
Abstract
Although the chemistry of uranium-ligand multiple bonding is burgeoning, analogous complexes involving other actinides such as thorium remain rare and there are not yet any terminal thorium nitrides outside of cryogenic matrix isolation conditions. Here, we report evidence that reduction of a thorium-azide produces a transient Th≡N triple bond, but this activates C-H bonds to produce isolable parent imido derivatives or it can be trapped in an N-heterocycle amine. Computational studies on these thorium-nitrogen multiple bonds consistently evidences a σ > π energy ordering. This suggests pushing-from-below for thorium, where 6p-orbitals principally interact with filled f-orbitals raising the σ-bond energy. Previously this was dismissed for thorium, being the preserve of uranium-nitrides or the uranyl dication. Recognising that pushing-from-below perhaps occurs with thorium as well as uranium, and with imido ligands as well as nitrides, suggests this phenomenon may be more widespread than previously thought.
Similar content being viewed by others
Introduction
In recent years there has been intense interest in early actinide-nitrides, [An≡N]n (An = uranium or thorium), in molecular and materials contexts1,2. From the patent for the Haber Bosch chemical process for ammonia synthesis in the early 1900s it has been known that uranium-nitrides are implicated in the formation of ammonia, where uranium was noted to be the best promoter for this heterogeneous process3. Binary nitride materials also have great potential as accident tolerant fuels, and although much focus has been on uranium-nitrides it is being increasingly recognised that proliferation-resistant thorium-nitride offers potential in next generation reactors due to a range of favourable physical properties4,5. It is thus important to study such materials, but these binary systems can be ill-defined and difficult to probe, and so there has been interest in studying well-defined molecular congeners6 where the nature of the chemical bonding and electronic structure of actinides can be probed7. This addresses one of the longstanding challenges of actinide chemistry, which is the study of covalency, since this could benefit recycling and clean up of nuclear fuels and wastes. Although binary materials are unlikely to contain much actinide-element multiple bonding, the study of discrete molecular linkages is important because they usually exhibit covalency8,9, and only by generating such linkages can the chemistry of those bonds be benchmarked in isolation, as well as perhaps generating molecular precursors that could be decomposed to binary systems.
The chemistry of uranium-ligand multiple bonds is now well developed10, but in comparison that of thorium is sparse. Apart from a few carbene11,12,13,14,15,16,17,18, pnictidene19,20,21,22,23,24,25,26,27,28,29,30,31,32,33 and chalcogenido34,35,36,37,38,39 complexes, thorium-ligand multiple bonding remains overall quite rare. A transient zero-valent thorium synthon produced a thorium-amide from dinitrogen40, but whether this forms via a nitride or a biomimetic-type sequence of separate protonation and reduction steps is unknown, and only a few elegant species such as ThN, F3ThN, NThN, Th(N)2Th, and NThO have been trapped under cryogenic matrix isolation conditions21,41,42. In contrast, molecular uranium-nitrides43 were first prepared under matrix isolation conditions42,44,45,46,47,48,49, then a range of bridging nitrides were isolated and characterised50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65, or inferred from the products of transient species66, and then terminal U≡N triply bonded species were finally secured67,68,69,70,71. During publication of this work a bridging thorium-nitride was reported72, but isolation of a terminal molecular Th≡N triple bond under normal experimental conditions remains an unmet challenge.
Recently, utilising the triamidoamine ligand {N(CH2CH2NSiMe2But)3}3− (TrenDMBS), we reported attempts to prepare bridging diactinide-nitrides73. Although diuranium-nitrides [{U(TrenDMBS)}2(μ-N)]0/1− in two different charge states could be prepared and isolated and were found to be robust, the corresponding dithorium-nitride [{Th(TrenDMBS)}2(μ-N)]1− proved elusive and bridging parent imido complexes, formed from C–H bond activation reactions, were the only isolable species. Given that these diactinide-nitride species derive from azide precursors and are produced in concerted reactivity the formation of any Th≡N triple bond seems quite unlikely. An interesting feature of these bridging [{An(TrenDMBS)}2(μ-N)]1− and terminal uranium-nitrides is that the σ-component of their An-N multiple bonds is higher in energy than the π-bonds, and this is also found for the uranyl dication but not bis(imido) analogues or other actinide-ligand multiple bonds more generally74,75. This phenomenon can be partly explained by anti-bonding interactions raising the energy of the σ-bond at short actinide-nitrogen distances. However, the bridging nitrides do not fit with that model. An alternative, more complete explanation may be the pushing-from-below phenomenon where the pseudo-core 6p-orbitals are engaged in repulsive interactions, principally with f-orbitals76. To date, pushing-from-below occurs with hard, charge-dense anions, such as O2− and N3−, but it is the preserve of high oxidation state uranium or actinyl complexes so whether it is a specific or more periodic in nature phenomenon has remained an open question. Thus, we focussed our attention on the more sterically demanding ligand {N(CH2CH2NSiPri3)3}3− (TrenTIPS) to extend our search for a thorium-nitride since this ligand is the only ligand so far to support isolable terminal nitride linkages at uranium.
Here, we report findings that suggest that reduction of a thorium-azide supported by TrenTIPS generates a transient Th≡N triple bond, but this is highly reactive and activates C–H bonds of aromatic solvents to give rare thorium parent imido complexes. However, we were able to effect nitride atom-transfer by incorporation into an organic amine heterocycle. Thus, we find that the Th≡N triple bond is inherently destabilised compared to U≡N triple bonds, which can be related to the greater covalency of uranium chemical bonding. The formation of parent imido complexes from a nitride with expulsion of alkyl during C–H activation presents some similarities to established actinide-nitride reactivity, but also introduces a contrasting mode of reactivity outcome since all prior related terminal uranium-nitride reactivity has resulted in 1,1-insertion to give an alkyl-amide or 1,2-addition to form an imido-alkyl combination. Calculations suggest that for the Th≡N bond the energy ordering of the Th–N bond components is σ > π, but for Th = NR it is generally σ < π with Th = NH potentially being either depending on the Th = NH geometry. This permits us to recognise that thorium as well as uranium might engage in pushing-from-below, suggesting that this phenomenon may play a wider and more common periodic role in actinide chemical bonding than previously thought.
Results
Synthesis and characterisation
Treatment of the thorium-chloride complex [Th(TrenTIPS)(Cl)] (1) with excess NaN3 affords the colourless thorium-azide complex [Th(TrenTIPS)(N3)] (2) in good (80%) isolated yield, Fig. 1a (see Supplementary Methods). The use of 1-15N-NaN3 gives the 15N-isotopologue (215N) where the 15N-label is distributed Th-15N14N14N:Th-14N14N15N in a 1:1 ratio. This can be observed in the ATR-IR data of 2 and 215N (see Supplementary Fig. 1), which exhibit azide asymmetric stretching absorptions at 2086 and 2082/2070 cm−1, respectively, which compare well to the azide stretch of 2086 cm−1 for [U(TrenTIPS)(N3)]70 and other thorium-azides generally77,78. All other characterisation data for 2 and 215N are unremarkable and support for the proposed formulations (see Supplementary Figs. 2–4).

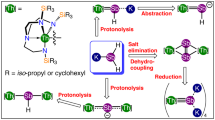
Synthesis of the compounds reported in this study. a Treatment of 1 with sodium azide produces the thorium-azide 2, which when reduced in toluene with potassium graphite gives the known thorium-benzyl 3. When the same reduction is conducted in benzene the thorium-imidos 4M are isolated, and these complexes can alternatively be prepared by deprotonation of the thorium-amide 5. Notably, when the deprotonation of 5 is conducted in toluene an imido/toluene-amide-benzyl equilibrium is established. Attempts to trap the thorium-nitride intermediate resulted in the isolation of the cyclic amine 7. b Treatment of the potassium-amide 8 with a thorium separated ion pair gives the thorium-methoxide 9 whereas use of a less sterically demanding thorium-iodide gives the isolable thorium-amide 10
Reduction of 2 with an excess of KC8 in toluene gives full consumption of 2 as evidenced by NMR and ATR-IR spectroscopies. However, surprisingly, after work-up the sole thorium-containing product that is repeatedly isolated is the known thorium-benzyl [Th(TrenTIPS)(CH2Ph)] (3) (45% crystalline yield)79, Fig. 1a. All characterisation, including a single-crystal cell check, are in accord with published data, and additionally the formation of KN3 was evidenced by the presence of an azide asymmetric stretch at 2032 cm−1 in the ATR-IR spectrum, which is identical to that found for an authentic sample of KN3.
In contrast, when 2 is reduced with an excess of KC8 in benzene, the dimeric thorium-parent-imido [{Th(TrenTIPS)(μ-NHK)}2] (4K) is isolated in 50% yield, Fig. 1a. Using 215N we also prepared the 50% labelled 15N-isotopologue 4K15N. Complex 4K is isolated in similar yield when the reaction is conducted under argon, which together with the 15N-labelling confirms azide as the source of the imido nitrogen in 4K. The N–H stretch could not be observed in the ATR-IR spectrum of 4K, presumably because its asymmetric stretching mode is coupled to other vibrational modes in the molecule, which is a common feature in heavy-atom structures including the uranium congeners [{U(TrenTIPS)(μ-NHM)}2] (M = Li, Na, K, Rb, Cs)80. However, we were able to identify the Th = N stretch of 4K at 575 cm−1, in perfect agreement with a computed value of 575 cm−1 from an analytical frequencies calculation, since in the corresponding spectrum of 4K15N the absorption at 575 cm−1 is reduced in intensity and a band at 565 cm−1 has grown in, consistent with the presence of a 50% 15N-label (see Supplementary Figs. 5 and 6). Once isolated, the dimeric nature of 4K renders it poorly soluble in non-polar solvents, and it decomposes in polar solvents, which has hampered its characterisation by NMR spectroscopy, but elemental analyses confirm the bulk formulation.
To establish the generality of the reaction that produces 4K, we treated 2 separately with RbC8 and CsC8, isolating the corresponding dimeric thorium-parent-imido complexes [{Th(TrenTIPS)(μ-NHM)}2] (M = Rb, 4Rb; Cs, 4Cs) in crystalline yields of 30 and 20%, respectively, Fig. 1a. Like 4K, 4Rb and 4Cs are poorly soluble once isolated which has prevented their full characterisation. In order to complete the 4M series we targeted the Li and Na analogues. Unfortunately, reductions with these metals are more difficult to control, and intractable product mixtures were obtained. However, using the new thorium-parent-amide [Th(TrenTIPS)(NH2)] (5) – which is only the third example of a structurally authenticated Th–NH2 bond40,72, prepared here by reaction of NH3 with the known cyclometallate [Th{N(CH2CH2NSiPri3)2(CH2CH2SiPri2CHMeCH2)}]79 (see Supplementary Figs. 7–9) – we find that 5 can be deprotonated with strong bases; treating 5 with ButLi or NaCH2Ph in benzene affords [{Th(TrenTIPS)(μ-NHM)}2] (M = Li, 4Li; Na, 4Na) in 56 and 38% isolated yields, respectively, Fig. 1a. Interestingly, if toluene is used as the solvent in the preparation of 4Li and 4Na, an equilibrium between 4M and 5/MCH2Ph is established, suggesting that the 4M series are capable of activating C–H bonds. Complexes 4Li and 4Na also suffer from low solubility once isolated, but in both cases the asymmetric imido N–H stretch with these lighter alkali metals can be observed in their respective ATR-IR spectra (4Li, 3393; 4Na, 3397 cm−1). With the deprotonation of 5 established for 4Li and 4Na, we find that 4K, 4Rb and 4Cs can also be prepared by deprotonation of 5 with MCH2Ph, but the yields are consistently low (av. 16%).
