
Abstract
Transition metal-catalyzed carbonylation with carbon nucleophiles is one of the most prominent methods to construct ketones, which are highly versatile motifs prevalent in a variety of organic compounds. In comparison to the well-established palladium catalytic system, the nickel-catalyzed carbonylative coupling is much underdeveloped due to the strong binding affinity of CO to nickel. By leveraging easily accessible tert-butyl isocyanide as the CO surrogate, we present a nickel-catalyzed allylic carbonylative coupling with alkyl zinc reagent, allowing for the practical and straightforward preparation of synthetically important β,γ-unsaturated ketones in a linear-selective fashion with excellent trans-selectivity under mild conditions. Moreover, the undesired polycarbonylation process which is often encountered in palladium chemistry could be completely suppressed. This nickel-based method features excellent functional group tolerance, even including the active aryl iodide functionality to allow the orthogonal derivatization of β,γ-unsaturated ketones. Preliminary mechanistic studies suggest that the reaction proceeds via a π-allylnickel intermediate.
Similar content being viewed by others
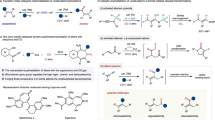
Introduction
Pioneering work developed by Heck in the 1970s catapulted palladium (Pd)-catalyzed three component reactions with carbon monoxide (CO) as a powerful strategy for introduction of carbonyl group1,2,3,4,5,6,7,8. Among these processes, the use of carbon-based nucleophiles allows for the convenient synthesis of ketones with broad applications9. Pd-catalyzed allylic reaction represents one of the most prominent carbon–carbon bond-forming reactions with wide synthetic applications in organic chemistry, including synthesis of various biologically active natural products, pharmaceuticals, and agrochemicals10,11,12,13. Thus, the allylic carbonylation would be an important strategy for incorporation of both alkene and carbonyl functionality in one synthetic protocol, enabling the expedient synthesis of the versatile β,γ-unsaturated ketones, which are ubiquitous motifs in bioactive compounds and utilized as valuable synthetic building blocks14,15,16,17,18,19,20. The Stille group has realized the Pd-catalyzed allylic carbonylative Stille coupling, while the organotin reagents largely limited on aryl, vinyl, and allyl stannanes21,22. The Tamaru group has previously developed a Pd-catalyzed allylic carbonylative Negishi coupling with CO to access β,γ-unsaturated ketones23,24. However, the site selectivity of this process is highly dependent on the electronics of the organozinc coupling partner, and a mixture of the linear and branched coupling products were usually obtained when simple alkyl Negishi reagents were used (Fig. 1a). Despite the progress in Pd-catalyzed three component reactions with CO gas, it is still highly imperative to develop practical carbonylation utilizing earth abundant transition metal25,26 for functionalized ketone synthesis, especially to circumvent the long-term limitations.

a Pd-catalyzed allylic carbonylative Negishi reaction. b Pd-catalyzed aryl carbonylative Negishi reaction. c Ni-catalyzed allylic carbonylative Negishi reaction.
Isocyanide is an important array of organic reagent widely used in transition metal catalyzed carbonylations as C-1 source and in heterocycle synthesis27,28,29,30,31,32,33,34,35,36,37,38. Despite the progress, the leverage of functionalized alkyl Negishi reagents for transition metal catalyzed carbonylative coupling with isocyanide still remains extremely limited33. Recently, Dechert-Schmitt and Blackmond reported a modular unsymmetrical 1,2-diketone synthesis via Pd-catalyzed four-component coupling between an aryl halide, an alkyl zinc reagent and two molecules of tBuNC. In this process, resting bisiminoyl Pd intermediate was formed between the Pd-iminoacyl species and 1-iminoalkyl zinc reagents (Fig. 1b)33.
