorobenzenesulfonic acid facilitated the imine formation, imine reduction and the C=C bond reduction. Noncoordinated counterion of the cationic rhodium complex hexafluoroantimonate improved both the reaction reactivity and the stereoselectivity. With our protocol, useful chiral amino compounds can be synthesized in a more convenient and effective manner.
Methods
General procedure for asymmetric reductive amination
In a nitrogen-filled glovebox, [Rh(cod)Cl]2 (5 μmol) and (R)-Segphos (11 μmol) was dissolved in anhydrous CH3COOCH3 (1.0 mL), stirred for 20 min, and equally divided into 10 vials charged with aldehyde (0.1 mmol) and aniline (0.1 mmol) in anhydrous CH3COOCH3 solution (0.5 mL). Then 4-Cl–PhSO3H (0.3 equiv.) and NaSbF6 (0.05 equiv) were added and the total solution was made to 2.0 mL (MeOAc:DMF = 4:1) for each vial. The resulting vials were transferred to an autoclave, which was charged with 50 atm of H2, and stirred at 60 °C for 24 h. The hydrogen gas was released slowly and the solution was quenched with aqueous sodium bicarbonate solution. The organic phase was concentrated and passed through a short column of silica gel to remove the metal complex to give the crude products, which were purified by column chromotography and then analyzed by chiral HPLC to determine the enantiomeric excesses.
Data availability
The experimental procedure and characterization data of new compounds are available within the Supplementary Information. Any further relevant data are available from the authors upon reasonable request.
References
Vineyard, B. D., Knowles, W. S., Sabacky, M. J., Bachman, G. L. & Weinkauff, D. J. Asymmetric hydrogenation. Rhodium chiral bisphosphine catalyst. J. Am. Chem. Soc. 99, 5946–5952 (1977).
Ohkuma, T., Kitamura, M. & Noyori, R. In Catalytic Asymmetric Synthesis 2nd edn (ed Ojima, I.) (Wiley, New York, 2000).
Tang, W. & Zhang, X. New chiral phosphorus ligands for enantioselective hydrogenation. Chem. Rev. 103, 3029–3269 (2003).
Xie, J.-H., Zhu, S.-F. & Zhou, Q.-L. Transition metal-catalyzed enantioselective hydrogenation of enamines and imines. Chem. Rev. 111, 1713–1760 (2011).
Wang, D.-S., Chen, Q.-A., Lu, S.-M. & Zhou, Y.-G. Asymmetric hydrogenation of heteroarenes and arenes. Chem. Rev. 112, 2557–2590 (2012).
Verendel, J. J., Pàmies, O., Diéguez, M. & Andersson, P. G. Asymmetric hydrogenation of olefins using chiral crabtree-type catalysts: scope and limitations. Chem. Rev. 114, 2130–2169 (2014).
Zhang, Z., Butt, N. A. & Zhang, W. Asymmetric hydrogenation of nonaromatic cyclic substrates. Chem. Rev. 116, 14769–14827 (2016).
Genet, J.-P. Asymmetric catalytic hydrogenation. Design of new Ru catalysts and chiral ligands: from laboratory to industrial applications. Acc. Chem. Res. 36, 908–918 (2003).
Shultz, C. S. & Krska, S. W. Unlocking the potential asymmetric hydrogenation at Merck. Acc. Chem. Res. 40, 1320–1326 (2007).
Blaser, H. & Federsel, H. (eds.) Asymmetric Catalysis On Industrial Scale: Challenges, Approaches And Solutions 2nd edn (Wiley-VCH, 2010).
Palmer, A. M. & Zanotti-Gerosa, A. Homogenous asymmetric hydrogenation: recent trends and industrial applications. Curr. Opin. Drug Discov. Devel. 13, 698–716 (2010).
Busacca, C. A., Fandrick, D. R., Song, J. J. & Senanayake, C. H. The growing impact of catalysis in the pharmaceutical industry. Adv. Synth. Catal. 353, 1825–1864 (2011).
Ager, D. J., de Vries, A. H. M. & de Vries, J. G. Asymmetric homogeneous hydrogenations at scale. Chem. Soc. Rev. 41, 3340–3380 (2012).
Magano, J. & Dunetz, J. R. Large-scale carbonyl reductions in the pharmaceutical industry. Org. Process Res. Dev. 16, 1156–1184 (2012).
Ager, D. J. Popular synthetic approaches to pharmaceuticals. Synthesis 47, 760–768 (2015).
Hayler, J. D., Leahy, D. K. & Simmons, E. M. A pharmaceutical industry perspective on sustainable metal catalysis. Organometallics 38, 36–46 (2019).
Seo, C. S. G. & Morris, R. H. Catalytic homogeneous asymmetric hydrogenation: successes and opportunities. Organometallics 38, 47–65 (2019).
Knowles, W. S. Asymmetric hydrogenations (Nobel lecture 2001). Angew. Chem. Int. Ed. 41, 1998–2007 (2002).