In related uranium chemistry, when [U(η5-C5Me5)2{(N(SiMe3)2}(N3)] is photolysed it produces the insertion product [U(η5-C5Me5)(η5:κ1-C5Me4CH2NH){N(SiMe3)2}] via a transient nitride66. On the other hand, reaction of [N3B(C6F5)3][NBun4] with [U{NButC6H3-3,5-Me2}3] produced [(C6F5)3BNU{NButC6H3-3,5-Me2}3], which can be formulated as either a borane-capped nitride or an imido-borate56. Therefore, we treated 2 with B(C6F5)3 in hexane and isolated [Th(TrenTIPS)(μ-N3)B(C6F5)3] (6) in 62% isolated yield, Fig. 1a. The characterisation data for 6 (see Supplementary Figs. 10–14) support its formulation, with a diagnostic azide stretch at 2171 cm−1 observed in its ATR-IR spectrum, which compares very well to an azide stretch of 2178 cm−1 reported for [U(η5-C5Me5)2{(N(SiMe3)2}(μ-N3)B(C6F5)3]66. However, 6 does not liberate N2 when reduced by KC8, with or without crown ethers or 2,2,2-cryptand, and instead borane-centred decomposition occurs, presumably involving fluoride extraction which is common for fluorinated-boranes. Treating 2 with excess KC8 in the presence of B(C6F5)3 to generate a nitride and then trap it results in the same outcome. Further, complex 6 does not react under photolytic conditions (125 W Hg-lamp) – which efficiently converts [U(TrenTIPS)(N)] to the nitride-insertion product [U{N(CH2CH2NSiPri3)2(CH2CH2SiPri2CMe2NH)}]68 – most likely reflecting the fact that 6 does not contain any localised metal valence electrons to transfer to the azide π*-orbitals. Seeking to provide an electron source, we photolysed 6 in the presence of KC8 but again borane-centred decomposition occurred. Thus, the borane capping would seem to deactivate the azide linkage in 2, as it does also for [U(η5-C5Me5)2{(N(SiMe3)2}(μ-N3)B(C6F5)3]66.
The isolation of the 4M series suggests that [Th(TrenTIPS)(NM2)] is initially formed, which is credible given the prior synthesis of the uranium-arsenido complex [{U(TrenTIPS)(AsK2)}4]81. For [{U(TrenTIPS)(AsK2)}4], abstraction of K ions results in K–H exchange to form arsinidiide, U = AsHK, linkages, presumably because the K ions perform a key, charge-stabilising role. It can be surmised that the same process occurs with [Th(TrenTIPS)(NM2)], since Th is generally expected to bond more ionically than uranium giving a more polar, charge-rich and reactive nitride, which then spontaneously exchanges K for H to give the imido complexes 4M. We therefore sought to trap [Th(TrenTIPS)(NK2)] using (ClSiMe2CH2)2, to eliminate KCl and produce [Th(TrenTIPS){N(SiMe2CH2)2}]. However, stepwise or one-pot reduction of 2 and treatment with (ClSiMe2CH2)2 resulted in the formation of 1 and HN(SiMe2CH2)2 (7), Fig. 1a, both identified by comparison of reaction mixture NMR data with characterisation data on authentic pure samples (see Supplementary Figs. 15 and 16). Nonetheless, the formation of 7 suggests that [Th(TrenTIPS)(NK2)] forms and then converts to [Th(TrenTIPS){N(SiMe2CH2)2}], but then the latter decomposes effecting, overall, nitride atom-transfer into an organic heterocycle. We suggest that the bulky TrenTIPS ligand would sterically clash with the four silyl methyl groups of the heterocycle in the putative [Th(TrenTIPS){N(SiMe2CH2)2}] excessively straining and thence rupturing the Th–Nheterocycle bond. Supporting evidence for this is provided by the observation that the known cyclometallate [Th{N(CH2CH2NSiPri3)2(CH2CH2SiPri2CHMeCH2)}]79 does not react with HN(SiMe2CH2)2, presumably on steric grounds given NH3 reacts with the former to give 5. Further, we prepared the potassium salt [{KN(SiMe2CH2)2}2] (8) (see Supplementary Figs. 17–19) and reacted it with [Th(TrenTIPS)(DME)][BPh4]31, which has proven itself an excellent precursor to forming thorium-group 15 element bonds. However, the only thorium-containing product that could be isolated was the methoxide [Th(TrenTIPS)(OMe)] (9) (see Supplementary Figs. 20–22), Fig. 1b; this indirectly suggests, though not definitively, that [Th(TrenTIPS){N(SiMe2CH2)2}] may form but be so reactive as to cleave the DME by-product. Lastly, we find that reaction of 8 with the sterically less encumbered [Th(TrenDMBS)I] results in the target amide complex [Th(TrenDMBS){N(SiMe2CH2)2}] (10) being isolated in 76% yield (see Suppupplementary Figs. 23–25), Fig. 1b, so there is nothing inherently unstable about the Th–Nheterocycle linkage in a Tren-ligand environment so long as the Tren ligand is not too sterically demanding. We therefore conclude that reduction of 2 generates the thorium-nitride [Th(TrenTIPS)(NK2)], but this either degrades to the parent imidos 4M, or can be converted to the sterically overloaded amide [Th(TrenTIPS){N(SiMe2CH2)2}], but this in turn decomposes to 7.
Solid state structures
We determined the solid state structures of 2, 4M, 5, 6, and 8–10 and 4K (see Supplementary Figs. 26–36), and 4K, 5, 6 and 10 are shown in Fig. 2. All of the thorium complexes contain trigonal bipyramidal thorium ions with a single functional group linkage in the pocket defined by the TrenTIPS ligand. For 2, the Th–Nazide distance of 2.365(3) Å is similar to the three Th–Namide distances (range: 2.301(3)–2.317(3) Å) and the Th–Namine distance is as expected longer at 2.673(3) Å, which for the Tren-related distances are similar to those observed for Th-TrenTIPS complexes with P- and As-donor ligand sets31,32. The Nα-Nβ and Nβ-Nγ distances of 1.200(4) and 1.150(4) Å, respectively, are only marginally distinct, suggesting a slight prevalence of the N≡N+–N2− resonance form of azide over the −N = N+ = N− form induced by coordination to the thorium ion, and hence only weak azide activation.

Molecular structures of 4K, 5, 6 and 10 with selective labelling. a the parent imido dimer 4K. b the parent amide 5. c the borane-capped azide 6. d the amide-heterocycle 10. The data for these complexes were collected at 150 K, displacement ellipsoids are presented at 40% probability and non-imido hydrogen atoms and minor disorder components are omitted for clarity
For the imido series 4M, as Group 1 is descended from Li to Cs the Th–Nimido distance becomes shorter overall, though after Li the heavier alkali metal distances are statistically indistinguishable (4Li, 2.209(2); 4Na, 2.158(5); 4K, 2.147(4); 4Rb, 2.149(6); 4Cs, 2.105(13) Å). This suggests that the donor capacity of the imido with respect to thorium is dictated in part by how charge attracting the alkali metal is, so with the most polarising metal Li the imido experiences the greatest charge depletion and is thus the poorest donor. A similar trend was observed for uranium analogues80. According to Pyykkö, the sum of the single and double bond covalent radii of thorium and nitrogen are 2.46 and 2.03 Å82, respectively; despite their bridging natures, the Th = N distances above are closer to the double than single bond values suggesting multiple bond interactions, and they compare favourably to terminal Th–Nimido distances that span the range 2.034–2.165 Å19,20,22,23,26,28,29,30. The strong donor nature of the imido units in 4M is also suggested by the Th–Namine bond lengths, which are now ~2.745 Å, some 0.07 Å longer than the analogous distance in 2. This most likely reflects a trans-influence in the Nimido–Th–Namine linkages, which are all ~175°, and thus close to optimally aligned for a trans-influence to exert itself. The Th–Namide distances in the 4M series now average 2.385 Å, nearly 0.09 Å longer than in 2, most likely reflecting the sterically congested nature of these dimers coupled to the presence of the strong imido donor ligands.
The parent amide character of 5 versus the imido nature of the 4M series is reflected by a Th–N distance of 2.290(4) Å in 5, which is 0.08–0.19 Å longer than the Th-imido distances, though the parent amide is clearly a stronger donor than the azide in 2 since the Th–Namine distance of 2.710(3) Å is between the Th–Namine distances for 2 and the 4M series.
Of all the complexes reported here, 6 reports perhaps the greatest structural changes. The coordination of the borane to azide clearly renders the azide an extremely poor donor as evidenced by the very long Th–Nazide distance of 2.551(6) Å. Likely in response to this, the Th–Namide (2.275(6)–2.286(5) Å) and Th–Namine (2.652(6) Å) distances are much shorter than the analogous distances in 2, the 4M series, and 5. The Nα-Nβ and Nβ-Nγ distances of 1.160(8) and 1.173(8) Å are now statistically indistinguishable from one another (c.f. 2) suggesting that the −N = N+ = N− resonance form now dominates in this bridging azide.
Lastly, the structure of 10 reveals a Th–Nheterocycle distance of 2.385(2) Å that is slightly longer than the other three Th–Namide distances (2.310(2)–2.335(2) Å). The Namine–Th–Nheterocycle angle is 165.68(7)°, distorted ~10° from the ~175° Namine–Th–Naxial-amide/-imido angle observed in 2, 4M, 5 and 9. Also, the N-heterocycle is coordinated in a skewed manner to the thorium ion, as evidenced by Si-Nheterocycle-Th angles of 113.77(10) and 141.23(11)°, with the remaining Si–Nheterocycle–Si angle of 104.42(11)° completing the trigonal planar geometry of the heterocycle amide. Taking these data together, along with the fact the TrenTIPS analogue has not been isolated, suggests that 10 is close to the limit of steric saturation and thence TrenTIPS would be beyond that limit.
Computational reaction profiling studies
The reaction of 2 with KC8 to give 3 has the appearance of KN3 elimination with a thorium(III) intermediate activating toluene, but it stands in contrast to the production of 4K when the reaction is carried out in benzene. The latter invokes a nitride intermediate, and since C–H bond activation occurs in both reactions we computed possible reaction mechanism profiles in simulated benzene or toluene solvent media at the DFT level (B3PW91) in order to ascertain whether distinct paths are followed or whether there is any commonality to these seemingly unrelated reaction outcomes.
