
Abstract
Salinity, pH, and redox states are fundamental properties that characterize natural waters. These properties of surface waters on early Mars reflect palaeoenvironments, and thus provide clues on the palaeoclimate and habitability. Here we constrain these properties of pore water within lacustrine sediments of Gale Crater, Mars, using smectite interlayer compositions. Regardless of formation conditions of smectite, the pore water that last interacted with the sediments was of Na-Cl type with mild salinity (~0.1–0.5 mol/kg) and circumneutral pH. To interpret this, multiple scenarios for post-depositional alterations are considered. The estimated Na-Cl concentrations would reflect hyposaline, early lakes developed in 104–106-year-long semiarid climates. Assuming that post-depositional sulfate-rich fluids interacted with the sediments, the redox disequilibria in secondary minerals suggest infiltration of oxidizing fluids into reducing sediments. Assuming no interactions, the redox disequilibria could have been generated by interactions of upwelling groundwater with oxidized sediments in early post-depositional stages.
Similar content being viewed by others


Introduction
There have been a number of attempts to estimate water chemistry (i.e., pH, salinity, redox state, and dissolved species) on early Mars by means of geochemical reaction-transport modeling1,2,3. These bottom-up modeling approaches can reproduce time and spatial variations of water chemistry based on the observed mineral assemblages. However, they need to assume initial solution compositions, reactant minerals, fluid permeability, water-rock ratios, and a series of chemical reactions. Thus, these studies involve a number of uncertainties depending on these a-priori parameters, which are always difficult to constrain on early Mars.
In contrast, a top-down approach for quantitative reconstruction of water chemistry of pore water in terrestrial clay-bearing rocks has been proposed by Gaucher et al.4. Using the mineralogical and chemical compositions of clay-bearing rocks and their cation exchange equilibria, this approach successfully reproduces the water chemistry of terrestrial pore waters without any a-priori parameters4,5,6. Smectite is unique in possessing exchangeable cations in interlayer sites7. Although there is a wide diversity of mineralogical characteristics of smectites including the chemical compositions and formation processes, all smectite possess the cation exchange capacity (Supplementary Fig. 1). The cation exchange reaction of smectite is very rapid process with sub-second timescale in suspension solutions8,9. The rate limiting step of the exchange reaction is diffusion and, thus, the activation energy is very low8,9. Therefore, the exchangeable cations in smectite can readily record the surrounding solution composition4,5,6 even at low temperatures (e.g., ~0 °C). On Mars, there is a debate about the formation process of smectite in sediments, i.e., in situ formation by early diagenesis10,11,12,13 vs. detrital deposition3. However, regardless of the chemical compositions and formation conditions of smectite, the top-down approach can provide a robust snapshot of the chemical properties of the pore water that finally interacts with the clay-bearing rocks4 if the clay-bearing rocks contain smectite.
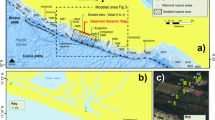
The Mars Science Laboratory rover Curiosity has analyzed the phase and chemical composition of fluvial-lacustrine sediments deposited in long-lived lakes within Gale Crater, a late Noachian to early Hesperian-aged crater14,15. Two drill core samples of lacustrine sediments were obtained from Yellowknife Bay Formation12,14. The Yellowknife Bay Formation was probably deposited by flow deceleration as water streams encountered a lake in Gale Crater14,15 (Fig. 1a). The Yellowknife Bay sediments contain ~20 wt% of smectite12. These smectites would have formed in the early diagenesis within the lake12,13 (Fig. 1b) and/or may have been of detrital origin3 (Fig. 1a). The Yellowknife Bay sediments were subjected to burial pressure and pervasively fractured after the deposition and early diagenesis14,15 (Fig. 1c). During the rewetting events, the late-diagenetic (post-depositional) fluids were introduced into the fractures, which are currently filled with calcium sulfates; presumably a substantial amount of time after the period of the early lakes12,14,16 (Fig. 1d). These rewetting events occurred within Gale even after the formation of Aeolis Mons17, a 5 km mound of layered sedimentary rock in Gale crater. After the final rewetting event, liquid water in the Yellowknife Bay sediments disappeared (Fig. 1e).

Illustrations of hydrogeological context of Yellowknife Bay Formation at Gale Crater (from a–e in time sequence). The dashed lines show the horizon of Yellowknife Bay Formation. In d, e, the full- and no-interaction scenarios are compared. See the main text for the details and references
The proposed hydrogeological context suggests that the Yellowknife Bay sediments would have interacted with waters with different compositions at different stages since its deposition. Bottom water in lakes is continuously trapped within pores of sediments and buried together18 (Fig. 1b). If groundwater largely upwelled into Gale Crater19, this could have also affected pore water chemistry in the early post-depositional stages. After loss of liquid water trapped in pores of the Yellowknife Bay sediments, dissolved components would have been left in the sediments, e.g., as trace of evaporites (Fig. 1c). The presence of sulfate-rich fractures strongly suggests that the post-depositional fluids provided additional SO42− into the sediments in the last wetting event11,16. If the post-depositional SO42−-rich fluids infiltrate into smectite-rich matrix of the sediments, the primary evaporites left in the matrix would have been re-dissolved and mixed with the additional components (Fig. 1d). The duration of the last wetting event determines whether the post-depositional fluids infiltrate into the matrix. Using both terrestrial analog permeability of dried marine clays (10−7–10−10 cm/s)20 and the typical distance between the calcium sulfate fractures in the Yellowknife Bay sediments (~1 cm)11, post-depositional fluids can chemically interact with the matrix of the Yellowknife Bay sediments in a wetting event for 1–102 years or longer through diffusion.
Here we apply the top-down approach4 to the Yellowknife Bay sediments at Gale Crater, Mars. To quantitatively estimate the water chemistry of the pore water that finally interacted with the sediments, we use the exchangeable cation compositions of smectite’s interlayer together with the presence of secondary minerals commonly found in Yellowknife Bay (see Methods). Our results show that the pore water is characterized as mildly saline, Na–Cl type water. We interpret that this salinity of the pore water originates from lake water trapped within sediments in early Gale lakes. The salinity of the lake water would have provided through hydrological cycles during warm and semiarid climate periods in the Hesperian.
Results and Discussion
Based on the hydrogeological context (Fig. 1), we consider two end-member scenarios concerning the interactions between the primary and additional components in the sediments (see Methods): In one, the post-depositional SO42−-rich fluids in the last wetting event fully interact with the sediments (the full-interaction scenario), while in the other we assume no chemical interactions between the post-depositional SO42−-rich fluids and the sediments in the last wetting event (the no-interaction scenario; Fig. 1e). In the full-interaction scenario, we consider that the pore water was reacted with not only the primary components in the matrix of the sediments but also with the additional components (i.e., calcium sulfate) in veins (Fig. 1d). Thereby, the estimated water chemistry in the full-interaction scenario would reflect mixtures of the primary and additional components. In the no-interaction scenario, we assume that the observed secondary minerals in the matrix of the sediments are not influenced by the post-depositional SO42−-rich fluids in the last wetting event (Fig. 1d). In the latter scenario, we estimate the chemical composition of the pore water using only the secondary minerals that are considered to be contained in the matrix of the sediments, i.e., akaganeite. The estimated pore water in the no-interaction scenario would reflect the primary components trapped within the sediments before the last wetting event.
The full-interaction scenario
We first examine the full-interaction scenario and discuss the water chemistry of the pore water at the last rewetting event immediately before the disappearance of liquid water (Fig. 1d). The obtained X-ray diffraction (XRD) patterns of smectite12,13,21 provide information about exchangeable cations in the interlayer (Fig. 2). Basal spacing of smectite depends on the cationic composition in the interlayer and the relative humidity (RH). The cationic composition of smectite of John Klein and Cumberland are estimated from the peak deconvolutions based on the previous experimental studies of smectite basal spacing by RH controlled XRD analyses (see Method). In addition to smectite, the presence of calcium sulfate and akaganeite12,14,16 constrain the composition of liquid water.