At the outset of our investigation, we recognized several issues that needed to be addressed in order to develop an effective nickel-catalyzed allylic carbonylation. First, the use of nickel catalysts in carbonylation reactions has been less explored likely due to the strong binding affinity of CO towards nickel39,40,41,42,43,44,45,46,47,48,49,50,51,52,53. As a rare example, the Skrydstrup group elegantly succeeded the catalytic carbonylation of primary benzylic electrophile using a nickel pincer complex with a CO-gen precursor via the slow addition of Negishi reagent to circumvent the direct Negishi coupling, although the use of allyl electrophiles received limited success with 8% ketone formation51. Additionally, the preference of imidoylnickel intermediates to undergo further migratory insertion with isocyanides to furnish poly(iminomethylene)s is a well-known process in polymer chemistry54,55,56,57, presenting a major hurdle to access monocarbonylated products. Furthermore, Zhu and co-workers58 illustrate that the allyl imidoylpalladium intermediate is extremely prone to undergo β-H elimination to provide the ketenimine intermediate, which could further hydrolyze to β,γ-unsaturated amide59. To overcome these challenges, we envisioned that the use of functional group compatible organozinc reagents that are highly effective for transmetallation might disfavor the overcarbonylation process. Rapid C−C bond formation via reductive elimination would provide the monocarbonylated coupling product60,61. Moreover, the β,γ-unsaturated ketone easily undergoes the undesired isomerization step to afford the thermodynamically stable α,β-unsaturated ketone in the presence of transition metal catalyst62. Herein, we report the highly regio- and chemoselective nickel-catalyzed allylic carbonylative Negishi reaction with tert-butyl isocyanide63, which allows the expedient synthesis of β,γ-unsaturated ketones with broad substrate scope under mild conditions (Fig. 1c).
Results
Optimization of reaction conditions
We started our investigation by studying the nickel-catalyzed reaction of phenyl allyl acetate 1a and nC8H17ZnBr (1.5 equiv). To our delight, when the commercially available tert-butyl isocyanide (1.5 equiv) was used as the CO equivalent, the reaction proceeded smoothly with 10 mol% bench-stable and easily accessible NiCl2·DME in dimethylacetamide (DMA) at 25 °C. The starting material was consumed in 30 min; affording the desired trans β,γ-unsaturated ketone 4a in 86% isolated yield after the simple acidic work up procedure, only small amount of direct coupling byproduct 6 (1%) could be observed at gas chromatography (GC) (Table 1, entry 1). This nickel-isocyanide system features excellent linear selectivity. Neither the branched nor the secondary allylic products as observed in previous work23 were detected. Ligand-free nickel species was found to be the most effective for this coupling process. The use of phosphine ligands including PPh3 and other bidentate phosphines such as dppf (1,1′-ferrocenediyl-bis(diphenylphosphine)) was detrimental to this reaction, β,γ-unsaturated amide byproduct 5 was detected by GC (Table 1, entries 2 and 3). The use of 1,10-phenathorine also lowered the yield (Table 1, entry 4), and the inclusion of N-heterocyclic carbene ligand was also not beneficial (Table 1, entry 5). Additionally, Ni(0) precatalysts such as Ni(cod)2 provided similar results (Table 1, entry 6). Interestingly, Pd(dba)2 only afforded 4% ketone product (4a), demonstrating the difference in reactivity between the two transition metals (Table 1, entry 7). Finally, control experiments showed that the nickel catalyst was essential for this three-component reaction (Table 1, entry 8). The use of CO gas was also ineffective for this coupling (Table 1, entry 9).
Substrate scope of alkyl zinc reagents
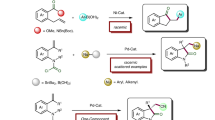
With the optimized conditions in hand, we next explored the substrate scope of alkylated zinc nucleophiles (Fig. 2). The reaction tolerates a wide range of organozinc reagents, affording β,γ-unsaturated ketones with complete trans-selectivity. Furthermore, the undesired isomerization of alkene moiety was not detected. Diethyl zinc could be successfully applied in this carbonylation process with 84% isolated yield (4b); however, requiring sacrifice one equivalent alkyl source. Simple alkyl groups including methyl (4c), benzyl (4d), and 2-phenylethyl (4e) could be incorporated into the products in excellent yield. The alkene moiety was compatible in this carbonylative process, and homoallyl (4f) and prenyl (4g) substituted β,γ-unsaturated ketones could be formed in good yield with excellent regio- and stereoselectivity. Additionally, various functionalized alkylzinc reagents, including those possessing a fluoride (4h), a chloride (4i), a nitrile (4j), an ester (4k), and an ether (4l, 4m), were successfully converted to the corresponding ketone product. Moreover, secondary cyclopentyl zinc bromide could also serve as the nucleophile to afford 4p in moderate yield. The utilization of nickel catalyst also tolerated the halogen-containing (hetero)benzyl nucleophiles (4n and 4q–4s), allowing further functionalization to be carried out. Unfortunately, the use of the tertiary alkylzinc and arylzinc reagent did not afford the desired carbonylative cross-coupling product under these conditions.

Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol), 3 (0.3 mmol), NiCl2·DME (0.02 mmol), DMA (2 mL), 25 °C, 0.5 h, and then 1 M HCl. aNi(cod)2 (0.02 mmol), 50 °C.
Substrate scope of allylic electrophiles
The scope of allylic electrophiles was also investigated, the reaction proceeded well with both aryl and alkyl substituted alkene, affording the carbonylation product in 43–96% yield with excellent regio- and stereoselectivities (Fig. 3). Aryl substituents including 2-Br (4v), 4-I (4w), and 4-Bpin (4x) were tolerated, providing the feasibility for subsequent derivatization. The employment of heteroaromatic ring including thiophene (4y), furan (4z), and indole (4aa) also provided the product in high isolated yield. When a methyl group was introduced at the C-2 position of the allyl electrophile (4ae), the reaction also proceeded with excellent E/Z selectivity. The migratory insertion of allylic nickel intermediate with isocyanide mainly proceeded in the least sterically hindered position, furnishing the trans ketone 4af–4ah in good yield.

Reaction conditions: 1 (0.2 mmol), 2 (0.3 mmol), 3 (0.3 mmol), NiCl2·DME (0.02 mmol), DMA (2 mL), 25 °C, 0.5 h, and then 1 M HCl. a50 °C; bNi(cod)2 (0.02 mmol); cthe ratio of regioisomer is 6:1; dthe ratio of regioisomer is 4:1.
To illustrate potential utility of this nickel-catalyzed three-component carbonylative coupling, we performed late-stage modifications on complex and/or biologically active compounds (Fig. 4). For instance, several pharmaceutical derivatives, naproxen (4aj), ibuprofen (4ak), hyodeoxycholic acid (4am), and indometacin (4an) could be readily applied in this reaction, affording the desired product in good yield. To our delight, the olefin containing citronellal (4ai) and oleic acid (4al) both could be effectively transformed to relatively β,γ-unsaturated ketones in regioselective way, while no alkene isomerization could be observed in the standard condition. Notably, vitamin-E (4ao) derivative is also amenable and the desired product could be obtained in moderate yield. These results clearly demonstrate that this carbonylation has promising applications in late-stage modification of complex molecules.

Reaction conditions: 1 (0.2 mmol), 2 (0.3 mmol), 3 (0.3 mmol), NiCl2·DME (0.02 mmol), DMA (2 mL), 25 °C, 0.5 h, and then 1 M HCl. aNi(cod)2 (0.02 mmol), 50 °C.
Synthetic applications
To further showcase the synthetic potential for this nickel catalyzed carbonylative Negishi reaction using tert-butyl isocyanide as carbonyl source, the gram-scale synthesis of 4t was carried out with 90% isolated yield (Fig. 5a). The imine intermediate could be reduced using NaBH4 to provide the useful building block homoallylic amine 7 (83% yield), further demonstrating the advantage of this isocyanide coupling technology (Fig. 5b). The tolerance of active aryl halide under current conditions allows the orthogonal cross-coupling strategy (Fig. 5c). Pd-catalyzed Suzuki coupling of (E)-3-(4-iodophenyl)allyl acetate 1w with phenylboronic acid afforded intermediate 1ap, which could further undergo the allylic carbonylation with benzyl zinc reagent to provide β,γ-unsaturated ketones 4ap in 58% overall yield. Meanwhile, β,γ-unsaturated ketones 4w could be obtained via the selective Ni-catalyzed carbonylative Negishi reaction, and then thiophene functionality could be incorporated through the Suzuki coupling to deliver the desired product 4aq in 51% yield.