The first step of the computed mechanism for the reaction in toluene, Fig. 3, involves the formation of a nitride through the KC8 mediated reduction. Adding the alkali metal leads to a low-lying transition state (TS) with an associated barrier of 1.3 kcal/mol that corresponds to the release of dinitrogen from 2. The involvement of potassium in this step is crucial. Indeed, when this same step is computed without potassium ions an extremely high energy barrier (85.6 kcal/mol) is found which clearly would impede nitride formation, and this is in-line with previous uranium-azide chemistry that promoted the elimination of N2 also with potassium as a reductant70 or by photolytic oxidation66. Moreover, potassium coordination not only changes the geometry of the coordinated azide ligand from linear to bent, needed to release N2, but also empty hybrid sp-orbitals of potassium are involved in the frontier orbitals of the TS, Fig. 4, which assist in stabilising charge accumulation within the azide unit as it is reduced.

Computed reaction profile for the reduction of 2 by potassium in toluene. This pathway accounts for the reduction of the azide to nitride then subsequent protonation from toluene solvent to give a parent imido which reacts further with toluene to give parent amide 5 along with benzyl 3 and potassium-azide in-line with experimental findings. The iso-propyl groups of the silyl substituents are omitted for clarity. In this reaction scheme potassium rather than potassium graphite was used to give a tractable calculation

Views of the potassium-azide stabilising interactions. These are both in the first transition state of Figs. 3 and 5 at 1.3 kcal/mol as the azide is activated and extrudes N2. These frontier orbitals alternatively highlight the interaction of the azide unit with vacant sp-hybrid orbitals of the two potassium ions following electron transfer from the latter into the π*-orbitals of the former. The iso-propyl groups of the silyl substituents are omitted for clarity
Following the reaction coordinate yields a thorium nitride capped by two K+ ions as the newly formed N2 molecule is expelled, with a global energy gain of 23.1 kcal/mol, Fig. 3. Natural Bond Order (NBO) analysis show the presence of a polarised Th≡N triple bond with one σ- and two π-bonding orbitals and a Wiberg Bond Index (WBI) of 1.56 (0.57 in the Th–N bond from the initial thorium-azide complex), indicative of a strongly polarised bond. The resulting nitride activates the C–H bond from toluene in an irreversible exothermic step with a 2-kcal/mol barrier. This step is best described as a proton transfer from the CH3 group of toluene to the nucleophilic nitride. Indeed, this reaction is an outer-sphere process indicating that only the nucleophilicity of the nitride is needed to achieve the process. Analogously, the resulting benzyl anion does not bind the metal centre, but instead extracts a potassium ion from the nitride, yielding an imido intermediate [Th(TrenTIPS)(NHK)]. NBO analysis of this imido complex indicates the presence of a Th = N double bond interaction and the WBI of 1.04 is in line with the results found for the formal polarised single and triple Th–N bonds. Interestingly, the imido complex can activate another C–H bond from a toluene molecule. This process involves a barrier of 22.6 kcal/mol that is ~20 kcal/mol higher than the first proton transfer. This is because the imido is less nucleophilic than the nitride, as evidenced by the geometry of the TS which resembles a classical σ-bond metathesis. In the same way, the final product of this step involves the coordination of the benzyl group to the metal centre rather than the extraction by the second potassium. This step is slightly endothermic by 6.8 kcal/mol and is in line with the experimental observation that the C–H bond activation of toluene is reversible in toluene, establishing an imido(4K)/toluene-amide(5)/benzyl equilibrium, when preparing 4K from benzyl potassium, for example. The system can evolve with another instance of the thorium-azide complex 2 that can extract the benzyl ligand to generate thorium-benzyl 3 and liberate the thorium-amide 5 and KN3.
Modelling the reaction with benzene instead of toluene gives similar results for the initial steps, Fig. 5, including a low activation barrier for the first C-H bond activation of 1.7 kcal/mol. However, the formation of KPh, whilst still exothermic, is not as favourable, with an energy gain of 11.1 kcal/mol as opposed to the 63.5 kcal/mol from the reaction with toluene. Mirroring the mechanism with toluene, a second C–H bond activation from another benzene molecule was studied, to form the thorium-amide 5, however, the intermediate, which retains the amide and phenyl ligands, is ~100 kcal/mol higher in energy, ruling out this reaction. Therefore, dimerisation of the imido intermediate [Th(TrenTIPS)(NHK)] is preferred, yielding the stabilised and experimentally obtained imido dimer 4K.

Computed reaction profile for the reduction of 2 by potassium in benzene. This pathway accounts for the reduction of the azide to nitride then subsequent protonation from benzene solvent to give a parent imido, which dimerises in-line with experimental findings. The iso-propyl groups of the silyl substituents are omitted for clarity. In this reaction scheme potassium rather than potassium graphite was used to give a tractable calculation
The formation of the imido series 4M partially mimics the known reactivity of terminal uranium-nitrides and a transient bridging dithorium-nitride. When uranium(VI)-nitrides react with C–H bonds, always in an intramolecular fashion with ancillary ligands to date, uranium(IV)-amides are formed where the nitride has undergone a 1,1-insertion with a C–H bond66,68. A transient uranium(IV)-nitride also activates ancillary-ligand C–H bonds, but instead of undergoing a 1,1-insertion a parent imido-alkyl is formed by 1,2-addition to avoid the formation, formally, of uranium(II)83. Our previous work on [{Th(TrenDMBS}2(μ-N)]1− showed that the transient nitride is converted to a bridging imido and either potassium alkyl is eliminated or intramolecular Tren-activation occurs depending on the availability of the potassium counter-ion73. The chemistry reported here is distinct to the prior uranium and thorium chemistry because although parent imido groups are formed the alkyl component is always eliminated with potassium; certainly thorium(II) is a very difficult oxidation state to access and together with the steric demands of TrenTIPS 1,1-insertion and 1,2-additions would seem to be disfavoured. This suggests that the transient thorium-nitride is a highly polar and thus reactive group, which is confirmed by the following computational studies, and the observed reactivity here complements but also contrasts to established modes of uranium-nitride reactivity introducing an alternative nitride reactivity outcome.
Computational modelling of the thorium-parent-imido and -nitride bonds
Since the reaction profile calculations suggest the presence of a common Th≡N triple bond intermediate and experimentally the imido series 4M is isolated we have carried out a detailed examination of the 4M complexes and the potassium-free dianion intermediate [Th(TrenTIPS)(N)]2− (11) using DFT calculations. In order for these calculations to be meaningfully consistent with our prior work these calculations were performed in the gas-phase at the BP86 TZP level with an all-electron basis set and ZORA. For completeness, we also provide a comparison to the hypothetical uranium(IV)-nitride [U(TrenTIPS)(N)]2− (12) along with previously reported [U(TrenTIPS)(N)]1− (13)67 and [U(TrenTIPS)(N)]0 (14)68 to enable meaningful benchmarking. Where experimental comparisons are possible, the geometry optimised structures of the computed complexes are in close agreement with the solid state structures, Table 1 and Supplementary Tables 1–38, so we conclude that these models provide qualitatively meaningful pictures of the electronic structure of these compounds.
For all complexes, the computed Mayer bond orders are ~0.7 and ~0.2 for the Th–Namide and Th–Namine linkages, respectively. In comparison, the Th–Nazide and Th–Namide bond orders of 0.55 and 0.54 for 2 and 5 confirm their formal single bond status. As expected, the Th–Nimido bond orders are substantially larger, at 0.87–1.29 reflecting their polarised multiple bond interactions. Notably, on moving from lithium to caesium the Th–Nimido bond order increases, consistent with the more polarising lithium depleting charge from the imido group as also suggested by the solid state Th–Nimido bond lengths. Even though 11 is a charge-rich dianion, a Th–Nnitride bond order of 2.88 suggests a Th≡N triple bond. Interestingly, the analogous bond order for 12 of 2.91 is only marginally larger than that of 11, and on moving from uranium(IV) (12) to (V) (13) to (VI) (14) the U–Nnitride bond order only modestly increases to 2.92 for the latter. The computed multipole derived (MDCq) charges for the metal and amide/imido/nitride centres are in-line with their formulations, though we note that the metal charges for 11 and 12 are higher than for their imido cousins, suggesting that the nitrides exhibit charge accumulation at the nitride centres, which for 11 is certainly consistent with the experimentally observed reactivity.
The Kohn Sham frontier molecular orbitals of these complexes (see Table 1, Supplementary Figs. 37–41, and Fig. 6) reveals several notable phenomena. As expected, 2 and 5 exhibit single covalent Th–Nazide/amide σ2-bonds that are supplemented by weak, dative π-interactions from the N-ligands. For 4M, 11 and 12 σ2-π2-π2-triple bond interactions are apparent; these are certainly polarised and not optimally aligned due to symmetry constraints for the 4M series, and although less polarised the triple bonds of 11 and 12 are clearly fairly N-centred consistent with the computed charges and the experimental reactivity. The frontier orbitals of 11 and 12 are visually strikingly similar to each other, and appear more polarised than 13 and 14 where more covalent interactions are present by virtue of the uranium(V) and (VI) ions, respectively, in those complexes.

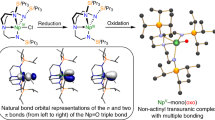
Frontier molecular orbitals of [M(TrenTIPS)(N)]2− (M = Th, 11; M = U, 12). a HOMO of 11 (221, 3.382 eV), b HOMO−1 of 11 (220, 2.757 eV), c HOMO−2 of 11 (219, 2.746 eV), d α-spin HOMO−2 of 12 (221a, 1.679 eV), e α-spin HOMO−3 of 12 (220a, 1.215 eV), f α-spin HOMO−4 of 12 (219a, 1.200 eV). The HOMO (223a, 3.921 eV) and HOMO−1 (222a, 3.824 eV) in the α-spin manifold of 12 are of essentially pure, non-bonding 5 f orbital single electron character. Hydrogen atoms in these all-electron DFT calculations are omitted from these visualisations for clarity
For computed 11 and 12 the An-N σ-bonds are clearly resolved and higher in energy than the two quasi-degenerate An-N π-bonding contributions. The situation is not as clear-cut for the 4M imidos because the deviation of the Th centres from the M2N2 rings leads to non-ideal bonding alignment, but in each case the HOMO approximates to a skewed Th–Nimido σ-bond with more clearly resolved Th–Nimido π-MOs lower in energy. The σ > π phenomenon is known for uranyl74,75,76, 13 and 1467,68, and [{An(TrenDMBS)}2(μ-N)]1− (An = U, Th)73, but it is not found in U = C, mono-U = O, and U = NR complexes generally, including closely related [U(TrenTIPS)(NH)][K(15C5)2]80. To ascertain whether 4M are outliers, we computed the electronic structures of [Th(TrenTIPS)(NR)]1− (R = H, 1-adamantyl, Ph, SiMe3) and in all instances the calculations return a π > σ energy ordering like uranium analogues and other thorium-imidos (see Supplementary Figs. 42–45). For a bond defined as being along a z-axis, a σ > π energy ordering is usually rationalised on the basis that at short U–E (E = N, O) distances the σ-orientated pz-orbital is at such close-approach that it experiences anti-bonding interactions with annular 5f- and 6d-orbital lobes thus raising it above the π-bonds. To date this has been the hallmark of high oxidation state uranium-element multiple bonds with high bond orders and short U-E distances. Whilst 11 and 12 contain high bond order Th-/U-N bonds, they are lower (IV) oxidation states than 13 and 14 and have longer nitride bond distances so would not be expected to have this σ > π energy ordering. It may be that the longer Th–N distances, compared to uranium analogues, may be off-set or even outweighed by the greater radial distribution of thorium 5f- and 6d-orbitals compared to uranium, on a like-for-like basis, due to a lower effective nuclear charge of the former compared to the latter.