Basal spacings of smectite from John Klein (black) and Cumberland (red), compared with Marimba2 (blue) in Murray formation, obtained by Curiosity’s CheMin. The 001 reflection peaks at 10 Å (blueish background) and 14 Å (orangish background) are mainly due to Na+ and Mg2+ in the interlayers, respectively. Yellowknife Bay smectites contain less Na+ and more Mg2+ in the interlayers than Murry smectites
Table 1 and Supplementary Fig. 2 show the compositions of the pore water that last interacted with the Yellowknife Bay sediments for the full-interaction scenario. The concentrations of all components from the Cumberland site overlap those from the John Klein site (Supplementary Fig. 2). This suggests that the pore-water compositions at the two adjacent sites would have been similar and may define the pore water chemistry throughout the Yellowknife Bay sediments. This is consistent with a view of the full-interaction scenario that the post-depositional fluids infiltrated into the sediments. The reconstructed similar compositions of pore water at these sites do not contradict with the fact that the top of the peaks of the 001 reflection of John Klein and Cumberland are different (Fig. 2). The peak deconvolution analyses show that the contribution of the predominant cations (Na+ for John Klein and Mg2+ for Cumberland) to the total interlayer cations are almost half (i.e., ~0.5) (Supplementary Table 1). This suggests that both of the smectites from John Klein and Cumberland are close to transition of the predominant cations in the interlayer sites, whereas that of Marimba2 is dominated by Na+. In a solution within a range of compositions, a transition of the predominant cation occurs in smectite22. Within this range of solution composition, smectites with different of cation occupancies can co-exist. Since the pore-water composition from the Cumberland site is more constrained than those from John Klein site, the former would be the representative water chemistry of the pore water prevailing in the Yellowknife Bay sediment.
There are two possible ways for the liquid pore water to have disappeared from the sediments after the final wetting event (Fig. 1d, e). The first is drying out of the liquid pore water due to complete evaporation in a warm hyperarid climate above freezing temperature. According to this, concentrations of dissolved components should increase upon drying and finally reach saturation levels of high-solubility salts23, such as halite. The estimated concentrations of Na+ correspond to weakly-to-moderately saline (hyposaline) (0.1 mol/kg) and are far below the saturation level of halite (e.g., 10 mol/kg at ambient temperature). This suggests that the scenario for the complete evaporation in warm climate can be ruled out. The other is freezing of the liquid pore water and subsequent sublimation of ground ice in a cold climate. In this case, concentrations of salinity in remained liquid water would have also occurred upon freezing. However, freezing of pore water most likely initiates at the interfaces with solid particles24. This suggests that upon freezing, highly concentrated brine would be unable to directly interact with smectite particles. Hence, the pore-water chemistry recorded in the Yellowknife Bay sediments most likely represents saturated pore water immediately before freezing in the last wetting event.
The dissolved components in Table 1 originate from both the primary components from the pore water in the bottom sediment of the early lake and the additional components from post-depositional fluids in the rewetting events. We believe that the predominant dissolved components of Na and Cl in the pore water would have largely originated from the primary component; namely, lake water trapped within the sediments (Fig. 1a, b). This is because both Na+ and Cl− behave as conservative species which are generally retained in subsequent chemical reactions (e.g., diagenesis), resulting in preservation of information about bottom water within co-buried smectite on Earth18. Additionally, the post-depositional SO42−-rich fluids in the last wetting event were depleted in Na16. Na contents are almost zero (<0.05 wt%) at sulfate-rich veins with the highest SO3 contents16. No enrichments of Na in sulfate-rich veins in the Murray and Stimson formations25 may also support depletion of Na in post-depositional SO42−-rich fluids, if the fluids are in common with Yellowknife Bay26.
Groundwater within terrestrial basalts typically contains low Na concentrations (10−4–10−3 mol/kg: see Methods). Given the possibility that the primary components could have been leached due to upwelling groundwater into Gale19, the estimated Na concentration would be a lower limit of the primary Na concentration in the bottom water of the early lakes. Nevertheless, we consider that the original Na concentration in the bottom water would not be remarkably higher than those in Table 1 because of lack of evidence for drying hypersaline lakes (e.g., desiccation cracks and rip-up chips)27 at Yellowknife Bay14. We consider that if the early lakes existed, its Na+ concentration would have been on the same order of magnitude as that of the pore water in the last wetting event.
Our results of the pore-water chemistry suggest that the water chemistry of the early Gale lakes would be characterized as hyposaline, at least, at the time of the Yellowknife Bay sediments deposition. The estimated Na–Cl concentrations are lower than terrestrial seawater ({Na} = 0.49 mol/kg and {Cl} = 0.56 mol/kg), but significantly higher than freshwater23. On Earth, hyposaline lakes are abundant in semiarid climate regions, such as inner-continental steppe areas28,29. Desiccation cracks and rip-up chips are not pervasive around terrestrial hyposaline lakes (see Supplementary Fig. 3). Thus, the absence of desiccation cracks at Yellowknife Bay is compatible with our conclusion of hyposaline early Gale lakes.
Most of the terrestrial hyposaline lakes in continental areas are terminal lakes without any outflowing rivers. Lake levels of terminal lakes are maintained by a balance between inflowing water and evaporation. Through their hydrological cycles, solutes accumulate within the lakes. Our conclusion of hyposaline water suggests that the outflow of water from the Gale lakes was restricted, which is consistent with the geomorphic interpretation of the Gale lakes as potentially being closed-basin30. Additionally, formation of trioctahedral smectite, including saponite, usually occurs in terrestrial alkaline-saline lakes as an alteration product by early diagenesis31 (Supplementary Table 2). Thus, the abundance of saponite in the Yellowknife Bay sediments is also supportive of hyposaline conditions for Gale lake water, although saponite formation may also occur through closed-system alterations of mafic minerals32. Furthermore, akaganeite, a chloride-bearing ferric oxyhydroxide, occurs with smectite in the Yellowknife Bay mudstone11. The formation of akaganeite requires >0.05 mol/kg of dissolved Cl concentrations in the solution33. Accordingly, the Cl concentrations (0.1–0.3 mol/kg from the Cumberland site) estimated by the present study are consistent with the formation conditions of akaganeite, although the akaganeite is most likely to have been formed during the rewetting events using primary Cl in the sediments in the full-interaction scenario (see below).
We constrain the pH of the pore water based on the stability relationship of pH-sensitive secondary minerals found in the sediments. Calcium carbonates are ubiquitous in terrestrial surface conditions23 and frequently observed in terrestrial lacustrine sediments29 regardless of their origins (authigenic or clastic). However, calcium carbonates are very low (<0.8 wt%) in the Yellowknife Bay sediments12,34,35. The estimated Ca concentrations in the pore water are 0.02–0.05 mol/kg from the Cumberland site (Table 1), which are high enough to produce calcium carbonates if sufficient amounts of CO32− are available. The absence of calcium carbonates must be ascribed to limitation of CO32− in the pore water in the last rewetting event, indicating that the pore water may also be characterized by low pH and/or low levels of dissolved ΣCO2 (ΣCO2 = {CO2} + {HCO3−} + {CO32−}, where {i} denotes concentration of the ith species in molal units)23. Gypsum, proposed as a late diagenetic product, is thought to have been ubiquitously present throughout the entire Yellowknife Bay sediments instead of calcium carbonates12,14,16. We believe that gypsum was the solubility-controlled phase of Ca at the time of the late diagenesis (see Methods). This means that gypsum is more stable than calcium carbonates in the water-sediment system. The stability condition between gypsum and calcium carbonates can be calculated as functions of pH and dissolved ΣCO2 levels (Fig. 3).

