
Abstract
The asymmetric cross-coupling reaction is developed as a straightforward strategy toward 1,1-diaryl alkanes, which are a key skeleton in a series of natural products and bioactive molecules in recent years. Here we report an enantioselective benzylic C(sp3)−H bond arylation via photoredox/nickel dual catalysis. Sterically hindered chiral biimidazoline ligands are designed for this asymmetric cross-coupling reaction. Readily available alkyl benzenes and aryl bromides with various functional groups tolerance can be easily and directly transferred to useful chiral 1,1-diaryl alkanes including pharmaceutical intermediates and bioactive molecules. This reaction proceeds smoothly under mild conditions without the use of external redox reagents.
Similar content being viewed by others

Introduction
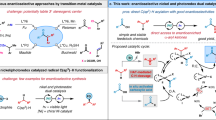
Enantioenriched 1,1-diaryl alkanes are a key skeleton in a series of natural products and bio-active molecules, such as sertraline1, tolterodine2,3, podophyllotoxins4, etc5,6,7,8. Due to the broad application of 1,1-diaryl alkanes in pharmaceutical industry, their asymmetric synthesis has attracted intensive interests in organic chemistry community and multiple strategies have been developed9,10,11,12,13,14,15,16,17. As a highly efficient and direct methodology for generating stereogenic centers in target molecules, transition-metal-catalyzed enantioselective cross-coupling reactions of electrophiles with organometallic reagents have been developed by Fu and colleagues18, and Molander and colleagues19,20 to furnish 1,1-diaryl alkanes using chiral bioxazolines (BiOX) as ligands. In addition, stereospecific cross-coupling reactions could also deliver this class of compounds21,22,23,24,25. Recently, nickel-catalyzed asymmetric reductive cross-coupling strategies of racemic benzylic electrophiles with aryl halides were reported by Weix and colleagues26, Reisman and colleagues27, Sigman and Doyle28 to provide an alternative strategy using chiral BiOXs as ligands and stoichiometric reductive transition metals (Fig. 1a). Compared with the well-established methologies with alkenes or electrophiles, using alkane as a substrate, the direct C–H arylation is considered a preferable step- and atom-economic method for the construction of C(sp3)–C(sp2) bonds29,30,31,32,33,34,35,36,37,38. During the preparation of this manuscript, the Cu/BOX-catalyzed radical relay strategy was used by Liu and colleagues39 to realize an elegant enantioselective arylation of C–H bonds on a methylene group adjacent to a naphthalene moiety40. By the merge of photocatalysis and transition-metal catalysis20,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56, the milestone of C–H arylation reactions via hydrogen atom transfer (HAT) process has been recently marked by Molander and colleagues57, Shields and Doyle58, MacMillan and colleagues59, and Martin and colleagues60 (Fig. 1b) to provide an alternative for the direct construction of 1,1-diaryl alkanes with readily available starting materials in a mild reaction condition. However, by lack of development of ligands able to differentiate between competing diastereomeric transition states, asymmetric cross-coupling reaction via this photocatalytic HAT process is quite challenging. So far, the best enantioselectivity of C–H arylation via photoredox/nickel dual catalysis is 77:23 enantiometric ratio (er)60. Our researches focus on asymmetric earth-abundant transition metal catalysis via chiral ligand design61,62,63,64,65,66. It is noted that the oxazoline derivated chiral ligands (BOX or BiOX), which have been well established in the cross-coupling strategies toward 1,1-diaryl alkanes, performed unsatisfactorily in controlling enantioselectivity in the visible-light-induced C–H arylation methology60. Thus, an effective chiral ligand is to be discovered for the enantioselective construction of 1,1-diaryl alkanes under photoredox/nickel dual catalysis.