a Gram-scale experiment. b Synthesis of homoallylic amine. c Orthogonal cross-coupling.
Mechanistic studies and proposed mechanism
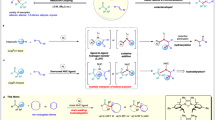
To shed light on the reaction mechanism, several experiments were performed (Fig. 6). When 1-phenylallyl acetate 1a′ was used as the allylic electrophile, the linear β,γ-unsaturated ketone 4t was obtained as the sole product in 88% yield. Additionally, when allyl acetate containing a cyclopropyl group (1ar) was subjected to the standard conditions, ring-opening product of the cyclopropyl group could not be detected (Fig. 6a). Together, these results indicate that the reaction might proceed via the π-allylnickel intermediate, and the radical intermediates is unlikely involved. The stereochemistry of this carbonylative coupling reaction was also examined by using (R)-1af as the starting material (Fig. 6b): the desired product 4af was obtained with 75% ee (enantiomeric excess) with the inversed configuration, which indicated that the initiate step most likely undergo SN2 type in the oxidative addition process similar to the Tsuji–Trost-type oxidation64,65,66. The erosion of ee may arise from the tautomerization between imine and enamine intermediate in the acidic work up procedure. The tert-butyl isocyanide possesses strong binding affinity to the nickel center, we prepared the tetrakis(tert-butyl isocyanide)nickel (II) perchlorate complex 8. Under standard condition, using 0.38 equiv 8 in the absence of additional tBuNC delivered the desired product in 57% yield. In contrast, using 10 mol% 8 with the addition of 1.5 equiv tBuNC increased the yield to 82%. These results suggest that the binding of multiple tBuNC as ligands to the nickel catalyst may proceed with ligand dissociation process in the catalytic cycles and slow down the coupling process (Fig. 6c). When we preform the [1-(tert-butylimino)-butyl]zinc chloride, which is prepared in situ via the reaction of tBuNC and nBuLi followed by transmetallation with ZnCl2 at room temperature, the desired product 4t was not observed under our conditions, in contrast to the previous findings of Ito and co-workers67 and Dechert-Schmitt et al.33, where high temperature is essential for carbonylative coupling (Fig. 6d). This result highlights the distinctive mechanism in the Ni-catalyzed carbonylative Negishi coupling reaction.

a The reaction proceeds with π-allyl nickel intermediate. b Stereochemistry of this carbonylative coupling reaction. c The use of Ni(tBuNC)4(ClO4)2 as carbonyl source. d The use of [1-(tert-butylimino) butyl] zinc reagent in the reaction.
Based on the preliminary mechanistic studies, a plausible reaction pathway is proposed in Fig. 7. Oxidative addition of allyl acetate 1 with the nickel catalyst affords the π-allylnickel(II) intermediate A. Migratory insertion of tert-butyl isocyanide then provides allyl imidoylnickel intermediate B, from which transmetallation and reductive elimination lead to the β,γ-unsaturated imine D. The desired β,γ-unsaturated ketone 4 could be obtained via acidic hydrolysis of D.

Possible reaction pathway of the carbonylative coupling reaction.
Discussion
In summary, nickel-catalyzed allylic carbonylative coupling with alkyl zinc reagent has been developed for the synthesis of β,γ-unsaturated ketones from allylic acetate and alkyl zinc reagent using commercially available tert-butyl isocyanide as a CO source. In this coupling process, the allyl imidoylnickel intermediate undergoes rapid transmetallation with the zinc nucleophile, thus avoiding the undesired polycarbonylation. This reaction features broad substrate scope with excellent regio- and chemoselectivity. Preliminary mechanistic studies reveal the reaction proceeds with the π-allyl nickel intermediate. Further effort on detailed mechanism and exploration of other electrophiles is currently underway in our laboratory will be reported in future.