Constrained pH-ΣCO2 conditions (white area) in the pore water for mean water composition of the full-interaction scenario (Cumberland). The red line shows the stability boundary between carbonate and gypsum. The orange and sky-blue areas correspond to aragonite stable conditions and saponite unstable conditions, respectively. The broken lines show ΣCO2 concentrations for \(P_{{\rm{CO}}_{2}}\) assuming dissolution equilibrium. See Methods for calculation methodology
Figure 3 shows that, unless dissolved ΣCO2 levels are extremely low, an alkaline pH can be ruled out for the pore water in the last wetting event (Fig. 1d). The estimated dissolved ΣCO2 in the pore water would be a mixture of both a supply of CO2 from the primary components (e.g., dissolution of carbonate) and CO2 dissolved in the post-depositional fluids. Thus, an assumption of no CO2 supply from the primary components (e.g., absence of carbonate)34 provides an upper limit of the pore water’s pH. Given the present level of partial pressure of atmospheric CO2 (\(P_{{\rm{CO}}_{2}}\)) on Mars as a conservative lower limit of ancient \(P_{{\rm{CO}}_{2}}\) levels36 and dissolution equilibrium between the atmosphere and post-depositional fluids, the pore water’s pH would have an upper limit of ~7.3.
Another constraint on the pore-water pH relates to smectite. The Yellowknife Bay sediments contain ~20 wt% smectite12. The detailed mineralogical investigations suggest that the smectite is composed mainly of ferrian (Fe3+) saponite13,37. Since Fe3+ saponite generally forms through oxidative alterations of ferrous (Fe2+) saponite, formation and deposition of Fe3+ saponite through oxidative weathering of basaltic rocks are unlikely. Fe3+ saponite needs to have been originally Fe2+ saponite in the sediments, whichever it is of diagenesis or detrital origin. In this case, a concentration of Fe2+ in the pore water in the last wetting event is likely to have been controlled by Fe2+ saponite because it would be the predominant Fe2+-bearing secondary mineral in the sediments, and because low water-rock ratios are expected in the water-sediment system. Fe2+ concentrations in the pore water must be low to keep the observed interlayer spacing and compositions (see Methods). Since more Fe2+ are provided from Fe2+-bearing saponite at lower pH, the observed interlayer composition provides a lower limit of the pore water’s pH at ~6.9. Together with the constraint from a carbonate-gypsum stability line, we propose a circumneutral pH for the pore water in the last wetting event (Fig. 1d).
The proposed neutral pH agrees with the iron-bearing secondary mineral assemblages found by Curiosity. The Yellowknife Bay sediments possess a wide variety of secondary iron-bearing minerals (akaganeite, magnetite, and Fe2+ saponite)12. Although magnetite and saponite might be of detrital origins, their relatively-high abundance suggests that the pore-water pH would be compatible with their thermochemical stability. Our quantitative reconstruction of water chemistry in the pore water enables us to construct the stability relationship of these iron minerals in regard to pH and Eh (Fig. 4). The fact that all of the observed secondary iron-bearing minerals appear in the pH range of 6.8–8.0 (Fig. 4a) strongly supports our conclusion of circumneutral pH.

Eh-pH diagram of relevant iron species for the pore-water composition of the full-interaction scenario (a) and the no-interaction scenario (b) at 0 °C and 1 atm assuming equilibrium with albite and amorphous silica. The mean water composition at Cumberland are used for the full-interaction scenario, whereas the upper limits of the concentrations are used for the no-interaction scenario
In contrast to a narrow range of pH constraints, the Eh covering these secondary minerals ranges widely from upper to lower limits for the presence of liquid water at circumneutral pH (Fig. 4a). To form akaganeite in particular, high-Eh oxidants should have existed in the water-sediment system (Eh > 0.3 V at pH 7: Fig. 4a). On Mars, potential oxidants capable of producing sufficient Eh include molecular oxygen, ozone, perchloric acid, and nitric acid. Given that all of these potential candidates are proposed to form via atmospheric processes38,39, we theorize that oxidants were transported from the surface, plausibly together with protons (i.e., H2SO4), so that the fluids can contain the photochemical products. Photochemically produced oxidants and volcanic acid fog40 may have become trapped in surface ice/frost at Gale and subsequently melted. Fluids with oxidants would have been transported to the Yellowknife Bay sediments through cracks and then diffused into surrounding pores1 (Fig. 1d). Persistence of the disequilibria in the mineral assemblages strongly suggests that redox reactions in the pore water were very slow because of low temperatures11 and proceeded only partially due to the relatively short existence time of fluids until freezing41, limited amounts of trapped oxidants, and/or high activation energy of Fe2+ oxidation42. Although acidic-oxidizing alterations would have been widespread on Hesperian Mars43, their durations and temperature conditions are poorly constrained. Our results of persistence of redox disequilibria suggest that acidic-oxidizing episodes of alteration would have occurred at low temperatures and were relatively short-lived at Gale.
The no-interaction scenario
We next consider the no-interaction scenario. In the no-interaction scenario, we consider that SO42−-rich fluids did not chemically interact with the matrix of the sediments (see above). In this case, the composition of pore water that finally interacted with the matrix reflects that in the early post-depositional stages immediately before the disappearance of liquid water (Fig. 1b). In the no-interaction scenario, we did not use dissolution equilibrium of calcium sulfate but used the presence of akaganeite in the matrix in addition to the exchangeable cations in the interlayer of the smectite (see Methods). In the no-interaction scenario, akaganeite needs to be formed before intrusion of SO42−-rich fluids given their occurrence in the matrix of the sediments11. The lower limit of pH are estimated from both the stability of Fe2+ saponite; the same as in the full-interaction scenario. The upper limit of pH is estimated from the conditions that allow the formation of akaganeite in the matrix of the sediments (see Methods).
Table 1 and Supplementary Fig. 2 shows the water chemistry for the no-interaction scenario. The concentrations of Na and Cl are broadly close to those for the full-interaction scenario. Since the estimated Na concentrations are also significantly lower than the solubility of halite, the liquid pore water most likely disappeared via freezing and subsequent sublimation (see above). The lowest salinity of the pore water in the estimated range is 2300 mg/kg, which can still be classified as hyposaline. Our results of Na–Cl-type hyposaline water for the early Gale lakes in both the full- and no-interaction scenarios suggest that this conclusion is robust.
The pH is estimated to be 6.8–8.3. The obtained pH range is consistent with that of the full-interaction scenario. The stability fields of akaganeite and Fe2+ saponite in regard to pH and Eh further provide constraints on the redox conditions of the pore water for the no-interaction scenario (Fig. 4b). Figure 4b shows that akaganeite is stable under low-to-neutral pH (<8) and highly oxidizing (Eh > 0.3 V) conditions (Fig. 4b). The pH-Eh conditions are consistent with the experimental investigations of the formation conditions of akaganeite38. The required high Eh (>0.3 V) infers an abundance of strong oxidants in the lake water in the no-interaction scenario. On the other hand, reducing Fe2+ saponite are also suggested to exist in the matrix of the Yellowknife Bay sediments12. In the no-interaction scenario, the observed redox disequilibria (i.e., co-existence of akaganeite, magnetite, and Fe2+ saponite) need to be formed other than intrusion of SO42−-rich fluids.
One possibility to generate the redox disequilibria in the no-interaction scenario is interactions of upwelling groundwater at Gale19 with initially oxidized sediments10 after deposition. Groundwater within Martian crust would become reducing and alkaline10. If reducing groundwater upwelled into oxidized sediments at Gale, Fe2+ saponite would have formed through reduction of oxidized smectite, e.g., nontronite, by groundwater. Akaganeite in the sediments could have been also reduced into magnetite by groundwater; however, this conversion might have proceeded only incompletely due to kinetics. The occurrence of the redox disequilibria due to groundwater upwelling would be also applicable to the full-interaction scenario.
The full-interaction scenario vs the no-interaction scenario
Although we cannot conclude whether post-depositional SO42−-rich fluids interacted with the matrix of the Yellowknife Bay sediments, we prefer the full-interaction scenario over the no-interaction scenario for the following reasons. First, the results of detailed analyses of Curiosity’s data show that even the Yellowknife Bay mudstones without visual calcium sulfate veins may contain small amounts of CaSO4 (1.4 ± 1.4 wt.% of SO3)44. Although this calcium sulfate in the matrix could have been of diagenetic origin, this suggests that SO42−-rich fluids would have diffused into the matrix of the mudstone in the last wetting event. Additionally, post-depositional fluids could seep into the matrix of the mudstones if the rewetting event persisted for >1–102 years (see above). The rewetting with this timescale could have been achieved by hydrological activity that formed small gullies and deltas at Gale after the formation of Aeolis Mons45. Finally, because of the similar elemental compositions between the altered and unaltered mudstones, the early diagenesis could have occurred under isochemical conditions11. Under the isochemical conditions, the simultaneous formation of the authigenic minerals with different stability fields is unlikely.