An alternative explanation that would account for our data is that these systems are manifesting the pushing-from-below phenomenon where the pseudo-core occupied 6p-orbitals are engaged in repulsive, polarising interactions with occupied f-orbitals or hybrids with f-character74,75,76. However, this has previously been restricted to uranium, and indeed it has been pointed out that its occurrence for thorium has been previously dismissed because of the bent nature of ThO2 vs linear uranyl76. It is unrealistic to expect thorium 6p-orbital contributions to be localised in any particular molecular orbital due to the delocalised nature of molecular orbital theory and so this aspect of the calculations cannot be extracted directly, but these calculations use an all-electron basis set and so the 6p-electrons are explicitly treated in the calculations. However, in order to probe the involvement, or not, of 6p-orbitals in the observed σ > π energy ordering we performed two further sets of calculations (see Supplementary Figs. 46–49). Firstly, instead of using exclusively all-electron basis sets we geometry optimised 11 and 12 using a frozen core up to and including the 5d-level for the Th and U ions. This results in virtually no change to valence orbital energies (Δmax ≤ 0.01 eV), and thus the σ–π energy gaps of 11 and 12, and so this establishes that the frozen core has not perturbed the valence electronic structure. We then geometry optimised the structures of 11 and 12 with Th and U frozen cores up to and including the 6p-level. For both 11 and 12 this results in the metal-nitride distances lengthening (by 0.09 and 0.05 Å for 11 and 12, respectively), and thus the σ- and π-components of the An≡N triple bonds are destabilised and rise in energy as a group. However, the σ-components of the An≡N triple bonds are stabilised relative to the π-components, and for 11 the σ–π gap decreases from 0.621 to 0.439 eV and for 12 it decreases from 0.450 to 0.270 eV. To put this another way, taking the 6p-orbitals out of the frozen core and placing them in the valence region results in shorter An≡N triple bonds with destabilised σ-bonds relative to the π-components. These observations, consistent with prior work on {UO2}2+ and related systems74,75,76, suggest that pushing-from-below involving 6p-orbitals may be operating in 11 and 12. This may seem strange for 11 at first given the dominance of 6d-orbital character in the bonding of thorium, but in 11 thorium bonds with 5f-6d hybrids (see NBO discussion below). It is interesting to note that the reduction in the σ–π gaps for 11 and 12 are 71 and 60%, respectively. This suggests that there is slightly greater pushing-from-below for uranium than thorium, and indeed the ~0.18 eV σ–π energy gap changes of tetravalent 11 and 12 compare to changes of ~1 eV for the σu HOMO of hexavalent {UO2}2+ when the 6p-orbitals of uranyl are frozen. This would suggest that the 6p-5f energy gap for Th(IV) and U(IV) might be expected to be larger than that of U(VI), and so whilst still present the pushing-from-below phenomenon would be diminished with the former ions. Indeed, calculations on U(VI), U(V), U(IV), and Th(IV) ions in the gas-phase return 6p-5f energy gaps of 19.0, 21.1, 22.9, and 23.2 eV, respectively, where the 6p-5f energy gap is taken as the difference between the fully occupied β-spin 6p and fully unoccupied 5f-orbitals. Whilst 6p-5d interactions cannot be ruled out from contributing to pushing-from-below, we note that it is the 6p-5f interactions that are most pronounced on the basis that f-p overlap tends to be better than d-p overlap for early actinides, and certainly 12 has more 5f character in its An≡N bond than 11 which has more 6d character74. We note, however, that 6p-orbital energies would be raised in anions such as 11 and 12 perhaps favouring 6p-5f interactions. We also note that pushing-from-below tends to occur when hard, polarising anions are coordinated to An-metals, and certainly O2− and N3− are hard, charge-dense anions. PhN2−, Me3SiN2−, and 1-adamantyl-N2− can be considered to be softer donors than nitrides and oxides. For linear HN2− the presence of the H atom along the σ-axis would be expected to diminish the 6p-5f interaction that raises the σ-MO energy in the same way as is found for {H3P-N = U = N-PH3}4+74. However, this linear arrangement is not found for the 4M series consistent with the observations above. A similar argument applies for the variation of σ and π frontier orbital energies for uranyl vs bis(imido) complexes84. Looking more widely, we note that certain group 6 nitrides are computed to have σ > π energy ordering68, the thorium-oxo [Th(η5-C5H2But3)2(O)] appears to have a weak Th-O σ-amplitude in its HOMO with π-interactions lower in energy34, and indeed more broadly this is experimentally determined to be the case for N2 also. Interestingly, for molecules like N2 the 2s-2p energy gap is ~12 eV, whereas the 6p-5f gaps for the naked actinide ions are computed to be ~22 eV. Of course these 6p-5f energy gaps will change upon complexation, but it is likely that they are still larger for the actinides, which suggests that the respective orbital overlap efficiencies could be a contributing factor. When taken together, noting the above examples span the p-, d-, and f-blocks, this emerging picture suggests that there may be a σ/π ligand field strength and metal identity/oxidation state energy ordering phenomenon that is more periodic and commonly applicable than currently is appreciated.
From an energetic perspective, we note that 11 contains the highest-lying and thus most destabilised An≡N interactions of 11–14. The σ/2π energies (eV) for 11–14 are 3.382/2.757/2.746, 1.679/1.215/1.200, −1.261/−1.547/−1.552, and −5.173/−5.644/−5.668, respectively. Whilst the decreasing energies for 12–14 are as would be expected for the decreasing formal charge of these nitrides and the increasing oxidation state of uranium, since nitrides inherently stabilise high oxidation state metal ions, and it follows that they are ill-suited to mid- and low-oxidation state metals, it is notable that for 11 vs 12 the Th–Nnitride σ orbital is destabilised by ~1.7 eV compared to the analogous interaction for 12 and the π-bonds are destabilised by ~1.5 eV for 11 compared to 12. A similar situation was also found for [{Th(TrenDMBS)}2(μ-N)]1− vs [{U(TrenDMBS)}2(μ-N)]1− 74, though in that case the destabilisation of thorium compared to uranium was not so great. The significant destabilisation of the frontier orbitals of the Th–Nnitiride bond in 11 is in-line with the experimental paucity of terminal Th≡N triple bonds generally, noting that there are few examples even in matrix isolation experiments21,35,36, the observed C–H activation chemistry73, and the generally more covalent nature of uranium bonding compared to thorium.
NBO analysis further supports the polarised bonding picture for the Th–N bonds in 4M and 11, but confirms the multiple bond formulations of these complexes. The 4M imido complexes have highly polar Th = Nimido bonds with ~10% thorium contribution to the σ- and π-bonds, and, perhaps surprisingly considering the often found dominance of 6d over 5f orbital character of thorium bonding, the thorium contributions to these bonds are reasonably balanced with ~1:1 5f:6d contributions making up the vast (>97%) majority of these components with only modest (<3%) 7s and 7p intrusions. For 11, the thorium now plays a substantially larger (25%) role in the σ-bonding to the nitride, but, mirroring the visualised Kohn Sham orbitals, only 13% in the π-bonds suggesting that the latter are quite polar and N-centred. Interestingly, for the nitride 11 thorium 5f character now starts to dominate over 6d in the σ-component though they are balanced ~1:1 in the π-bonds. This contrasts to the situation for 12 where the anticipated greater covalency of uranium is reflected by greater uranium contributions to the σ- and π-bonds and within those contributions dominant 5f character of the bonding. As expected, as the uranium oxidation state continues to increase the U–Nnitride bonds become more covalent with greater uranium contributions.
DFT and NBO approaches are orbital-based, so to examine the bonding of these complexes further we used QTAIM since this probes the bonding topology, i.e. the electron density, at the bond critical point (BCP) which occurs when a bonding interaction is present85. For 4M, 11, and 12 An-Nimido and An-Nnitride 3,−1-BCPs were found in all cases. At a BCP the electron density, ρ(r) tends to be >0.2 for a covalent bond, and 0.1–0.2 for a polarised-covalent bond; a lower value tends to represent an ionic interaction. Thus, the BCP ρ(r) values for 2 and 5 are consistent with predominantly electrostatic bonds, whereas for the 4M series, they are polarised-covalent. The nitrides 11–14 are all >0.2 indicating covalent bonds in-line with their covalent triple bond formulations. The more covalent a bond is the more negative its electronic energy density term, H(r), should be at the BCP. Here, we find moderately negative values for 3 and 5, with slightly more negative values for 4M, but substantially negative values for the nitrides. Lastly, the bond ellipticity, ε(r), is definitive for determining whether bonds are single, double, or triple in nature, being zero or close to zero for single or triple bonds, which present symmetrical distributions of electron density when viewed down the inter-nuclear axis, or up to ~0.5 for double bonds which are asymmetric. The 4M series presents ε(r) values close to zero that are indicative of triple bonds but the deviation from zero reflects the fact they are not pure triple bonds being distorted by the fact the imido units in those complexes are bridging. Interestingly, the nitrides 11 and 12 deviate from zero, most probably reflecting the fact that whilst of triple bond parentage they are quite polarised and so not of pure triple bond character like the higher valent uranium complexes 13 and 14 which are clearly strong triple bonds.