Noyori, R. Asymmetric catalysis: science and opportunities (Nobel lecture 2001). Adv. Synth. Catal. 345, 15–32 (2003).
Xie, J.-H., Liu, X.-Y., Xie, J.-B., Wang, L.-X. & Zhou, Q.-L. An additional coordination group leads to extremely efficient chiral iridium catalysts for asymmetric hydrogenation of ketones. Angew. Chem. Int. Ed. 50, 7329–7332 (2011).
Nugent, T. C. & El-Shazly, M. Chiral amine synthesis–recent developments and trends for enamide reduction, reductive amination, and imine reduction. Adv. Synth. Catal. 352, 753–819 (2010).
Gomez, S., Peters, J. A. & Maschmeyer, T. The reductive amination of aldehydes and ketones and the hydrogenation of nitriles: mechanistic aspects and selectivity control. Adv. Synth. Catal. 344, 1037–1057 (2002).
Tripathi, R. P., Verma, S. S., Pandey, J. & Tiwari, V. K. Recent development on catalytic reductive amination and applications. Curr. Org. Chem. 12, 1093–1115 (2008).
Abdel-Magid, A. F. & Mehrman, S. J. A review on the use of sodium triacetoxyborohydride in the reductive amination of ketones and aldehydes. Org. Process Res. Dev. 10, 971–1031 (2006).
Storer, R. I., Carrera, D. E., Ni, Y. & MacMillan, D. W. C. Enantioselective organocatalytic reductive amination. J. Am. Chem. Soc. 128, 84–86 (2006).
Steinhuebel, D., Sun, Y., Matsumura, K., Sayo, N. & Saito, T. Direct asymmetric reductive amination. J. Am. Chem. Soc. 131, 11316–11317 (2009).
Wakchaure, V. N., Zhou, J., Hoffmann, S. & List, B. Catalytic asymmetric reductive amination of α-branched ketones. Angew. Chem. Int. Ed. 49, 4612–4614 (2010).
Savile, C. K. et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 329, 305–309 (2010).
Wang, C., Pettman, A., Bacsa, J. & Xiao, J. A versatile catalyst for reductive amination by transfer hydrogenation. Angew. Chem. Int. Ed. 49, 7548–7552 (2010).
Xiao, X., Xie, Y., Su, C., Liu, M. & Shi, Y. Organocatalytic asymmetric biomimetic transamination: from α-keto esters to optically active α-amino acid derivatives. J. Am. Chem. Soc. 133, 12914–12917 (2011).
Strotman, N. A. et al. Reaction development and mechanistic study of a ruthenium catalyzed intramolecular asymmetric reductive amination en route to the dual orexin inhibitor Suvorexant (MK-4305). J. Am. Chem. Soc. 133, 8362–8371 (2011).
Werkmeister, S., Junge, K. & Beller, M. Copper-catalyzed reductive amination of aromatic and aliphatic ketones with anilines using environmental-friendly molecular hydrogen. Green Chem. 14, 2371–2374 (2012).
Simon, R. C., Richter, N., Busto, E. & Kroutil, W. Recent developments of cascade reactions involving ω-transaminases. ACS Catal. 4, 129–143 (2014).
Pavlidis, I. V. et al. Identification of (S)-selective transaminases for the asymmetric synthesis of bulky chiral amines. Nat. Chem. 8, 1076–1082 (2016).
Jagadeesh, R. V. et al. MOF-derived cobalt nanoparticles catalyze a general synthesis of amines. Science 358, 326–332 (2017).
Zhou, H., Liu, Y., Yang, S., Zhou, L. & Chang, M. One-pot N-deprotection and catalytic intramolecular asymmetric reductive amination for the synthesis of tetrahydroisoquinolines. Angew. Chem. Int. Ed. 56, 2725–2729 (2017).
Aleku, G. A. et al. A reductive aminase from Aspergillus oryzae. Nat. Chem. 9, 961–969 (2017).
Gallardo-Donaire, J. et al. Direct asymmetric ruthenium-catalyzed reductive amination of alkyl−Aryl ketones with ammonia and hydrogen. J. Am. Chem. Soc. 140, 355–361 (2018).
Tan, X. et al. Asymmetric synthesis of chiral primary amines by ruthenium-catalyzed direct reductive amination of alkyl aryl ketones with ammonium salts and molecular H2. J. Am. Chem. Soc. 140, 2024–2027 (2018).
Lou, Y. et al. Dynamic kinetic asymmetric reductive amination: synthesis of chiral primary β-amino lactams. Angew. Chem. Int. Ed. 57, 14193–14197 (2018).
Mayol, O. et al. A family of native amine dehydrogenases for the asymmetric reductive amination of ketones. Nat. Catal. 2, 324–333 (2019).