Below, we discuss the implications of the estimated water chemistry for climate and redox interactions on early Mars, as well as a possible trigger for the last wetting event. Early climate and redox interactions are discussed based on the estimated salinity of the early lakes. Given the similar values of salinity, the former discussion is applicable to both the full- and no-interaction scenarios. In contrast, we can only constrain the water chemistry of post-depositional fluids for the full-interaction scenario; accordingly, the latter discussion is only for the case in which the fluids fully interacted with the mudstone.
Implications for early Martian climate and redox states
Water chemistry revealed by the present study provides constraints on the palaeoclimate and palaeohydrology at the time when early major lake systems were active. Although the Gale impact occurred at around the Noachian/Hesperian boundary14, the timing and duration of the early lakes are poorly constrained15. The estimated Na concentrations of the early lakes allow us to estimate a total duration of hydrological activity at Gale until the time of the Yellowknife Bay sediments’ deposition. For both of the full- and no-interaction scenarios, the Na concentration of the early lakes is estimated to be ~0.03–0.2 mol/kg (Table 1) from Cumberland site. Combined with the proposed lake volume14, a total amount of Na in the lake is obtained when the Yellowknife Bay sediments were deposited (see Methods). A total duration of hydrological activity can be estimated by division of the total amount of Na in the lake by a Na influx into the lake. Thus, short-term intermittency of lakes without removal of Na as halite burial does not reset the total duration. Assuming a steady-state lake level, the Na influx is equivalent to the product of Na concentrations in groundwater and an evaporation (or sublimation if lake surface was frozen) rate of lake water (see Methods). Using Na concentrations in groundwater in terrestrial basalts and evaporation rates derived from three-dimensional general circulation model (GCM) results, a total duration of hydrological activity until the deposition of the Yellowknife Bay sediments would be 105–106 years for the mean surface temperature near the H2O freezing point (see Methods). Higher temperatures (~25 °C) result in shorter durations (~104 years) due to efficient evaporation (see Methods), although higher-temperature conditions are harder to achieve climatically46,47. This duration is broadly consistent with the duration of the lacustrine environment (104–107 years) estimated from the thickness of the Gale sediments and typical sediment accumulation rates on Earth15. Our results also show that the early Gale lakes would have been hyposaline and closed, where evaporation dominated fluid input. Together with the occurrence of similar hyposaline lakes in terrestrial steppe area28 and results of hydrological modeling to make a closed-basin lake at Gale19, we also suggest that the early Gale lakes developed under semiarid climatic conditions, at least, during the deposition of the Yellowknife Bay sediments, which is consistent with the low values of the chemical index of alteration for these sediments10,11. Our results of water chemistry suggest that Gale would have experienced prolonged (104–106 years) episodes of warm and semiarid climates in total, although these episodes might have been intermittent. Similar to soils on present-day Mars, Na-bearing salts would have contained in soils within Gale Crater before the appearance of the lakes2. In warm periods, surface runoff could have transported them to the lake2, providing salinity efficiently to the lake water. With this regard, our estimate of lake duration would be an upper limit. Assuming ~1 wt% of the salt content based on present-day soils48, such an accumulation of surface salts through surface runoff could explain ~50% of the estimated salinity of the lakes in maximum (see Methods). Thus, the required lake duration could be shortened if saline surface runoff occurred.
The estimated total duration of the early Gale lakes agrees with some formation timescales proposed for valley networks49 and Al-rich surface clays50 that formed in the late Noachian to the early Hesperian, although the timing of formation of valley networks and Al-rich surface clays may not be coincident with the existence of the early lakes. Although an ancient Martian ocean might have also existed in the early Hesperian51, three-dimensional GCM results show that its presence is not required to explain fluvial activity at Gale Crater52. The GCM results52 also suggest that when the total Martian surface water inventory is high, climate conditions at the Aeolis quadrangle are humid rather than semiarid to arid (e.g., see Fig. 9 of ref. 52), although locally semiarid climates at Gale can be achieved for some orbital and atmospheric conditions. These suggest that the early Gale lakes, valley networks, and ancient shorelines might not have formed at the same time in a single climate, but they would have formed gradually through dynamic climates of prolonged warming on a cold early Mars46,50.
Given the difficulty of sufficient warming on early Mars solely by CO2-H2O greenhouse gases, the total duration for warm episodes may reflect residence times of additional greenhouse gases46,47 (e.g., H2 and CH4) in the atmosphere, repeated periods of high solar insolation at snowy low-latitude highlands upon obliquity cycles52,53, or combinations of both53. Although outgassing H2/CH4 flux is uncertain, once released, CO2-H2-CH4 greenhouse gases would persist longer than 105 years against the diffusion-limited escape and photodissociation46. In a manner similar to acidic-oxidizing alterations at Gale in later stages for the full-interaction scenario, we can infer that oxidants, e.g., perchlorate, chlorate, ozone, and nitrate, might have also accumulated in surface ice/frost or upstream soils during a cold period in the early Hesperian prior to warming, which in turn could have been released into aqueous environments upon warming, e.g., triggered by H2/CH4 outgassing46. The released oxidants might have been consumed via reactions with abundant reductants from rock components during prolonged warm episodes in the early Hesperian; whereas redox disequilibria would have been preserved due to short-term warming in the late Hesperian or later (see below). We propose that dynamic climates on early-Hesperian Mars46,50 might have been a thermochemical drive to promote redox interactions in aqueous environments, which could have provided free energy to drive the onset of chemical evolution on this planet54.
Timing and trigger for acidic-oxidizing alterations
We then turn to a climatic trigger for the last warming event at Gale that caused post-depositional, acidic-oxidizing alterations after sedimentation of the Yellowknife Bay mudstone in the full-interaction scenario. Our results of low levels of dissolved CO2 in the pore water (Table 1) require both low CO2 supply from the primary components and low dissolved CO2 in the post-depositional fluids (see above). The former suggests the absence of carbonate in the matrix of the sediments34. Given that post-depositional fluids would have originated from the surface in the full-interaction scenario, the latter may reflect low PCO2 at the time of the last wetting event at Gale. Alternatively, dissolution equilibrium between post-depositional SO42−-rich fluids with atmospheric CO2 was not achieved (e.g., due to rapid melting of ice). Drawdown of CO2 from fluids during transportation to Yellowknife Bay via carbonate formation is unlikely given the acidity of fluids. Based on a lower limit of pH, we propose an upper limit of \(P_{{\rm{CO}}_{2}}\) of ~100 mbar if atmospheric CO2 was in equilibrium with the fluids (Table 1). Although formation of Fe carbonate can be kinetically inhibited even at high CO2 levels55, a previous bottom-up approach to estimate dissolved CO2 provided an upper limit of \(P_{{\rm{CO}}_{2}}\) levels as tens of mbar34. The consistency between these independent approaches shows robustness of low CO2 in the pore water. We consider that the estimated \(P_{{\rm{CO}}_{2}}\) would reflect a recent atmosphere at the time of the last wetting event at Gale. The estimated \(P_{{\rm{CO}}_{2}}\) suggests that the last wetting event may have occurred in the late Hesperian or even the Amazonian, when atmospheric pressure would have already declined closer to the present-day level36. The estimated timing of this last wetting event is consistent with the youthful age of jarosite in the Mojave2 sample (2.12 ± 0.36 Ga)17, drilled from a lacustrine mudstone ~60 m higher in the stratigraphic section than the Yellowknife Bay sediments. The last wetting event might be caused by a relatively short warming pulse from the obliquity variations56 would last 1–103 years, rather than episodic warming due to the release of CH4 by clathrate destabilization46 into a dense CO2 atmosphere.
Methods
Mineralogical constraints on water chemistry
We estimate water chemistry of pore water based on the following constraints from the samples at the Cumberland and John Klein drilling sites: (a) the basal spacing of smectite, (b) the presence of saponite, (c) the mineral composition of sulfate, and d) the presence of akaganeite.