Discussion
We have assembled evidence that reduction of a thorium-azide generates a transient thorium-nitride with a Th≡N triple bond, however this linkage is highly reactive and activates C–H bonds of aromatic solvents to give isolable parent thorium-imido complexes that are themselves capable of activating C–H bonds. When the thorium-azide is coordinated to a Lewis acidic borane its reduction chemistry is blocked. Attempts to trap the thorium-nitride intermediate by incorporating it into a thorium-N-heterocycle were unsuccessful, but instead due to steric overload the N-heterocycle is formed and liberated confirming nitride formation and atom-transfer. Calculations consistently suggest that the Th≡N triple bond is more reactive than uranium analogues, and the formation of parent imidos with concomitant expulsion of alkali metal alkyl complements existing uranium(IV/VI)-nitride reactivity but also contrasts by introducing an alternative nitride reactivity outcome. For all the computed Th(IV)/U(IV)/U(V)/U(VI)-nitrides the An-N σ-bonds are found to be higher in energy than the two quasi-degenerate An-N π-bonding contributions even though this has previously been recognised only with high oxidation state uranium-element multiple bonds with high bond orders and short U-E distances. Whilst this can be in part accounted for by anti-bonding interactions raising the energy of the σ-bond at short actinide-nitrogen distances, an alternative explanation that would account for our data is that these systems are manifesting the pushing-from-below phenomenon where the pseudo-core 6p-orbitals are engaged in repulsive interactions, principally with 5f-orbitals but also likely to some extent with 6d-orbitals. Whilst recognising that pushing-from-below involving 6p-orbitals is almost certainly not the only factor contributing to σ > π energy ordering the calculations suggest that the σ-components of our computed An≡N triple bonds are stabilised relative to the π-components when the 6p-orbitals are frozen. Pushing-from-below occurs with hard, charge-dense and polarising anions and O2− and N3− fit this description, but RN2− is softer, though HN2− may be a special cross-over case. Such arguments also account for the different energy ordering of σ and π bonds in uranyl (σ > π) vs bis(imido) complexes (π > σ). Taken together, now recognising that thorium as well as uranium might be subject to pushing-from-below suggests that there may be a σ/π ligand field and metal identity/oxidation state energy ordering phenomenon that is far more periodic and thus commonly applicable than currently is appreciated.
Methods
General
Experiments were carried out under a dry, oxygen-free dinitrogen atmosphere using Schlenk-line and glove-box techniques. All solvents and reagents were rigorously dried and deoxygenated before use. Compounds were characterised by single-crystal X-ray diffraction, multi-nuclear NMR spectroscopy, infra-red spectroscopy, and elemental analyses, and DFT, NBO, QTAIM, and reaction profile theoretical calculations. Further details are provided in the Supplementary Information.
Preparation of [Th(TrenTIPS)(N3)] (2)
Cold THF (100 ml, –78 °C) was added to solid 1 (8.80 g, 10.00 mmol) and NaN3 (1.30 g, 20.00 mmol). The resulting mixture was stirred at –78 °C for 1 h and then left to warm to room temperature and stirred for an additional 24 h. All volatiles were subsequently removed in vacuo. The remaining solids were extracted into hot hexanes (100 ml) and filtered. All volatiles were removed in vacuo, and the residue was washed with pentane (2 × 10 ml) and dried in vacuo to yield 2 as an analytically pure colourless solid (4.81 g). An additional 2.30 g of colourless crystals were obtained from the combined washings upon cooling to –30 °C. Yield: 7.11 g, 80%. Anal. Calcd for C33H75N7Si3Th: C, 44.72; H, 8.53; N, 11.06%. Found: C, 44.50; H, 9.03; N, 10.87%. 1H NMR (400 MHz, C6D6, 298 K): δ (ppm) 1.20–1.33 (m, 63 H, CH(CH3)2), 2.55 (t, 1 H, 3JHH = 4.9 Hz, 6 H, CH2CH2), 3.52 (t, 3JHH = 4.9 Hz, 6 H, CH2CH2). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ (ppm) 13.12 (CH(CH3)3), 19.48 (CH(CH3)3), 46.69 (CH2CH2), 62.89 (CH2CH2). 29Si{1H} NMR (79 MHz, C6D6, 298 K): δ (ppm) 4.15 (Si(CH(CH3)2)3). FTIR v/cm−1: 2938 (br, m), 2861 (br, m), 2086 (vs, N3−), 1462 (m), 1362 (s), 1269 (w), 1133 (m), 1060 (w), 1009 (w), 987 (s), 921 (s), 880 (w), 819 (vs), 736 (s), 673 (s), 631 (w), 548 (m), 514 (m), 450 (w), 423 (w). The 15N-isotopologue [Th(TrenTIPS)(15/14N14N14/15N)] was prepared analogously using Na(15N14N2). The asymmetric νazide stretching frequency for the 15N-isotopologue was measured at 2082 and 2070 cm–1 by FTIR.
Reduction of 2 with KC8 in Toluene
Toluene (30 ml) was added slowly to a pre-cooled (–78 °C) mixture of 2 (0.89 g, 1.00 mmol) and KC8 (0.41 g, 3.00 mmol). The resulting mixture was stirred at –78 °C for 1 h and then left to warm to room temperature with stirring for 24 h, during which time the formation of black graphite was observed. The suspension was filtered to remove graphite and unreacted KC8, affording a colourless solution. All volatiles were removed in vacuo, yielding 3 as an off-white solid, which was crystallised from toluene to yield 3 as a colourless crystalline solid. Yield: 0.42 g, 45%. All characterisation data matched those of an authentic sample79.
Preparation of [{Th(µ-NHLi)(TrenTIPS)}2] (4Li)
Benzene (40 ml) was added slowly to a pre-cooled (–78 °C) mixture of 5 (0.86 g, 1.00 mmol) and tBuLi (0.064 g, 1.00 mmol). The frozen mixture was left to thaw to room temperature and stirred for 2 h, during which time the formation of colourless precipitate was observed. Removal of all volatiles in vacuo yielded a white solid, which was washed with pentane (2 × 10 ml) and dried in vacuo to afford analytically pure 4Li. Yield: 0.48 g, 56%. Colourless crystals suitable for an X-ray diffraction study were obtained by cooling a saturated solution in hot benzene (80 °C) to room temperature. Anal. Calcd for C66H152Li2N10Si6Th2: C, 45.76; H, 8.84; N, 8.08%. Found: C, 46.05; H, 9.12; N, 7.52%. FTIR v/cm−1: 3393 (br, w, NH), 2937 (s), 2860 (s), 1460 (m), 1380 (w), 1275 (w), 1252 (w), 1136 (w), 1056 (s), 1009 (w), 927 (vs), 880 (s), 733 (vs), 669 (s), 628 (m), 574 (m), 539 (w), 508 (w), 476 (w). Once isolated as a pure sample, this compound is poorly soluble in non-polar solvents, and rapidly decomposes in the polar solvents THF and pyridine, and is therefore not amenable to NMR spectroscopic analysis.
Preparation of [{Th(µ-NHNa)(TrenTIPS)}2] (4Na)
Benzene (80 ml) was added slowly to a pre-cooled (–78 °C) mixture of 5 (0.86 g, 1.00 mmol) and NaCH2Ph (0.23 g, 2.00 mmol). The frozen mixture was left to thaw to room temperature and stirred for 24 h, forming an orange suspension. The mixture was heated to 80 °C and filtered, colourless crystals started to form upon cooling the filtrate, which was left to further crystallise at 10 °C for 15 h. After decanting the mother liquor, crystalline 4Na was dried in vacuo. Yield: 0.34 g, 38%. Anal. Calcd for C66H152N10Na2Si6Th2: C, 44.92; H, 8.68; N, 7.94%. Found: C, 43.84; H, 8.81; N, 7.79%. FTIR v/cm−1: 3397 (br, w, NH), 2939 (s), 2861 (s), 1461 (m), 1380 (w), 1275 (w), 1252 (w), 1128 (w), 1058 (m), 1010 (w), 928 (vs), 880(s), 736 (vs), 669 (s), 626(m), 562 (m), 539 (w), 508 (w), 438 (w). Once isolated as a pure sample, this compound is poorly soluble in non-polar solvents, and rapidly decomposes in the polar solvents THF and pyridine, and is therefore not amenable to NMR spectroscopic analysis.
Preparation of [{Th(TrenTIPS)(µ-NHK)}2] (4K)
Benzene (120 ml) was added slowly to a pre-cooled (–78 °C) mixture of 2 (1.77 g, 2.00 mmol) and KC8 (0.81 g, 6.00 mmol). The frozen mixture was left to thaw to room temperature and stirred for 24 h, during which time the formation of black graphite was observed. The mixture was then heated to 90 °C to dissolve the poorly soluble product, and filtered to remove insoluble graphite and unreacted KC8. Colourless crystals started to form upon cooling the clear filtrate, which was left to further crystallise at 10 °C for 15 h. After decanting the mother liquor, crystalline 4K was dried in vacuo. Yield: 0.90 g, 50%. Anal. Calcd for C66H152K2N10Si6Th2: C, 44.12; H, 8.53; N, 7.80%. Found: C, 44.32; H, 8.89; N, 7.48%. FTIR v/cm−1: 2936 (br, m), 2859 (s), 1458(m), 1378 (w), 1338 (w), 1275 (w), 1253 (w), 1137 (w), 1060 (w), 1023 (s), 1010 (w), 988 (w), 928 (vs), 880 (s), 809 (w), 735 (vs), 668 (s), 626 (m), 575 (w, Th = NH), 563 (w), 539 (w), 507 (w), 463 (w). The N–H stretching resonance was not observed in the IR spectrum (see Rb analogue). Once isolated as a pure sample, this compound is poorly soluble in non-polar solvents, and rapidly decomposes in the polar solvents THF and pyridine, and is therefore not amenable to NMR spectroscopic analysis. The 15N-isotopologue [{Th(TrenTIPS)(µ-15N[H]K)}2] (50 atom % 15N) was prepared analogously from [Th(TrenTIPS)(15/14N14N14/15N)]. The Th = NH asymmetric stretching frequency for [{Th(TrenTIPS)(µ-N[H]K)}2] was experimentally measured at 575 cm–1 by FTIR, which corresponds exactly to the calculated value (575 cm–1). For the 15N-isotopologue (50 atom% 15N), the Th = 14NH stretch at 575 cm–1 stretch was reduced in intensity, whilst an additional stretch was observed at 565 cm–1, corresponding to the isotopically shifted Th = 15NH stretch.
Preparation of [{Th(TrenTIPS)(µ-NHRb)}2] (4Rb)
Benzene (80 ml) was added slowly to a pre-cooled (–78 °C) mixture of 2 (0.89 g, 1.00 mmol) and KC8 (0.41 g, 3.00 mmol). The frozen mixture was left to thaw to room temperature and stirred for 48 h, during which time the formation of black graphite was observed. After filtration to remove graphite and unreacted RbC8, a yellow solution was obtained. All volatiles were removed in vacuo yielding a yellow solid, which was washed with pentane (2 × 10 ml) and dried in vacuo to afford analytically pure 4Rb as a colourless solid. Yield: 0.28 g, 30%. Colourless crystals suitable for an X-ray diffraction study were obtained by slow evaporation of a saturated solution in benzene. Anal. Calcd for C66H152N10Rb2Si6Th2: C, 41.95; H, 8.11; N, 7.41%. Found: C, 42.17; H, 8.44; N, 7.53%. FTIR v/cm−1: 2937 (s), 2860 (s), 1460 (m), 1400 (w), 1380 (w), 1275 (w), 1251 (w), 1128 (m), 1063 (w), 1010 (w), 989 (w), 930 (s), 879 (s), 809 (w), 736 (vs), 667 (s), 628 (w), 582 (w), 563 (w), 539(w), 507 (w), 459 (w). The N–H stretching resonance was not observed in the IR spectrum. Once isolated as a pure sample, this compound is poorly soluble in non-polar solvents, and rapidly decomposes in the polar solvents THF and pyridine, and is therefore not amenable to NMR spectroscopic analysis.