Chen, Y., Pan, Y., He, Y.-M. & Fan, Q.-H. Consecutive intermolecular reductive amination/asymmetric hydrogenation: facile access to sterically tunable chiral vicinal diamines and N‐heterocyclic carbenes. Angew. Chem. Int. Ed. 58, 16831–16834 (2019).
Huang, H., Liu, X., Zhou, L., Chang, M. & Zhang, X. Direct asymmetric reductive amination for the synthesis of chiral β-arylamines. Angew. Chem. Int. Ed. Engl. 55, 5309–5312 (2016).
Huang, H., Zhao, Y., Yang, Y., Zhou, L. & Chang, M. Direct catalytic asymmetric reductive amination of aliphatic ketones utilizing diphenylmethanamine as coupling partner. Org. Lett. 19, 1942–1945 (2017).
Wu, Z. et al. Secondary amines as coupling partners in direct catalytic asymmetric reductive amination. Chem. Sci. 10, 4509–4514 (2019).
Vogl, E. M., Groeger, H. & Shibasaki, M. Towards perfect asymmetric catalysis: additives and cocatalysts. Angew. Chem. Int. Ed. 38, 1570–1577 (1999).
Yu, Z., Jin, W. & Jiang, Q. Brønsted acid activation strategy in transition-metal catalyzed asymmetric hydrogenation of N-unprotected imines, enamines, and N-heteroaromatic compounds. Angew. Chem. Int. Ed. 51, 6060–6072 (2012).
Hong, L., Sun, W., Yang, D., Li, G. & Wang, R. Additive effects on asymmetric catalysis. Chem. Rev. 116, 4006–4123 (2016).
Blaser, H.-U., Buser, H.-P., Jalett, H.-P., Pugin, B. & Spindler, F. Iridium ferrocenyl diphosphine catalyzed enantioselective reductive alkylation of a hindered aniline. Synlett. 1999, 867−868 (1999).
Li, C., Villa-Marcos, B. & Xiao, J. Metal-Brønsted acid cooperative catalysis for asymmetric reductive amination. J. Am. Chem. Soc. 131, 6967–6969 (2009).
Smidt, S. P., Zimmermann, N., Studer, M. & Pfaltz, A. Enantioselective hydrogenation of alkenes with iridium–PHOX catalysts: a kinetic study of anion effects. Chem. Eur. J. 10, 4685–4693 (2004).
Moreno, A. et al. PGSE NMR diffusion overhauser studies on [Ru(Cp*)(η6‐arene)][PF6], plus a variety of transition-metal, inorganic, and organic salts: an overview of ion pairing in dichloromethane. Chem. Eur. J. 14, 5617–5629 (2008).
Sridharan, V., Suryavanshi, P. & Men´endez, J. C. Advances in the chemistry of tetrahydroquinolines. Chem. Rev. 111, 7157–7259 (2011).
Scott, J. D. & Williams, R. M. Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics. Chem. Rev. 102, 1669–1730 (2002).
Bentley, K. W. β-Phenylethylamines and the isoquinoline alkaloids. Nat. Prod. Rep. 23, 444–463 (2006).
Barton, D. H. R., Nakanishi, K. & Meth-Cohn, O. (eds.) Comprehensive Natural Products Chemistry (Elsevier, Oxford, 1999).
Zhou, Y. Asymmetric hydrogenation of heteroaromatic compounds. Acc. Chem. Res. 40, 1357–1366 (2007).
Wang, D.-W. et al. Highly enantioselective iridium-catalyzed hydrogenation of 2-benzylquinolines and 2-functionalized and 2,3-disubstituted quinolones. J. Org. Chem. 74, 2780–2787 (2009).
Paul, F., Patt, J. & Hartwig, J. F. Palladium-catalyzed formation of carbon-nitrogen bonds. Reaction intermediates and catalyst improvements in the hetero cross-coupling of aryl halides and tin amides. J. Am. Chem. Soc. 116, 5969–5970 (1994).
Guram, A. S. & Buchwald, S. L. Palladium-catalyzed aromatic aminations with in situ generated aminostannanes. J. Am. Chem. Soc. 116, 7901–7902 (1994).
Ruiz-Castillo, P. & Buchwald, S. L. Applications of palladium-catalyzed C-N cross-coupling reactions. Chem. Rev. 116, 12564–12649 (2016).
Acknowledgements
Financial support from the National Natural Science Foundation of China (21772155) is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
S.Y. established the reaction conditions. G.G. and C.L. prepared the substrates. S.Y., L.Wang., L.Wan., H.H. and H.G. expanded the substrate scope. M.C. conceived and supervised the project and wrote the manuscript. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks John Brown and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yuan, S., Gao, G., Wang, L. et al. The combination of asymmetric hydrogenation of olefins and direct reductive amination. Nat Commun 11, 621 (2020). https://doi.org/10.1038/s41467-020-14475-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-14475-x
This article is cited by
-
Practical N-alkylation via homogeneous iridium-catalyzed direct reductive amination
Science China Chemistry (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