The interlayer cationic compositions of smectites were estimated from the 001 reflections. The peak intensities of the 001 reflections are related to the abundance of the cationic components in the interlayer, the atomic structure factors of each components, and the Lorentz-polarization (LP) factors57. The peak deconvolutions of the 001 reflections of John Klein and Cumberland sites after LP factor and the background corrections were conducted using the Pseudo-Voigt function to estimate the cation compositions (Supplementary Fig. 4). The combinations of the three peaks at 13.7, 11.5–11.7, and 10.0–10.1 Å can reasonably reproduce the 001 reflections for both smectites.
The basal spacing of smectite depends on a cation composition in the interlayer and RH. The RH remains <1% inside CheMin under temperatures 5–25 °C (ref. 13). Supplementary Fig. 5 summarizes the basal spacings of several smectites with different cations (Na+, K+, Ca2+, Mg2+, Fe2+ and Fe3+) at RH = 0%. The basal spacings of Na+ and K+ smectites are ~10 Å; whereas, those of Ca2+ smectite are scattered, but most of the smectites exhibit peaks at ~12 Å. The basal spacing of Fe2+ and Fe3+ smectite at RH = 0% (ranging 11–12.5 Å) are similar to that of Ca2+ smectite. Those of Mg smectite vary from 11.5 to 13.9 Å. These values are generally higher than those of Ca smectite because the hydration states of Mg2+ in smectite are kept even at low relative humidity58. According to the relationship, the peak with 10.0–10.1 Å from the two smectites represent the Na+ and/or K+. That with 12 Å represent the Ca2+ and Fe (Fe2+ and/or Fe3+). That with 13.7 Å most likely represents the Mg2+. Partially intercalated hydroxylated Mg (i.e., MgOH+) in the interlayer site of the smectite at Cumberland has been proposed as an explanation for the expanded structure12,13. The dissolved MgOH+ species becomes important at extremely alkaline conditions (pH > 12: pKa = 12.8 at 0 °C) at ambient to low temperatures. However, concentration of ΣMg2+ (=Mg2+ + MgOH+) also becomes extremely low at high pH (pH > 10) because of the formation of Mg-bearing secondary minerals (saponite and brucite). Therefore, given the low {MgOH+} at alkaline pH, the exchange of the dissolved MgOH+ species into the interlayer occurs very inefficiently at ambient to low temperatures unless the Mg2+/(Na+ + K+) ratio in the pore water is extremely high. Assuming the peak from 10 Å represents Na+ and/or K+, the extremely high Mg2+/(Na+ + K+) ratio contradicts with Na+/K+ interlayers. Although the reported basal spacings of Mg-saturated saponite vary from 11.5 to 12.0 Å, we believe that Mg2+-saturated Mg smectite is responsible for the spacing of ~14 Å of the smectites. This is the case because the swelling behaviors of interlayer cations mainly depend on the layer charge and charge location59. Vermiculite, high-layer-charge montmorillonite, and well-crystalline synthesized smectite exhibit higher basal spacings of ~14 Å (Supplementary Fig. 5). These specimens are expected to possess high layer charges. The smectite found in the Yellowknife Bay sediments is considered to be Fe3+ saponite, which would have been formed by the alteration of Fe2+ saponite37. Since the oxidation of Fe2+ to Fe3+ should have resulted in an increase in the layer charge by substation of O2− for OH−37, the smectite in the Yellowknife Bay sediments would have larger spacings, e.g., ~14 Å.
Mass balance calculations using bulk-rock chemical data from alpha particle X-ray spectrometer (APXS) and the chemistry of crystalline components indicate that the Na+/K+ ratios of the clay mineral and amorphous material were 4 and 5 for John Klein and Cumberland, respectively60. Thus, the observed basal spacings of the smectites suggest their cation compositions in cationic electrical charge of Na+:K+ to be ~4.5:1. Based on the peak deconvolutions of 001 reflections (Supplementary Fig. 4) and the Na+:K+, the cationic composition of smectites are determined. The concentration of dissolved ferric iron is extremely low except for the extremely low or high pH conditions. The direct intercalation of Fe3+ into the interlayer of smectite from the solution was not likely. Therefore, Fe in the interlayer of smectite is most likely originated from Fe2+, although the part of or all Fe2+ possibly oxidized to Fe3+ in the interlayer. The equivalent ratios of the cationic composition from John Klein and Cumberland site are given in Supplementary Table 1.
Under terrestrial conditions, saponite depositions are observed in saline lakes, low-temperature upwelling solution from seafloor, and modern oceans (Supplementary Table 2). The pH of the solutions is commonly neutral to alkaline. The predominate cations in these natural waters are always Na+ rather than Mg2+. Under such alkaline conditions, concentrations of divalent cations, such as Mg2+ and Ca2+, must be very low due to formation of Mg/Ca-bearing secondary minerals23. Consequently, the low concentrations of divalent cations lead to the dominance of Na+ in the solutions. Thus, the presence of saponite in the Yellowknife Bay sediments strongly suggests that the predominant cation in the pore water is most likely Na+.
CheMin detected anhydrite (anhydrous CaSO4) and bassanite (CaSO4⋅0.5H2O) from both of the drill core samples12,61. Both of the minerals rarely form under terrestrial surface conditions. Bassanite is considered to be a likely product of gypsum (CaSO4⋅2H2O) dehydration61. We consider that bassanite and anhydrite were alteration products after the disappearance of pore water and assume that Ca2+ and SO42− concentrations of the pore water were controlled by dissolution and precipitation equilibrium of gypsum62.
The Yellowknife Bay sediments contain akaganeite, a chloride-bearing ferric oxyhydroxide. Akaganeite is associated with smectite in the matrix of the mudstone11. The formation of akaganeite requires >0.05 mol/kg of dissolved Cl− concentrations in the solution33. The presence of akaganeite in the sediments suggests that the dissolved Cl− concentrations in the solution must be higher than 0.05 mol/kg. Given the fact that akaganeite can accommodate the anions other than chloride, the presence of Cl− type akaganeite in Yellowknife Bay strongly suggests that the akaganeite was in contact with Cl−dominant solutions compared with other anions, such as sulfate63. By combining the dominant cation type of the water constrained from the presence of saponite, the original pore water in the sediments would be Na–Cl water type.
Thermodynamic calculations
To estimate water chemistry based on the above constraints, the modeling was performed using REACT from the Geochemist’s Workbench (GWB)64. We assume a temperature of 0 °C and a pressure of 1 bar13,34. Thermodynamic data for aqueous species and mineral solubility, except for the akaganeite, ferrihydrite and Fe2+-saponite, were from the “thermo.dat” in the GWB. The detailed descriptions for the database construction of these minerals are given in Supplementary Note 1. The activity coefficient was calculated using the Helgeson, Kirkham and Flower version of the extended Debye–Hückel equation65 for NaCl media at 0 °C. In the modeling, we used the molal (mol/kg H2O) unit notified in {i}, in ith chemical species in solution. The solution compositions are given by the component concentrations, which are the total concentrations of the species containing each component. In the following, we used the notation of {X} to express the molal concentrations in a solution of component X.
Constraining major components
Under terrestrial conditions, the major cations of natural waters are Na+, K+, Mg2+ and Ca2+, while major anions are Cl−, SO42−, and HCO3−(ref. 23). The concentration of Fe2+ becomes important only under reducing conditions23. The concentration of H+ and OH− are important only at extremely low or high pH. We assume that the H+, OH−, and HCO3− are negligible in the charge balance calculations compared with other major species. The validity of the assumption will be quantitatively examined (Table 1). Perchlorate (ClO4−) has been detected in the Yellowknife Bay sediments35. Both Cl− and ClO4− are monovalent anions, which do not take part in the cation exchange reactions. According to the present approach to constrain major components, the chemical behavior of ClO4− can be treated as same as that of Cl−. This means that the concentrations of Cl− obtained from the present study actually represent the concentrations of total chloride ({Cl−} + {ClO4−}) and that the results of cation compositions, pH, and salinity of the pore water do not change even when the pore water contained ClO4−. However, the presence of the Cl− bearing akaganeite in the Yellowknife Bay sediments suggests the concentration of Cl− must be higher than that of ClO4−(ref. 63).