Preparation of [{Th(TrenTIPS)(µ-NHCs)}2] (4Cs)
Benzene (80 ml) was added slowly to a pre-cooled (–78 °C) mixture of 2 (0.89 g, 1.00 mmol) and KC8 (0.41 g, 3.00 mmol). The frozen mixture was left to thaw to room temperature and stirred for 48 h, during which time the formation of black graphite was observed. After filtration to remove graphite and unreacted CsC8, a yellow solution was obtained. All volatiles were removed in vacuo yielding a yellow solid, which was washed with pentane (2 × 10 ml) and dried in vacuo to afford analytically pure 4Cs as a colourless solid. Yield: 0.20 g, 20%. Colourless crystals suitable for an X-ray diffraction study were obtained by slow evaporation of a saturated solution in benzene. Anal. Calcd for C66H152N10Cs2Si6Th2: C, 39.95; H, 7.72; N, 7.06%. Found: C, 39.87; H, 7.85; N, 6.75%. FTIR v/cm−1: 2935 (s), 2858 (s), 1458 (m), 1386 (w), 1339 (w), 1275 (w), 1253 (w), 1138 (w), 1063 (s), 1024 (w), 1012 (w), 931 (vs), 880 (s), 738 (vs), 669 (s), 628 (m), 580 (s), 539 (w), 508 (w), 439 (w). The N–H stretching resonance was not observed in the IR spectrum. Once isolated as a pure sample, this compound is poorly soluble in non-polar solvents, and rapidly decomposes in the polar solvents THF and pyirdine, and is therefore not amenable to NMR spectroscopic analysis.
Preparation of [Th(TrenTIPS)(NH2)] (5)
A NH3 solution (50 ml, 20 mmol) in THF (0.4 M) was added into a pre-cooled (–78 °C) colourless suspension of [Thcyclomet(TrenTIPS)] (8.43 g, 10.00 mmol) in DME (50 ml), and then the mixture was allowed to warm to the room temperature and stirred for 48 h. All volatiles were removed in vacuo yielding a colourless solid, which was extracted with hexanes (80 ml) and filtered. Removal of the volatiles in vacuo afforded a colourless solid, which was then washed with cold pentane (–78 °C, 2 × 10 ml) and dried. Yield: 6.62 g, 77%. Colourless crystals of 5 suitable for an X-ray diffraction study were obtained by slow evaporation of a saturated solution in pentane in the glove box. Anal. Calcd for C33H77N5Si3Th: C, 46.07; H, 9.02; N, 8.14%. Found: C, 45.64; H, 9.25; N, 7.92%. 1H NMR (400 MHz, C6D6, 298 K): δ (ppm) 1.26 (d, 3JHH = 7.1 Hz, 54 H, CH(CH3)2), 1.33–1.43 (m, 9 H, CH(CH3)2), 2.58 (t, 3JHH = 4.7 Hz, 6 H, CH2CH2), 3.21 (br, s, 2 H, NH2), 3.56 (t, 3JHH = 4.7 Hz, 6 H, CH2CH2). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ (ppm) 13.00 (CH(CH3)3), 19.43 (CH(CH3)3), 45.63 (CH2CH2), 63.06 (CH2CH2). 29Si{1H} NMR (79 MHz, C6D6, 298 K): δ (ppm) 3.44. FTIR v/cm−1: 2939 (br, s), 2862 (m), 1512 (w), 1461 (m), 1379 (w), 1341 (w), 1273 (w), 1136 (w), 1052 (s), 1010 (m), 925 (s), 880 (s), 817 (w), 737 (vs), 670 (s), 625 (m), 565 (w), 465 (w), 443 (w). The N–H stretching resonance was not observed in the IR spectrum.
Preparation of [Th(TrenTIPS)(N3(B(C6F5)3))] (6)
Hexanes (30 ml) were added slowly to a pre-cooled (–78 °C) mixture of 2 (0.89 g, 1.00 mmol) and B(C6F5)3 (0.52 g, 1.00 mmol). The resulting mixture was left to warm to room temperature and stirred for 24 h, during which time a white precipitate formed. The precipitate was isolated by filtration and washed with pentane (2 × 10 ml) and dried in vacuo to yield 6 as an analytically pure colourless solid. Yield: 0.87 g, 62%. Colourless crystals suitable for an X-ray diffraction study were obtained by cooling a concentrated toluene solution at –30 °C for 2 days. Anal. Calcd C51H75BF15N7Si3Th·(C7H8): C, 46.74; H, 5.61; N, 6.58%. Found: C, 46.34; H, 5.66; N, 6.61%. 1H NMR (400 MHz, C6D6, 298 K): δ (ppm) 0.99–1.07 (m, 63 H, CH(CH3)2), 2.48 (t, 3JHH = 4.9 Hz, 6 H, CH2CH2), 3.41 (t, 3JHH = 4.9 Hz, 6 H, CH2CH2). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ (ppm) 12.48 (CH(CH3)3), 18.93 (CH(CH3)3), 47.13 (CH2CH2), 62.07 (CH2CH2), 135.95 (Ar-C), 138.74 (Ar-C), 141.50 (Ar-C), 147.74 (Ar-C), 149.54 (Ar-C). 29Si{1H} NMR (79 MHz, C6D6, 298 K): δ (ppm) 4.86 (Si(CH(CH3)2)3). 11B{1H} NMR (128 MHz, C6D6, 298 K): δ (ppm) –5.79 (br, B(C6F5)3. 19F{1H} NMR (376 MHz, C6D6, 298 K): δ (ppm) –134.24 (dd, 3JFF = 23.8, 4JFF = 8.7 Hz, 6 F, o-F), –157.49 (t, 3JFF = 20.6 Hz, 3 F, p-F), –163.93 (td, 3JFF = 23.8, 4JFF = 8.7 Hz, 6 F, m-F). FTIR v/cm−1: 2946 (br, m), 2863 (br, m), 2171 (vs, N3−), 1643 (m), 1515 (s), 1458 (vs), 1382 (w), 1284 (m), 1104 (s), 1050 (w), 972 (w), 919 (s), 894 (s), 880 (s), 787 (w), 772 (w), 736 (vs), 673 (vs), 631 (w), 573 (w), 55(w).
Reduction of 2 with KC8 in the Presence of (ClSiMe2CH2)2
C6D6 (15 ml) was added slowly to a pre-cooled (–78 °C) mixture of 2 (0.27 g, 0.30 mmol), KC8 (0.12 g, 0.90 mmol), and (ClSiMe2CH2)2 (0.07 g, 0.30 mmol). The frozen mixture was left to thaw to room temperature and stirred for 24 h, during which time the formation of black graphite was observed. 1H NMR analysis of the filtered solution revealed the formation of 1, along with a small amount of HN(Si(Me)2CH2)2, identified by comparison to an authentic sample (see Supplementary Figs. 15 and 16). Due to the small quantity of HN(Si(Me)2CH2)2 produced, it was not possible to isolate this compound from the mixture.
Preparation of [{KN(SiMe2CH2)2}2] (8)
A solution of HN(SiMe2CH2)2 (0.64 g, 4.00 mmol) in THF (5 ml) was added slowly to a pre-cooled (–78 °C) suspension of PhCH2K (0.52 g, 4.00 mmol) in THF (10 ml). The resulting mixture was stirred at –78 °C for 1 h and then left to warm to room temperature and stirred for an additional 24 h. All volatiles were subsequently removed in vacuo. The remaining solids were extracted into toluene (15 ml) and filtered through celite® to afford a pale yellow solution. Toluene was then removed in vacuo, and the resulting off-white solid was washed with pentane (2 × 5 ml) and dried in vacuo to yield KN(Si(Me)2CH2)2 as a colourless solid. Yield: 0.40 g, 50%. Colourless crystals suitable for an X-ray diffraction study were obtained by cooling a saturated toluene solution at –30 °C. Anal. Calcd for C6H16KNSi2: C, 36.49; H, 8.17; N, 7.09%. Found: C, 36.67; H, 8.32; N, 6.89%. 1H NMR (400 MHz, C6D6, 298 K): δ (ppm) 0.00 (s, 12 H, Si(CH3)2), 1.04 (s, 4 H, CH2CH2). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ (ppm) 5.14 (Si(CH3)2), 12.75 (CH2CH2). 29Si{1H} NMR (79 MHz, C6D6, 298 K): δ (ppm) 1.01(Si(CH3)2). FTIR v/cm−1: 2933 (w), 2875 (br, w), 1577 (w), 1304 (w), 1232 (vs), 1184 (w), 1124 (w), 980 (vs), 877 (s), 832 (w), 807 (w), 758 (s), 650 (s), 519 (w), 422 (w).
Preparation of [Th(TrenTIPS)(OMe)] (9)
DME (30 ml) was added slowly to a pre-cooled (–78 °C) mixture of [Th(TrenTIPS)(DME)][BPh4] (1.25 g, 1.0 mmol) and KN(SiMe2CH2)2 (0.20 g, 1.0 mmol). The resulting mixture was stirred at –78 °C for 1 h and then left to warm to room temperature and stirred for 24 h, forming a colourless solution. All volatiles were removed in vacuo, and the residue was extracted with toluene (30 ml) and filtered, yielding a colourless solution. Then all volatiles were removed in vacuo again, and the oily residue was extracted with pentane (10 ml) and subsequently concentrated to 4 ml. Colourless crystals of 9 suitable for an X-ray diffraction study were obtained by cooling the concentrated pentane solution at –30 °C. Yield: 0.22 g, 24%. Anal. Calcd for C34H78N4OSi3Th·0.5(C5H12): C, 48.05; H, 9.39; N, 6.14%. Found: C, 48.28; H, 9.39; N, 6.18%. 1H NMR (400 MHz, C6D6, 298 K): δ (ppm) 1.26–1.27 (m, 63 H, CH(CH3)2), 2.61 (t, 1 H, 3JHH = 4.9 Hz, 6 H, CH2CH2), 3.56 (t, 3JHH = 4.9 Hz, 6 H, CH2CH2), 3.72 (s, 3 H, OCH3). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ (ppm) 12.73 (CH(CH3)3), 19.21 (CH(CH3)3), 45.58 (CH2CH2), 61.23 (OCH3), 62.58 (CH2CH2). 29Si{1H} NMR (79 MHz, C6D6, 298 K): δ (ppm) 3.10 (Si(CH(CH3)2)3). FTIR v/cm−1: 2938 (br, m), 2860 (br, m), 1462 (m), 1271 (w), 1104 (s), 1057 (m), 1008 (w), 925 (s), 880 (s), 739 (vs), 671 (s), 628 (m), 544 (w), 511 (w), 445 (w).