Methods
General procedure A for the allylic carbonylative reaction
An oven-dried Schlenk tube charged with NiCl2·DME (10 mol%) was evacuated and backfilled with N2. (This process was repeated for three times.) DMA (0.1 M) was added into the reaction mixture. To this solution was subsequently added allylic acetate (1.0 equiv), tBuNC (1.5 equiv), and Negishi reagent (1.5 equiv). The tube was equipped with a balloon filled with N2 at 25 °C until complete consumption of the starting material. The mixture was added 1 M HCl aq. and stirred at room temperature for 0.25 h. The mixture was then extracted with EtOAc and separated organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure to yield the crude product, which was purified by silica gel flash column chromatography.
Data availability
The authors declare that all the data supporting the findings of this work are available within the article and its Supplementary Information files, or from the corresponding author upon request.
References
Schoenberg, A. & Heck, R. F. Palladium-catalyzed formylation of aryl, heterocyclic, and vinylic halides. J. Am. Chem. Soc. 96, 7761–7764 (1974).
Schoenberg, A. & Heck, R. F. Palladium-catalyzed amidation of aryl, heterocyclic, and vinylic halides. J. Org. Chem. 39, 3327–3331 (1974).
Schoenberg, A., Bartoletti, I. & Heck, R. F. Palladium-catalyzed carboalkoxylation of aryl, benzyl, and vinylic halides. J. Org. Chem. 39, 3318–3326 (1974).
Brennführer, A., Neumann, H. & Beller, M. Palladium-catalyzed carbonylation reactions of aryl halides and related compounds. Angew. Chem. Int. Ed. 48, 4114–4133 (2009).
Wu, L. et al. Palladium-catalyzed carbonylative transformation of C(sp3)–X bonds. ACS Catal. 4, 2977–2989 (2014).
Barnard, C. F. J. Palladium-catalyzed carbonylations-a reaction come of age. Organometallics 27, 5402–5422 (2008).
Grigg, R. & Mutton, S. P. Pd-catalysed carbonylations: versatile technology for discovery and process chemists. Tetrahedron 66, 5515–5548 (2010).
Tsuji, J. Palladium Reagents and Catalysts: Innovations in Organic Synthesis (Wiley, New York, 1995).
Wu, X.-F., Neumann, H. & Beller, M. Palladium-catalyzed carbonylative coupling reactions between Ar–X and carbon nuclephiles. Chem. Soc. Rev. 40, 4986–5009 (2011).
Tsuji, J., Takahashi, H. & Morikawa, M. Organic syntheses by means of noble metal compounds XVII. reaction of π-allylpalladium chloride with nucleophiles. Tetrahedron Lett. 6, 4387–4388 (1965).
Trost, B. M. & Fullerton, T. J. New synthetic reactions. allylic alkylation. J. Am. Chem. Soc. 95, 292–294 (1973).
Trost, B. M. & Van Vranken, D. L. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 96, 395–422 (1996).
Trost, B. M. & Crawley, M. L. Asymmetric transition-metal-catalyzed allylic alkylations: applications in total synthesis. Chem. Rev. 103, 2921–2944 (2003).
Houk, K. N. The photochemistry and spectroscopy of βγ-unsaturated carbonyl compounds. Chem. Rev. 76, 1–74 (1976).
Ranu, B. C., Majee, A. & Das, A. R. A convenient synthesis of βγ-unsaturated ketones through zinc-mediated allylation of acid chlorides. Tetrahedron Lett. 37, 1109–1112 (1996).
Hoffmann, H. R. M. & Tsushima, T. Acylation of olefins by acetyl hexachloroantimonate. Selective formation of βγ-unsaturated ketones under kinetic control and mechanistic rationale as an ene reaction. J. Am. Chem. Soc. 99, 6008–6011 (1977).
Obora, Y., Ogawa, Y., Imai, Y., Kawamura, T. & Tsuji, Y. Palladium complex catalyzed acylation of allylic esters with acylsilanes. J. Am. Chem. Soc. 123, 10489–10493 (2001).
Yasuda, S., Ishii, T., Takemoto, S., Haruki, H. & Ohmiya, H. Synergistic N-heterocyclic carbene/palladium-catalyzed reactions of aldehyde acyl anions with either diarylmethyl or allylic carbonates. Angew. Chem. Int. Ed. 57, 2938–2942 (2018).