The cation compositions in the interlayer of smectite were calculated using the cation exchange equilibria as follows:
In those equations, Di represents the divalent metal (Ca, Mg and Fe), >X denotes the exchangeable site. ai denotes activity of ith species. βj stands for the activity of jth species complexed with an exchange site. Since surface activity coefficients are not well known, we assume that they are unity. Based on the Gaines–Thomas convention7, βj is given in terms of the fraction jth species in an exchangeable site of the total electrical equivalents of exchange capacity occupied by cations;
The interlayer compositions of smectites observed from the sediments (Supplementary Table 1) provide the constrains for βj. KNa_i represents the selectivity coefficients of the exchange of ith species to Na+ in the interlayer. KNa_i is highly dependent on the exchanger composition22. We estimated the KNa_i for smectite as a function of exchanger compositions (Supplementary Table 1) by using the generic empirical model22. The uncertainties of the estimated log KNa_i are ±0.222. The temperature dependences of selectivity coefficients were reported to be small within these uncertainties6.
The charge balance of the solution can be expressed as:
Although we consider aqueous complexes in the calculation, the charge balance expressions in the manuscript ignore the aqueous complexes for illustrative purpose. {Ca2+} and {SO42−} are constrained from dissolution equilibrium of gypsum (see above) for full-interaction scenario:
The corresponding mass action expression is expressed as follows:
where, γi represents the activity coefficients of ith species. Based on the constrain from the water type of Na–Cl, the equivalent concentrations of Na+ and Cl− are higher than the major species such as Mg2+, Ca2+ and SO42−. Therefore, the endmember compositions of solutions at an interlayer cationic composition under a single set of selectivity coefficients can be determined from following two boundary conditions:
where {Di2+} represents the concentrations of divalent cations, of which concentrations are highest next to that of Na+. We calculated the two endmember solution compositions from Eqs. (7) and (8) under the ranges of the selectivity coefficients with the uncertainties.
In the no-interaction scenario, the matrix of the mudstone is assumed to be isolated from post-depositional fluids enriched in sulfate. Therefore, the charge balance of the solution can be expressed as:
The constraint on {Cl−} with regard to the existence of akaganeite in the matrix of the mudstone is applied to the no-interaction scenario (i.e., {Cl−} ≥ 0.05 mol/kg)33 instead of the constrain from Eq. (8).
Constraining pH and dissolved ΣCO2 in the full-interaction scenario
The formation of calcite, most stable calcium carbonate phase, is known to be kinetically inhibited in the presence of significant amount of Mg2+ in solutions7. Instead, metastable aragonite is favored to form from the solutions7. The stability relationship between aragonite and gypsum is written as:
The Gibbs free energy (ΔGR) of the reaction can be written as follows:
ΔGR0 represents the standard free energies of the reactions and is related to the equilibrium constants23. The equilibrium constant of Eq. (10) was calculated using the RXN program in the GWB. The activity of SO42− comes from the estimated major components (Table 1). In Eq. (11), gypsum is thermochemically stable atΔGR < 0. The stability conditions between gypsum and aragonite were calculated as functions of pH and \(a_{{{\mathrm{CO}}_{2}} ({\mathrm{aq}})}\). Once \(a_{{{\mathrm{CO}}_{2}} ({\mathrm{aq}})}\) and pH are obtained, {HCO3−} and {CO32−} are calculated from their dissociation constants. Thus, the stability conditions between gypsum and aragonite can be expressed as functions of pH and ΣCO2 (Fig. 3).
Saponite is trioctahedral smectite bearing Mg and Fe, which is unstable at low pH31. Smectite usually occurs as fine particles and the dissolution rates must be higher than the primary minerals. It can be assumed that the concentrations of Mg and Fe in the solution were equilibrium with respect to saponite. The dissolution reaction of Fe-saponite can be written as follows;
{Al3+} and {SiO2(aq)} would be equilibrated with respect to albite (NaAlSi3O8) and amorphous silica (SiO2(am)), which are found in the Yellowknife Bay sediments34 through the following reaction;
The mass action expression is given as follows:
The equilibrium constants of K13 were calculated using the RXN program in the GWB. The activities of Na+, Fe2+ and Mg2+ derive from the estimated major components (Table 1). An lower limit of pH was calculated from the higher limits of Fe2+ and Mg2+ of the estimated major components.
Constraining pH in the no-interaction scenario
Because we exclude gypsum from the assumed system, the ΣCO2 cannot be constrained in the no-interaction scenario. The lower limit pH is estimated from the stability of smectite as described above. The upper limit of pH is estimated based on the formation conditions of akaganeite. Peretyazhko et al.33 showed that the upper limit pH allowing akaganeite formation is pH 8, above which ferrihydrite is formed instead of akaganeite. The transformation reaction of akaganeite to ferrihydrite (Fe(OH)3) can be written as follows:
The mass action expression of Eq. (15) is given as follows:
The activity of Cl− is derived from the estimated major components (Table 1) assuming Cl− is predominant species relative to ClO4−. The obtained activity of H+ from Eq. (16) represents the upper limit of pH, which enables the formation of akaganeite.
Estimates of total duration for hydrological activities at Gale
We assume a simplified hydrological system in a closed-basin lake, in which Na supplied from the surface/groundwater accumulate in the lake (see the main text and Supplementary Note 2 for a terrestrial analog). In this system, a mass balance of supplied Na+ can be expressed as follows;
where Cground and Clake are Na concentrations (mol/kg) in the surface/groundwater and lake, respectively, Fground is the flux (kg/sec) of inflowing water, t is the duration (sec), and Mlake is the mass (kg) of lake water. Although the mass of lake water at the time of deposition of Yellowknife Bay has been poorly constrained, Mlake may become (3–6) × 1015 kg for a Gale basin-sized lake (i.e., diameter of ~100 km with the central peak with ~45 km diameter15, mean lake depth of a few hundred meters). We use typical Na concentrations in terrestrial surface/groundwater within terrestrial basaltic rocks (10−4–10−3 mol/kg for circumneutral-pH waters in Iceland66, Australia67, and North America68) for Cground. Lower-temperature groundwater67 tends to contain lower Na; yet, Na concentrations66,67,68 vary only in a factor of ~5 for groundwater temperatures of 4–25 °C. Accumulation of volcanic acids during intermittency of hydrological cycles would increase Na concentration in groundwater due to high rates of chemical weathering2; however, low solution pH and acidity would maintain only for ~102 years owing to neutralization by interactions with rocks1 (also see the main text). In a steady state, Fground is balanced with the total mass flux of evaporating H2O, Fevap (kg/sec). The potential evaporation (or sublimation if lake surface is frozen) flux, Spot (kg/m2/sec), per unit surface area of the lake can be expressed as follows52:
where cD is the drag coefficient52 ( = 2.75 × 10−3), |v| is the surface wind velocity, \(R_{{{\mathrm{H}}_{2}}{\mathrm{O}}}\) is the specific gas constant for H2O, Ta is the atmospheric temperature near the surface, and psat(Ts) is the H2O saturation vapor pressure at the surface temperature, Ts. We used 3–4 m/s for |v| at Gale based on results of a three-dimensional GCM for a 1-bar atmosphere and obliquity of 25 degrees52 (Supplementary Fig. 6) (c.f., |v| = 5–6 m/sec at Gale on present-day Mars69) and assumed Ts ≈ Ta. Using the above values, Spot becomes (1–4) × 10−5 kg/m2/sec for Ts = 260–273 K. Thus, Fevap (=Fground) becomes ~105 kg/sec for a Gale-sized lake. By introducing Clake ~ 0.1 mol/kg (Table 1) into Eq. (18), t becomes ~105–106 years. The Gale lakes could have salinated more quickly under warmer and drier conditions due to high Spot. For instance, t is reduced to ~104 years for Ts = 298 K. Nevertheless, higher temperatures would have been harder to achieve on early Mars47,52; accordingly, the estimated duration for Ts = 298 K can serve as a conservative lower limit. Spot is the maximum evaporation rate assuming relative humidity of 0% (ref. 52). However, given the proposed semiarid to arid climates11,19, the assumption of a relative humidity of 0% would not affect our conclusions significantly. The estimated duration includes an order(s) of magnitude of uncertainty mainly derived from an uncertainty of the lake depth. Validity and uncertainties of our methodology can be evaluated through estimating the ages of hyposaline closed-basin lakes developed in semiarid areas on Earth (see Supplementary Note 2).