Preparation of [Th(TrenDMBS)(N(Si(Me)2CH2)2)] (10)
Toluene (15 ml) was added slowly to a pre-cooled (–78 °C) mixture of [Th(TrenDMBS)(I)] (0.42 g, 0.50 mmol) and KN(SiMe2CH2)2 (0.10 g, 0.50 mmol). The resulting mixture was stirred at –78 °C for 1 h and then left to warm to room temperature and stirred for 24 h, forming a suspension, which was filtered through celite® to remove the KI salt. All volatiles were removed in vacuo, and the residue was extracted with pentane (10 ml) and subsequently concentrated to 2 ml. Colourless crystals of 10 suitable for an X-ray diffraction study were obtained by cooling the concentrated pentane solution at –30 °C. Yield: 0.33 g, 76%. Anal. Calcd for C30H73N5Si5Th: C, 41.11; H, 8.40; N, 7.99%. Found: C, 41.65; H, 8.66; N, 7.94%. 1H NMR (400 MHz, C6D6, 298 K): δ (ppm) 0.37 (s, 18 H, Si((CH3)2(tBu)), 0.52 (s, 12 H, Si(CH3)3), 0.89 (s, 4 H, CH2CH2), 1.03 (s, 27 H, Si(Me2(C(CH3)3))), 2.57 (t, 3JHH = 5.1 Hz, 6 H, CH2CH2), 3.34 (t, 3JHH = 5.1 Hz, 6 H, CH2CH2). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ (ppm) –3.47 (Si((CH3)2(tBu)), 2.81 (Si((CH3)2), 11.09 (CH2CH2), 20.59 (SiMe2(C(CH3)3))), 27.83 (Si(Me2(C(CH3)3)), 46.51 (CH2CH2), 66.96 (CH2CH2). 29Si{1H} NMR (79 MHz, C6D6, 283 K): δ (ppm) 4.12 (SiMe2), –2.31 (SiMe2tBu). FTIR v/cm−1: 2951 (m), 2926 (m), 2882 (m), 2851 (m), 1467 (m), 1345 (w), 1247 (s), 1066 (s), 1025 (w), 924 (s), 875 (s), 801 (s), 771 (vs), 743 (vs), 712 (s), 654 (m), 566 (m), 548 (m), 443 (w).
Data availability
The X-ray crystallographic coordinates for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition nos. 1869060-1869070. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre (www.ccdc.cam.ac.uk/data_requst/cif). All other data can be obtained from the authors on request.
References
Streit, M. & Ingold, F. Nitrides as a nuclear fuel option. J. Eur. Ceram. Soc. 25, 2687–2692 (2005).
Chinthaka Silva, G. W. et al. Reaction sequence and kinetics of uranium nitride decomposition. Inorg. Chem. 48, 10635–10642 (2009).
Haber, F. Verfahren zur Herstellung von Ammoniak durch katalytische Vereinigung von Stickstoff und Wasserstoff, zweckmäßig unter hohem Druch. German patent DE229126 (1909).
Trianti, N., Su’ud, Z. & Riyana, E. S. Design study of thorium cycle based long life modular boiling water reactors. Ind. J. Phys. 22, 133–137 (2011).
Bagla, P. Thorium seen as nuclear’s new frontier. Science 350, 726–727 (2015).
Jones, M. B. & Gaunt, A. J. Recent developments in synthesis and structural chemistry of nonaqueous actinide complexes. Chem. Rev. 113, 1137–1198 (2013).
Liddle, S. T. The renaissance of non-aqueous uranium chemistry. Angew. Chem. Int. Ed. 54, 8604–8641 (2015).
Neidig, M. L., Clark, D. L. & Martin, R. L. Covalency in f-element complexes. Coord. Chem. Rev. 257, 394–406 (2013).
Kaltsoyannis, N. Does covalency increase or decrease across the actinide series? Implications for minor actinide partitioning. Inorg. Chem. 52, 3407–3413 (2013).
Hayton, T. W. Recent developments in actinide-ligand multiple bonding. Chem. Commun. 49, 2956–2973 (2013).
Ren, W., Deng, X., Zi, G. & Fang, D.-C. The Th=C double bond: an experimental and computational study of thorium poly-carbene complexes. Dalton Trans. 40, 9662–9664 (2011).
Ma, G., Ferguson, M. J. & Cavell, R. G. Actinide metals with multiple bonds to carbon: synthesis, characterization, and reactivity of U(IV) and Th(IV) bis(iminophosphorano)methandiide pincer carbene complexes. Inorg. Chem. 50, 6500–6508 (2011).
Lu, E., Lewis, W., Blake, A. J. & Liddle, S. T. The ketimide ligand is not just an inert spectator: heteroallene insertion reactivity of an actinide-ketimide linkage in a thorium carbene amide ketimide complex. Angew. Chem. Int. Ed. 53, 9356–9359 (2014).
Gregson, M. et al. Emergence of comparable covalency in isostructural cerium(IV) and uranium(IV)-carbon multiple bonds. Chem. Sci. 7, 3286–3297 (2016).
Gregson, M. et al. The inverse-trans-influence in tetravalent lanthanide and actinide bis(carbene) complexes. Nat. Commun. 8, 14137 (2017).
Smiles, D. E., Wu, G., Hrobárik, P. & Hayton, T. W. Synthesis, thermochemistry, bonding, and 13C NMR chemical shift analysis of a phosphorano-stabilized carbene of thorium. Organometallics 36, 4519–4524 (2017).
Rungthanaphatsophon, P. et al. Methane vs benzene elimination in the reactions of (C5Me5)2Th(CH3)2 with [CH3PPh3]X, X = Cl, Br, I, to form thorium-alkyl or thorium-carbene complexes. Angew. Chem. Int. Ed. 56, 12925–12929 (2017).
Rungthanaphatsophon, P., Huang, P. & Walensky, J. R. Phosphorano-stabilized carbene complexes with short thorium(IV) and uranium(IV)-carbon bonds. Organometallics 37, 1884–1891 (2018).
Haskel, A., Straub, T. & Eisen, M. S. Organoactinide-catalyzed intermolecular hydroamination of terminal alkynes. Organometallics 15, 3773–3775 (1996).
Straub, T. et al. Intermolecular hydroamination of terminal alkynes catalyzed by organoactinide complexes. Scope and mechanistic studies. Organometallics 20, 5017–5035 (2001).
Wang, X. & Andrews L. Infrared spectra and density functional theory calculations of triplet pnictinidene N÷ThF3, P÷ThF3 and As÷ThF3 molecules. Dalton Trans. 14, 9260–9265 (2009).
Ren, W., Zi, G., Fang, D.-C. & Walter, M. D. A base-free thorium-terminal-imido metallocene: synthesis, structure, and reactivity. Chem. Eur. J. 17, 12669–12682 (2011).
Ren, W., Zi, G. & Walter, M. D. Synthesis, structure, and reactivity of a thorium metallocene containing a 2,2’-bipyridyl ligand. Organometallics 31, 672–679 (2012).
Ren, W. et al. Experimental and computational studies on the reactivity of a terminal thorium imidometallocene towards organic azides and diazoalkanes. Angew. Chem. Int. Ed. 53, 11310–11314 (2014).
Ren, W. et al. Si-H addition followed by C-H bond activation induced by a terminal thorium imido metallocene: a combined experimental and computational study. Chem. Sci. 5, 3165–3172 (2014).
Bell, N., Maron, L. & Arnold, P. L. Thorium mono- and bis(imido) complexes made by reprotonation of cyclo-metalated amides. J. Am. Chem. Soc. 137, 10492–10495 (2015).
Zhou, E. et al. Small molecule activation mediated by a thorium terminal imido metallocene. Organometallics 34, 3637–3647 (2015).
Yang, P. et al. Experimental and computational studies on the formation of thorium-copper heterobimetallics. Chem. Eur. J. 22, 13845–13849 (2016).
Garner, M. E., Hohloch, S., Maron, L. & Arnold, J. A new supporting ligand in actinide chemistry leads to reactive bis(NHC)borate-supported thorium complexes. Organometallics 35, 2915–2922 (2016).
Zhang, C. et al. Reactivity of a Lewis base supported thorium terminal imido metallocene toward small organic molecules. Organometallics 36, 4525–4538 (2017).
Wildman, E. P., Balázs, G., Wooles, A. J., Scheer, M. & Liddle, S. T. Thorium-phosphorus triamidoamine complexes containing Th-P single- and multiple-bond interactions. Nat. Commun. 7, 12884 (2016).
Wildman, E. P., Balázs, G., Wooles, A. J., Scheer, M. & Liddle, S. T. Triamidoamine thorium-arsenic complexes with parent arsenide, arsinidiide and arsendio structural motifs. Nat. Commun. 8, 14769 (2017).
Vilanova, S. P., Alayoglu, P., Heidarian, M., Huang, P. & Walensky, J. R. Metal-ligand multiple bonding in thorium-phosphorus and -arsenic complexes. Chem. Eur. J. 23, 16748–16752 (2017).
Ren, W., Zi, G., Fang, D.-C. & Walter, M. D. Thorium oxo and sulfido metallocenes: synthesis, structure, reactivity, and computational studies. J. Am. Chem. Soc. 133, 13183–13196 (2011).
Gong, Y., Wang, X., Andrews, L., Schlöder, T. & Riedel, S. Infrared spectroscopic and theoretical investigations of the OUF2 and OThF2 molecules with triple oxo bond character. Inorg. Chem. 51, 6983–6991 (2012).
Wang, X., Andrews, L., Thanthiriwatte, K. S. & Dixon, D. A. Infrared spectra of H2ThS and H2US in noble gas matrixes: enhanced H-As-S covalent bonding. Inorg. Chem. 52, 10275–10285 (2013).
Smiles, D. E., Wu, G., Kaltsoyannis, N. & Hayton, T. W. Thorium-ligand multiple bonds via reductive deprotection of a trityl group. Chem. Sci. 6, 3891–3899 (2015).
Vent-Schmidt, T., Andrews, L., Thanthiriwatte, K. S., Dixon, D. A. & Riedel, S. Reaction of laser-ablated uranium and thorium atoms with H2Se: a rare example of selenium multiple bonding. Inorg. Chem. 54, 9761–9769 (2015).
Smiles, D. W., Wu, G., Hrobárik, P. & Hayton, T. W. Use of 77Se and 125Te NMR spectroscopy to probe covalency of the actinide-chalcogen bonding in [Th(En){N(SiMe3)2}3]− (E = Se, Te; n = 1, 2) and their oxo-uranium(VI) congeners. J. Am. Chem. Soc. 138, 814–825 (2016).
Korobkov, I., Gambarotta, S. & Yap, G. P. A. Amide from dinitrogen by in situ cleavage and partial hydrogenation promoted by a transient zero-valent thorium synthon. Angew. Chem. Ed. 42, 4958–4961 (2003).