Haruki, H., Yasuda, S., Nagao, K. & Ohmiya, H. Dehydrative allylation between aldehydes and allylic alcohols through synergistic N-heterocyclic carbene/Palladium catalysis. Chem. Eur. J. 25, 724–727 (2019).
Takemoto, S., Ishii, T., Yasuda, S. & Ohmiya, H. Synergistic N-heterocyclic carbene/palladium-catalyzed allylation of aldehydes with allylic carbonates. Bull. Chem. Soc. Jpn. 92, 937–940 (2019).
Sheffy, F. & Stille, J. Palladium-catalyzed cross-coupling of allyl halides with organotins. J. Am. Chem. Soc. 105, 7173–7175 (1983).
Sheffy, F., Godschalx, J. & Stille, J. Palladium-catalyzed cross coupling of allyl halides with organotin reagents: a method of joining highly functionalized partners regioselectively and stereospecifically. J. Am. Chem. Soc. 106, 4833–4840 (1984).
Yasui, K., Fugami, K., Tanaka, S. & Tamaru, Y. Unsymmetrical ketone synthesis via a three-component connection reaction of organozincs, allylating agents, and carbon monoxide. J. Org. Chem. 60, 1365–1380 (1995).
Tamaru, Y., Yasui, K., Takanabe, H., Tanaka, S. & Fugami, K. Palladium(0)-catalyzed three-component coupling reaction of allyl benzoates, carbon monoxide, and zincioesters. Angew. Chem. Int. Ed. 31, 645–646 (1992).
Peng, J.-B., Wu, F.-P. & Wu, X.-F. First-row transition-metal-catalyzed carbonylative transformations of carbon electrophiles. Chem. Rev. 119, 2090–2127 (2019).
Li, Y., Hu, Y. & Wu, X.-F. Non-noble metal-catalysed carbonylative transformations. Chem. Soc. Rev. 47, 172–194 (2018).
Boyarskiy, V. P., Bokach, N. A., Luzyanin, K. V. & Kukushkin, V. Y. Metal-mediated and metal-catalyzed reactions of isocyanides. Chem. Rev. 115, 2698–2779 (2015).
Qiu, G., Ding, Q. & Wu, J. Recent advances in isocyanide insertion chemistry. Chem. Soc. Rev. 42, 5257–5269 (2013).
Lang, S. Unravelling the labyrinth of palladium-catalysed reactions involving isocyanides. Chem. Soc. Rev. 42, 4867–4880 (2013).
Vlaar, T., Ruijter, E., Maes, B. U. W. & Orru, R. V. A. Palladium‐catalyzed migratory insertion of isocyanides: an emerging platform in cross‐coupling chemistry. Angew. Chem. Int. Ed. 52, 7084–7097 (2013).
Dömling, A. & Ugi, I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. 39, 3168–3210 (2000).
Otsuka, S., Nogi, K. & Yorimitsu, H. Palladium-catalyzed insertion of isocyanides into the C–S bonds of heteroaryl sulfides. Angew. Chem. Int. Ed. 57, 6653–6657 (2018).
Dechert-Schmitt, A.-M. et al. Highly modular synthesis of 1,2-diketones via multicomponent coupling reactions of isocyanides as CO equivalents. ACS Catal. 9, 4508–4515 (2019).
Jiang, H., Liu, B., Li, Y., Wang, A. & Huang, H. Synthesis of amides via palladium-catalyzed amidation of aryl halides. Org. Lett. 13, 1028–1031 (2011).
Jiang, X. et al. Palladium-catalyzed formylation of aryl halides with tert-butyl isocyanide. Org. Lett. 16, 3492–3495 (2014).
Fei, X.-D., Ge, Z.-Y., Tang, T., Zhu, Y.-M. & Ji, S.-J. Palladium-catalyzed synthesis of isocoumarins and phthalides via tert-butyl isocyanide insertion. J. Org. Chem. 77, 10321–10328 (2012).
Tang, T. et al. Palladium-catalyzed carbonylative sonogashira coupling of aryl bromides via tert-butyl isocyanide insertion. J. Org. Chem. 78, 3170–3175 (2013).