Martian soils, in general, contain abundant Na-bearing salts (e.g., chloride and perchlorate)48. As mentioned in the main text, surface runoff could have transported surface salts accumulated within soils efficiently to the early Gale lake in warm periods2. We estimate a quantitative effect of this process. Present-day soils on Mars typically contain ~1 wt% of salts48. Given ~(2–3) × 1010 m2 of the catchment area of the early lake (=the surface area of Gale Crater’s cavity) and ~3 × 103 kg/m3 of bulk density of soils, the maximum mass of transported salts through surface runoff can be ~5 × 1011 kg assuming a 1 m-thick soil layer and ~5 × 1012 kg for a 10 m-thick soil layer. According to the infiltration theory of rain into dried soils70, persistent inputs of surface water due to rain fall and/or snow melting needs to continue for ~20 h and ~2000 h to reach the soils (assuming volcanic loam) in depth of 1 and 10 m, respectively. Since this timescale would roughly correspond to those of precipitation/snow melting on early Mars52, we consider that the wetting depth within soils upon rain fall and/or snow melting over Gale Crater would be at most ~10 m. On the other hand, given ~0.03–0.2 mol/kg of lake salinity (see the main text) and (3–6) × 1015 kg of lake water mass (see Eq. (17)), the total salt mass in the lakes would become ~(1–10) × 1013 kg. This suggests that if the soils in the catchment area of the lakes contained salts comparable to that of today’s soils, saline surface runoff could provide ~50% of the total salinity of the lakes in maximum. Assuming the saline surface runoff2, thereby, the required lake duration could be reduced in a factor of ~2 or less.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information File.
Code availability
Code for all analyses will be made available on request.
References
Zolotov, M. Y. & Mironenko, M. V. Timing of acid weathering on Mars: a kinetic-thermodynamic assessment. J. Geophys. Res. Planets 112, E07006 (2007).
Melwani Daswani, M. & Kite, E. S. Paleohydrology on Mars constrained by mass balance and mineralogy of pre-Amazonian sodium chloride lakes. J. Geophys. Res. Planets 122, 1802–1823 (2017).
Schieber, J. et al. Encounters with an unearthly mudstone: understanding the first mudstone found on Mars. Sedimentology 64, 311–358 (2017).
Gaucher, E. C. et al. A robust model for pore-water chemistry of clayrock. Geochim. Cosmochim. Acta 73, 6470–6487 (2009).
Wersin, P. et al. Constraining porewater chemistry in a 250 m thick argillaceous rock sequence. Chem. Geol. 434, 43–61 (2016).
Gailhanou, H. et al. Effects of a thermal perturbation on mineralogy and pore water composition in a clay-rock: an experimental and modeling study. Geochim. Cosmochim. Acta 197, 193–214 (2017).
Appelo, C. A. J. & Postma, D. Geochemistry, Groundwater and Pollution. (CRC Press, 2005).
Ogwada, R. A. & Sparks, D. L. A critical evaluation on the use of kinetics for determining thermodynamics of ion exchange in soils. Soil Sci. Soc. Am. J. 50, 300–305 (1986).
Crooks, J. E., El-Daly, H., El-Sheikh, M. Y., Habib, A. M. & Zaki, A. B. Kinetics of ion-exchange on montmorillonite clays. Int. J. Chem. Kinet. 25, 161–168 (1993).
Hurowitz, J. A. et al. Redox stratification of an ancient lake in Gale crater, Mars. Science 356, eaah6849 (2017).
McLennan, S. M. et al. Elemental geochemistry of sedimentary rocks at Yellowknife. Science 343, 1244734 (2014).
Vaniman, D. T. et al. Mineralogy of a mudstone at Yellowknife Bay, gale crater, mars. Science 343, 1243480 (2014).
Bristow, T. F. et al. The origin and implications of clay minerals from Yellowknife Bay, Gale crater, Mars. Am. Mineral. 100, 824–836 (2015).
Grotzinger, J. P. et al. A habitable fluvio-lacustrine environment at yellowknife bay, gale crater, mars. Science 343, 1242777 (2014).
Grotzinger, J. P. et al. Deposition, exhumation, and paleoclimate of an ancient lake deposit, Gale crater, Mars. Science 350, aac7575 (2015).
Nachon, M. et al. Calcium sulfate veins characterized by ChemCam/Curiosity at Gale crater, Mars. J. Geophys. Res. Planets 119, 1991–2016 (2014).
Martin, P. E. et al. A two-step K-Ar experiment on mars: dating the diagenetic formation of jarosite from amazonian groundwaters. J. Geophys. Res. Planets 122, 2803–2818 (2017).
Soulet, G. et al. Glacial hydrologic conditions in the Black Sea reconstructed using geochemical pore water profiles. Earth Planet. Sci. Lett. 296, 57–66 (2010).
Horvath, D. G. & Andrews-Hanna, J. C. Reconstructing the past climate at Gale crater, Mars, from hydrological modeling of late-stage lakes. Geophys. Res. Lett. 44, 8196–8204 (2017).
Davis, S. N. Porosity and permeability of natural materials. in Flow Through Porous Media (ed. De Wiest, R. J. M.) 54–89 (Academic Press, 1969).
Bristow, T. F. et al. Clay mineral diversity and abundance in sedimentary rocks of Gale crater, Mars. Sci. Adv. 4, eaar3330 (2018).
Tournassat, C. et al. Cation exchange selectivity coefficient values on smectite and mixed-layer illite/smectite minerals. Soil Sci. Soc. Am. J. 73, 928–942 (2008).
Drever, J. I. The Geochemistry of Natural Waters: Surface and Groundwater Environments. (Prentice Hall, 1997).
Arenson, L. U. & Sego, D. C. The effect of salinity on the freezing of coarse-grained sands. Can. Geotech. J. 43, 325–337 (2006).
Yen, A. S. et al. Multiple stages of aqueous alteration along fractures in mudstone and sandstone strata in Gale Crater, Mars. Earth Planet. Sci. Lett. 471, 186–198 (2017).
Rampe, E. B. et al. Mineralogy of an ancient lacustrine mudstone succession from the Murray formation, Gale crater, Mars. Earth Planet. Sci. Lett. 471, 172–185 (2017).
Gutiérrez, M. Desert lakes: playa lakes and sebkhas. in Climatic Geomorphology 383–405 (Elsevier, 2005).
Lehmkuhl, F., Grunert, J., Hülle, D., Batkhishig, O. & Stauch, G. Paleolakes in the Gobi region of southern Mongolia. Quat. Sci. Rev. 179, 1–23 (2018).
Fukushi, K. & Matsumiya, H. Control of water chemistry in alkaline lakes: solubility of monohydrocalcite and amorphous magnesium carbonate in CaCl2–MgCl2–Na2CO3 solutions. ACS Earth Sp. Chem. 2, 735–744 (2018).
Forsythe, R. D. & Blackwelder, C. R. Closed drainage crater basins of the Martian highlands: constraints on the early Martian hydrologic cycle. J. Geophys. Res. Planets 103, 31421–31431 (1998).
Bristow, T. F. et al. Mineralogical constraints on the paleoenvironments of the Ediacaran Doushantuo Formation. Proc. Natl Acad. Sci. USA 106, 13190–13195 (2009).
Peretyazhko, T. S. et al. Fe/Mg smectite formation under acidic conditions on early Mars. Geochim. Cosmochim. Acta 173, 37–49 (2016).