Kushto, G. P., Souter, P. F. & Andrews, L. An infrared spectroscopic and quasirelativistic theoretical study of the coordination and activation of dinitrogen by thorium and uranium atoms. J. Chem. Phys. 108, 7121–7130 (1998).
Zhou, M. & Andrews, L. Infrared spectra and pseudopotential calculations for NUO+, NUO, and NThO in solid neon. J. Chem. Phys. 111, 11044–11049 (1999).
King, D. M. & Liddle, S. T. Progress in molecular uranium-nitride chemistry. Coord. Chem. Rev. 266-267, 2–15 (2014).
Green, D. W. & Reedy, G. T. The identification of UN in Ar matrices. J. Chem. Phys. 65, 2921–2922 (1976).
Hunt, R. D., Yustein, J. T. & Andrews, L. Matrix infrared spectra of NUN formed by the insertion of uranium atoms into molecular nitrogen. J. Chem. Phys. 98, 6070–6074 (1993).
Andrews, L., Wang, X., Lindh, R., Roos, B. O. & Marsden, C. J. Simple N≡UF3 and P≡UF3 molecules with triple bonds to uranium. Angew. Chem. Int. Ed. 47, 5366–5370 (2008).
Wang, X., Andrews, L., Vlaisavljevich, B. & Gagliardi, L. Combined triple and double bonds to uranium: the N≡U=N‒H uranimine nitride molecule prepared in solid argon. Inorg. Chem. 50, 3826–3831 (2011).
Heinemann, C. & Schwarz, H. NUO+, a new species isoelectronic to the uranyl dication UO2 2+. Chem. Eur. J. 1, 7–11 (1995).
Pyykkö, P., Li, J. & Runeberg, N. Quasirelativistic pseudopotential study of species isoelectronic to uranyl and the equatorial coordination of uranyl. J. Phys. Chem. 98, 4809–4813 (1994).
Korobkov, I., Gambarotta, S. & Yap, G. P. A. A highly reactive uranium complex supported by the calix[4]tetrapyrrole tetraanion affording dinitrogen cleavage, solvent deoxygenation, and polysilanol depolymerization. Angew. Chem. Int. Ed. 41, 3433–3436 (2002).
Evans, W. J., Kozimor, S. A. & Ziller, J. W. Molecular octa-uranium rings with alternating nitride and azide bridges. Science 309, 1835–1838 (2005).
Evans, W. J., Miller, K. A., Ziller, J. W. & Greaves, J. Analysis of uranium azide and nitride complexes by atmospheric pressure chemical ionization mass spectrometry. Inorg. Chem. 46, 8008–8018 (2007).
Nocton, G., Pécaut, J. & Mazzanti, M. A nitrido-centered uranium azido cluster obtained from a uranium azide. Angew. Chem. Int. Ed. 47, 3040–3042 (2008).
Fox, A. R., Arnold, P. L. & Cummins, C. C. Uranium-nitrogen multiple bonding: isostructural anionic, neutral, and cationic uranium nitride complexes featuring a linear U=N=U core. J. Am. Chem. Soc. 132, 3250–3251 (2010).
Fortier, S., Wu, G. & Hayton, T. W. Synthesis of a nitrido-substituted analogue of the uranyl ion, [N=U=O]+. J. Am. Chem. Soc. 132, 6888–6889 (2010).
Fox, A. R. & Cummins, C. C. Uranium-nitrogen multiple bonding: the case of a four-coordinate uranium(VI) nitridoborate complex. J. Am. Chem. Soc. 131, 5716–5717 (2009).
Todorova, T. K., Gagliardi, L., Walensky, J. R., Miller, K. A. & Evans, W. J. DFT and CASPT2 analysis of polymetallic uranium nitride and oxide complexes: how theory can help when X-ray analysis is inadequate. J. Am. Chem. Soc. 132, 12397–12403 (2010).
Maria, L., Santos, I. C., Sousa, V. R. & Marçalo, J. Uranium(III) redox chemistry assisted by a hemilabile bis(phenolate) cyclam ligand: uranium-nitrogen multiple bond formation comprising a trans-{RN=U(IV)=NR}2+ complex. Inorg. Chem. 54, 9115–9126 (2015).
Tsoureas, N., Kilpatrick, A. F. R., Inman, C. J. & Cloke, F. G. N. Steric control of redox events in organo-uranium chemistry: synthesis and characterisation of U(V) oxo and nitrido complexes. Chem. Sci. 7, 4624–4632 (2016).
Falcone, M., Chatelain, L. & Mazzanti, M. Nucleophilic reactivity of a nitride-bridged diuranium(IV) complex: CO2 and CS2 functionalization. Angew. Chem. Int. Ed. 55, 4074–4078 (2016).
Falcone, M., Kefalidis, C. E., Scopelliti, R., Maron, L. & Mazzanti, M. Facile CO cleavage by a multimetallic CsU2 nitride complex. Angew. Chem. Int. Ed. 55, 12290–12294 (2016).
Chatelain, L., Scopelliti, R. & Mazzanti, M. Synthesis and structure of nitride-bridged uranium(III) complexes. J. Am. Chem. Soc. 138, 1784–1787 (2016).
Falcone, M., Chatelain, L., Scopelliti, R., Zivkovic, I. & Mazzanti, M. Nitrogen reduction and functionalization by a multimetallic uranium nitride complex. Nature 547, 332–335 (2017).
Falcone, M., Poon, L. N., Tirani, F. F. & Mazzanti, M. Reversible dihydrogen activation and hydride transfer by a uranium nitride complex. Angew. Chem. Int. Ed. 57, 3697–3700 (2018).
Barluzzi, L., Chatelain, L., Fadaei-Tirani, F., Zivkovic, I. & Mazzanti, M. Facile N-functionalization and strong magnetic communication in a diuranium(V) bis-nitride complex. Chem. Sci. 10, 3543–3555 (2019).
Thomson, R. K. et al. Uranium azide photolysis results in C-H bond activation and provides evidence for a terminal uranium nitride. Nat. Chem. 2, 723–729 (2010).
King, D. M. et al. Synthesis and structure of a terminal uranium nitride complex. Science 337, 717–720 (2012).
King, D. M. et al. Isolation and characterisation of a uranium(VI)-nitride triple bond. Nat. Chem. 5, 482–488 (2013).
Cleaves, P. A. et al. Two-electron reductive carbonylation of terminal uranium(V) and uranium(VI) nitrides to cyanate by carbon monoxide. Angew. Chem. Int. Ed. 53, 10412–10415 (2014).
King, D. M. et al. Molecular and electronic structure of terminal and alkali metal-capped uranium(V)-nitride complexes. Nat. Commun. 7, 13773 (2016).
Cleaves, P. A. et al. Terminal uranium(V/VI)-nitride activation of carbon dioxide and carbon disulphide: factors governing diverse and well-defined cleavage and redox reactions. Chem. Eur. J. 23, 2950–2959 (2017).
Staun, S. L., Sergentu, D.-C., Wu, G., Autschbach, J. & Hayton, T. W. Use of 15N NMR spectroscopy to probe covalency in a thorium nitride. Chem. Sci. 10, 6431–6436 (2019).
Du, J. et al. Thorium- and uranium-azide reductions: a transient dithorium-nitride versus isolable diuranium-nitrides. Chem. Sci. 10, 3738–3745 (2019).
Kaltsoyannis, N. Computational study of analogues of the uranyl ion containing the –N=U=N– Unit: density functional theory calculations on UO2 2+, UON+, UN2, UO(NPH3)3+, U(NPH3)2 4+, [UCl4{NPR3}2] (R = H, Me), and [UOCl4{NP(C6H5)3}]−. Inorg. Chem. 39, 6009–6017 (2000).
Kaltsoyannis, N. Recent developments in computational actinide chemistry. Chem. Soc. Rev. 32, 9–16 (2003).
Pepper, M. & Bursten, B. E. The electronic structure of actinide-containing molecules: a challenge to applied quantum chemistry. Chem. Rev. 91, 719–741 (1991).
Herve, A., Garin, N., Thuery, P., Ephritikhine, M. & Berthet, J.-C. Bent thorocene complexes with the cyanide, azide and hyride ligands. Chem. Commun. 49, 6304–6306 (2013).
Monreal, M. J. et al. New twists and turns for actinide chemistry: organometallic infinite coordination polymers of thorium diazide. Angew. Chem. Int. Ed. 55, 3631–3636 (2016).
Gardner, B. M. et al. The role of 5f-orbital participation in unexpected inversion of the σ-bond metathesis reactivity trend of triamidoamine thorium(IV) and uranium(IV) alkyls. Chem. Sci. 5, 2489–2497 (2014).
King, D. M. et al. Synthesis and characterization of an f-block terminal parent imido [U=NH] complex: a masked uranium(IV)-nitride. J. Am. Chem. Soc. 136, 5619–5622 (2014).
Gardner, B. M. et al. Triamidoamine uranium(IV)-arsenic complexes containing one-, two-, and three-fold U-As bonding interactions. Nat. Chem. 7, 582–590 (2015).
Pyykkö, P. Additive covalent radii for single-, double-, and triple-bonded molecules and tetrahedrally bonded crystals: a summary. J. Phys. Chem. A 119, 2326–2337 (2015).
Mullane, K. C. et al. C-H bond addition across a transient uranium nitride and formation of a parent uranium imido complex. J. Am. Chem. Soc. 140, 11335–11340 (2018).
Hayton, T. W. et al. Synthesis of imido analogs of the uranyl ion. Science 310, 1941–1943 (2005).
Bader, R. F. W., Slee, T. S., Cremer, D. & Kraka, E. Description of conjugation and hyperconjugation in terms of electron distributions. J. Am. Chem. Soc. 105, 5061–5068 (1983).
Acknowledgements
We gratefully acknowledge funding and support from the UK Engineering and Physical Sciences Research Council (grants EP/K024000/1, EP/M027015/1, and EP/P001386/1), European Research Council (grant CoG612724), Royal Society (grant UF110005), Humboldt Foundation, CalMip, The University of Manchester, and COST Action CM1006.
Author information
Authors and Affiliations
Contributions
J.D. and E.P.W. synthesised and characterised the compounds. C.A-L. computed the reaction profiles. A.J.W. carried out the single-crystal X-ray diffraction work. L.M. supervised the reaction profile calculations and interpreted the results. S.T.L. originated the central idea, supervised the work, analysed all the data, and wrote the manuscript with contributions from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, J., Alvarez-Lamsfus, C., Wildman, E.P. et al. Thorium-nitrogen multiple bonds provide evidence for pushing-from-below for early actinides. Nat Commun 10, 4203 (2019). https://doi.org/10.1038/s41467-019-12206-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-12206-5
This article is cited by
-
Actinide inverse trans influence versus cooperative pushing from below and multi-center bonding
Nature Communications (2023)
-
A terminal neptunium(V)–mono(oxo) complex
Nature Chemistry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