Li, Z. et al. Synthesis of 1,4-enyne-3-ones via palladium-catalyzed sequential decarboxylation and carbonylation of allyl alkynoates. Org. Chem. Front. 4, 1363–1366 (2017).
Corey, E. J. & Hegedus, L. S. 1,4 Addition of acyl groups to conjugated enones. J. Am. Chem. Soc. 91, 4926–4928 (1969).
Jolly, P. W., Wilkinson, G., Stone, F. G. A. & Abel, E. W. Comprehensive Organometallic Chemistry Ch. 5 (Pergamon Press, New York, 1982).
Camps, F., Coll, J., Moretó, J. M. & Torras, J. Studies for control of the Ni(CO)4-promoted carbonylative cycloaddition allyl halides and acetylenes. J. Org. Chem. 54, 1969–1978 (1989).
Nadal, M. L. et al. The Ni-mediated cyclocarbonylation of allyl halides and alkynes made catalytic. Evidence supporting the involvement of pseudoradical NiI species in the mechanism. J. Am. Chem. Soc. 127, 10476–10477 (2005).
Wu, X., Zhao, Y. & Ge, H. Direct aerobic carbonylation of C(sp2)–H and C (sp3)–H bonds through Ni/Cu synergistic catalysis with DMF as the carbonyl source. J. Am. Chem. Soc. 137, 4924–4927 (2015).
Zhao, H.-Y., Gao, X., Zhang, S. & Zhang, X. Nickel-catalyzed carbonylation of difluoroalkyl bromides with arylboronic acids. Org. Lett. 21, 1031–1036 (2019).
Yu, H., Gao, B., Hu, B. & Huang, H. Charge-transfer complex promoted C–N bond activation for Ni-catalyzed carbonylation. Org. Lett. 19, 3520–3523 (2017).
Tjutrins, J., Shao, J. L., Yempally, V., Bengali, A. A. & Arndtsen, B. A. A nickel-based, tandem catalytic approach to isoindolinones from imines, aryl iodides, and CO. Organometallics 34, 1802–1805 (2015).
Peng, J.-B. et al. Nickel-catalyzed molybdenum-promoted carbonylative synthesis of benzophenones. J. Org. Chem. 83, 6788–6792 (2018).
Shi, R. & Hu, X. From alkyl halides to ketones: nickel-catalyzed reductive carbonylation utilizing ethyl chloroformates as the carbonyl source. Angew. Chem. Int. Ed. 58, 7454–7458 (2019).
Wang, Q. & Chen, C. Nickel-catalyzed carbonylative Negishi cross-coupling reactions. Tetrahedron Lett. 49, 2916–2921 (2008).
Shaifali, R. S., Thakur, V. & Das, P. Synthesis of α,β-alkynyl ketones via the nickel-catalysed carbonylative Sonogashira reaction using oxalic acid as a sustainable C1 source. Org. Biomol. Chem. 17, 7036–7036 (2019).
Andersen, T. L., Donslund, A. S., Neumann, K. T. & Skrydstrup, T. Carbonylative coupling of alkyl zinc reagents with benzyl bromides catalyzed by a nickel/NN2 pincer ligand complex. Angew. Chem. Int. Ed. 57, 800–804 (2018).
Donslund, A. S. et al. Access to β-ketonitriles through nickel-catalyzed carbonylative coupling of α-bromonitriles with alkylzinc reagents. Chem. Eur. J. 25, 9856–9860 (2019).
Ravn, A. K. et al. Carbon isotope labeling strategy for β-amino acid derivatives via carbonylation of azanickellacycles. J. Am. Chem. Soc. 141, 11821–11826 (2019).
Suginome, M. & Ito, Y. Polymer Synthesis Ch. 171 (Springer, Berlin, Heidelberg, 2004).
Kamer, P. C. J., Nolte, R. J. M. & Drenth, W. Screw sense selective polymerization of achiral isocyanides catalyzed by optically active nickel(II) complexes. J. Am. Chem. Soc. 110, 6818–6825 (1988).