Peretyazhko, T. S., Ming, D. W., Rampe, E. B., Morris, R. V. & Agresti, D. G. Effect of solution pH and chloride concentration on akaganeite precipitation: implications for akaganeite formation on mars. J. Geophys. Res. Planets 123, 2211–2222 (2018).
Bristow, T. F. et al. Low Hesperian P CO2 constrained from in situ mineralogical analysis at Gale Crater, Mars. Proc. Natl Acad. Sci. USA 114, 2166–2170 (2017).
Ming, D. W. et al. Volatile and organic compositions of sedimentary rocks in Yellowknife Bay, Gale Crater, Mars. Science 343, 1245267 (2014).
Hu, R., Kass, D. M., Ehlmann, B. L. & Yung, Y. L. Tracing the fate of carbon and the atmospheric evolution of Mars. Nat. Commun. 6, 10003 (2015).
Treiman, A. H. et al. Ferrian saponite from the Santa Monica Mountains (California, U.S.A., Earth): characterization as an analog for clay minerals on Mars with application to Yellowknife Bay in Gale Crater. Am. Mineral. 99, 2234–2250 (2014).
Smith, M. L., Claire, M. W., Catling, D. C. & Zahnle, K. J. The formation of sulfate, nitrate and perchlorate salts in the martian atmosphere. Icarus 231, 51–64 (2014).
Lasne, J. et al. Oxidants at the Surface of Mars: a review in light of recent exploration results. Astrobiology 16, 977–996 (2016).
Fairén, A. G., Fernández-Remolar, D., Dohm, J. M., Baker, V. R. & Amils, R. Inhibition of carbonate synthesis in acidic oceans on early Mars. Nature 431, 423–426 (2004).
Kreslavsky, M. A. & Head, J. W. Fate of outflow channel effluents in the northern lowlands of Mars: the vastitas borealis formation as a sublimation residue from frozen ponded bodies of water. J. Geophys. Res. Planets 107, 4-1–4-25 (2002).
Burns, R. G. Mineralogical Applications of Crystal Field Theory. (Cambridge Univ. Press, 1993).
Bibring, J.-P. et al. Global mineralogical and aqueous mars history derived from OMEGA/Mars express data. Science 312, 400–404 (2006).
Schmidt, M. E. et al. Dusty Rocks in Gale Crater: assessing areal coverage and separating dust and rock contributions in APXS analyses. J. Geophys. Res. Planets 123, 1649–1673 (2018).
Palucis, M. C. et al. Sequence and relative timing of large lakes in Gale crater (Mars) after the formation of Mount Sharp. J. Geophys. Res. Planets 121, 472–496 (2016).
Wordsworth, R. D. et al. Transient reducing greenhouse warming on early Mars. Geophys. Res. Lett. 44, 665–671 (2017).
Ramirez, R. M. et al. Warming early Mars with CO2 and H2. Nat. Geosci. 7, 59–63 (2013).
Hecht, M. H. et al. Detection of perchlorate and the soluble chemistry of martian soil at the phoenix lander site. Science 325, 64–67 (2009).
Hoke, M. R. T., Hynek, B. M. & Tucker, G. E. Formation timescales of large Martian valley networks. Earth Planet. Sci. Lett. 312, 1–12 (2011).
Bishop, J. L. et al. Surface clay formation during short-term warmer and wetter conditions on a largely cold ancient Mars. Nat. Astron. 2, 206–213 (2018).
Citron, R. I., Manga, M. & Hemingway, D. J. Timing of oceans on Mars from shoreline deformation. Nature 555, 643–646 (2018).
Wordsworth, R. D., Kerber, L., Pierrehumbert, R. T., Forget, F. & Head, J. W. Comparison of “warm and wet” and “cold and icy” scenarios for early Mars in a 3-D climate model. J. Geophys. Res. Planets 120, 1201–1219 (2015).
Palumbo, A. M., Head, J. W. & Wordsworth, R. D. Late Noachian Icy Highlands climate model: exploring the possibility of transient melting and fluvial/lacustrine activity through peak annual and seasonal temperatures. Icarus 300, 261–286 (2018).
Russell, M. J. et al. The drive to life on wet and icy worlds. Astrobiology 14, 308–343 (2014).
Tosca, N. J., Ahmed, I. A. M., Tutolo, B. M., Ashpitel, A. & Hurowitz, J. A. Magnetite authigenesis and the warming of early Mars. Nat. Geosci. 11, 635–639 (2018).
Christensen, P. R. Formation of recent martian gullies through melting of extensive water-rich snow deposits. Nature 422, 45–48 (2003).
Walker, J. R. An Introduction to computer modeling of X-Ray powder diffraction patterns of clay minerals: a guided tour of NEWMOD©. Computer Appl. X-Ray Powder Diffr. Anal. Clay Miner. 5, 1–17 (1993).
Morodome, S. & Kawamura, K. In situ X-ray diffraction study of the swelling of montmorillonite as affected by exchangeable cations and temperature. Clays Clay Min. 59, 165–175 (2011).
Sato, T., Watanabe, T. & Otsuka, R. Effects of layer charge, charge location, and energy change on expansion properties of dioctahedral smectites. Clays Clay Miner. 40, 103–113 (1992).
Morrison, S. M. et al. Crystal chemistry of martian minerals from Bradbury Landing. Am. Mineral. 103, 857–871 (2018).
Vaniman, D. T. et al. Gypsum, bassanite, and anhydrite at Gale crater, Mars. Am. Miner. 103, 1011–1020 (2018).
Hardie, L. A. & Eugster, H. P. The evolution of closed-basin brines. Miner. Soc. Am. Spec. Pap. 3, 273–290 (1970).
Peretyazhko, T. S. et al. Reaction of akaganeite with Mars-relevant anions. ACS Earth Sp. Chem. 3, 314–323 (2019).
Bethke, C. M. The Geochemist’s Workbench Users Guide. (University of Illinois, 1998).
Helgeson, H. C., Kirkham, D. H. & Flowers, G. C. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at high pressures and temperatures: IV. Calculation of activity coefficients, osmotic coefficients, and apparent molal and standard and relative partial molal properties to 600°. Am. J. Sci. 281, 1249–1516 (1981).
Kristmannsdóttir, H. & Ármannsson, H. Groundwater in the Lake Myvatn area, northern Iceland: chemistry, origin and interaction. Aquat. Ecol. 38, 115–128 (2004).
Tweed, S. O., Weaver, T. R. & Cartwright, I. Distinguishing groundwater flow paths in different fractured-rock aquifers using groundwater chemistry: Dandenong Ranges, southeast Australia. Hydrogeol. J. 13, 771–786 (2005).
Coombs, M. J. Threats to water quality in the deep basalt aquifer, city of Walla Walla, Washington. (Whitman College, 2000).
Martínez, G. M. et al. The modern near-surface martian climate: a review of in-situ meteorological data from viking to curiosity. Space Sci. Rev. 212, 295–338 (2017).
Brutsaert, W. Hydrology: An introduction. (Cambridge University Press, 2005).
Acknowledgements
Discussions with T. Kasama, T. Sato, T. Shibuya, and S. Yokoyama are greatly appreciated. Financial support was provided by KAKENHI from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) (Grant Nos. JP17H06458, JP17H06454, and JP17H06456) and a cooperative research program of the Institute of Nature and Environmental Technology, Kanazawa University (No. 19035).
Author information
Authors and Affiliations
Contributions
K.F., H.S. and K.M. modeled the water chemistry. Y.S. interpreted the estimated water chemistry. The estimate of the duration of the early Gale lakes was conceptualized by R.W. and Y.S., and calculated by Y.S. and R.W. All authors wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Edwin Kite, Elizabeth Rampe, Mikhail Zolotov and other anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fukushi, K., Sekine, Y., Sakuma, H. et al. Semiarid climate and hyposaline lake on early Mars inferred from reconstructed water chemistry at Gale. Nat Commun 10, 4896 (2019). https://doi.org/10.1038/s41467-019-12871-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-12871-6
This article is cited by
-
Constraining the preservation of organic compounds in Mars analog nontronites after exposure to acid and alkaline fluids
Scientific Reports (2020)
-
Relict Ocean Worlds: Ceres
Space Science Reviews (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