Deming, T. J. & Novak, B. M. Polyisocyanides using [(η3-C3H5)Ni(OC(O)CF3)]2: rational design and implementation of a living polymerization catalyst. Macromolecules 24, 6043–6045 (1991).
Deming, T. J. & Novak, B. M. Mechanistic studies on the nickel-catalyzed polymerization of isocyanides. J. Am. Chem. Soc. 115, 9101–9111 (1993).
Qiu, G., Mamboury, M., Wang, Q. & Zhu, J. Ketenimines from isocyanides and allyl carbonates: palladium-catalyzed synthesis of βγ-unsaturated amides and tetrazoles. Angew. Chem. Int. Ed. 55, 15377–15381 (2016).
Ito, Y., Hirao, T., Ohta, N. & Saegusa, T. Synthesis of ketenimine via (N-alkylimino)acylpalladium complex intermediate. Tetrahedron Lett. 8, 1009–1012 (1977).
Son, S. & Fu, G. C. Nickel-catalyzed asymmetric Negishi cross-couplings of secondary allylic chlorides with alkylzincs. J. Am. Chem. Soc. 130, 2756–2757 (2008).
Tao, J.-L., Yang, B. & Wang, Z.-X. Pincer-nickel-catalyzed allyl–aryl coupling between allyl methyl ethers and arylzinc chlorides. J. Org. Chem. 80, 12627–12634 (2015).
Ma, W., Xue, D., Yu, T., Wang, C. & Xiao, J.-L. Carbonylative coupling of allylic acetates with aryl boronic acids. Chem. Commun. 51, 8797–8800 (2015).
Fei, X.-D., Tang, T., Ge, Z.-Y. & Zhu, Y.-M. New method for the synthesis of lactones via Nickel-catalyzed isocyanides insertion. Synth. Commun. 43, 3262–3271 (2013).
Kobayashi, Y. & Ikeda, E. Nickel-catalysed substitution reactions of allylic carbonates with aryl- and alkenyl-borates. J. Chem. Soc. Chem. Commun. 1994, 1789–1790 (1994).
Usmani, S. B., Takahisa, E. & Kobayashi, Y. Coupling reaction of 4-cyciopentene-l,3-diol monoacetate and lithium alkenylborates and its application to chiral synthesis of prostaglandin intermediates. Tetrahedron Lett. 39, 601–604 (1998).
Trost, B. M. & Spagnol, M. D. Nickel catalysed coupling of allylamines and boronic acids. J. Chem. Soc. Perkin Trans. 1, 2083–2097 (1995).
Murakami, M., Ito, H., Bakar, W. Ab. W. A., Baba, A. Bb. & Ito, Y. Transition metal-catalyzed coupling of [1-(arylimino)alkyl]zinc with aromatic iodide. Chem. Lett. 18, 1603–1603 (1989).
Acknowledgements
This work was supported by the Youth 1000-Talent Plan Program, East China University of Science and Technology for startup funding, NSFC/China (21421004, 21702060), Shanghai Municipal Science and Technology Major Project (grant no. 2018SHZDZX03) and the Program of Introducing Talents of Discipline to Universities (B16017), and the Fundamental Research Funds for the Central Universities (WJ1814012). We thank Research Center of Analysis and Test of East China University of Science and Technology for the help on NMR analysis. Y.C. thanks Dr. Yang Yang (California Institute of Technology) in proofreading this manuscript.
Author information
Authors and Affiliations
Contributions
Y.C. conceived the project and wrote the paper with the feedback of the other authors. J.Q. and Y.C. directed the project. Y.W., C.Z. and Z.T. performed the experiments and analyzed the data. M.S. and W.H. contributed to the discussion and commented of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Troels Skrydstrup and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weng, Y., Zhang, C., Tang, Z. et al. Nickel-catalyzed allylic carbonylative coupling of alkyl zinc reagents with tert-butyl isocyanide. Nat Commun 11, 392 (2020). https://doi.org/10.1038/s41467-020-14320-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-14320-1
This article is cited by
-
Nickel-catalysed regio- and stereoselective acylzincation of unsaturated hydrocarbons with organozincs and CO
Nature Synthesis (2023)
-
Nickel-catalyzed acylzincation of allenes with organozincs and CO
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