Strategies for nickel-catalyzed asymmetric arylation of benzylic position or C–H bond reactions. a Asymmetric reductive cross-coupling strategies toward 1,1-diaryl alkanes with BiOX ligands. b Achiral C(sp3)−H arylation via photo/nickel dual catalysis and example for the asymmetric form. c The enantioselective benzylic C(sp3)−H arylation based on designed biimidazoline ligand
Here we report the enantioselective benzylic C–H arylation of readily available alkyl benzene with commercially available aryl bromides by using our designed chiral biimidazoline (BiIM) ligand (Fig. 1c). In addition, this protocol is redox neutral without using any additional single-electron oxidant or reductant.
Results
Reaction optimization
At the beginning of our study, the reaction of ethyl benzene 1a with methyl 4-bromobenzoate 2a using iridium photocatalyst with bis(4-methoxyphenyl)methanone (DMBP) as a co-photocatalyst57 promoting the yield of 3aa (see Supplementary Table 3) under the irradiation of blue LEDs, and nickel dichloride-dimethoxyethane complex and chiral ligand as the cross-coupling catalyst in the presence of K2HPO4 as a base in a solution of dioxane/ethyl benzene was chosen as a model reaction (Table 1). Inspired by previous reports on nickel-catalyzed asymmetric cross-coupling reactions using chiral BiOX ligand, we were so excited to find that the chiral BiOX ligands LS1 could accelerate the reaction to deliver 3aa in 79% yield, however, with a moderate enantiomeric ratio (69:31 er) (Table 1, entry 1). The more electron-rich chiral BiIM ligands67 were then applied as an alternative for the improvement of enantioselectivity due to the easy modification of electronic and steric effects. The reaction using N-isopropyl protected N-iPrBiIM (LS2) as a ligand afforded 3aa in 33% yield and 62.5:37.5 er (Table 1, entry 2). To our delight, when the N-aryl BiIM ligand L1a was used as a ligand, the reaction afforded the products 3aa in 44% yield with 92.5:7.5 er (Table 1, entry 3). The steric hindrance and possible π–π effect of the phenyl group on nitrogen atom increased the inflexibility of the BiIM, which might improve the enantioselectivity65,66. The homocoupling product from ethylbenzene was also observed, which illustrated that the reaction might undergo radical pathway. After screening various substitution effects on BiIM ligands (Table 1, entries 4–7), the sterically hinder N-3-tBuPh-iPrBiIM ligand (L1e) was designed as the best ligand that delivered 3aa in 44% yield and 95:5 er. When the reaction time was extended to 34 h, the reaction afforded 3aa in 62% yield with 94.5:5.5 er, which was established as standard conditions A (Table 1, entry 8). The reaction using 4.0 equivalent of ethyl benzene for 96 h afforded 3aa in 62% yield with a slightly lower er (92.5:7.5), which was established as standard conditions B (Table 1, entry 9). Control experiments (see Supplementary Table 3, entries 1–3) indicated that the iridium photocatalyst, nickel complex, and light was essential. Reactions were demonstrated to occur successfully with a lower yield in the absence of DMBP. In addition, the mixed solvent of dioxane and ethylbenzene is proved effective by inhibiting homocoupling of ethylbenzne (see Supplementary Table 3, entry 7)
Substrate scope
With optimized conditions in hands, we explored the substrate scope of the reaction with both aryl bromides and alkanes. As shown in Fig. 2, under standard conditions, the visible-light-induced asymmetric C−H arylation of ethyl benzene underwent smoothly with various coupling partners. Aryl bromides with either electron-donating (3ab–3ad) or electron-withdrawing functional groups (3ae–3ag) at the para-position were suitable in this reaction, delivering the corresponding chiral 1,1-diaryl ethanes in 45–84% yields with 93:7 to 95.5:4.5 ers (3ab–3ag). The reaction of aryl bromides with meta-substituents such as methyl and isopropyl groups gave 3ah and 3ai in 82% and 87% yields with 95.5:4.5 and 94:6 er. It is worth noting that 1° or 3° benzylic C(sp3)−H bonds could be differentiated, as only secondary benzylic C(sp3)−H bonds were directly activated under these conditions68. Various functional groups, such as methoxyl, trifluoromethyl, cyano, ester, thio ether, aceto, hydroxy, and Boc-protected amino groups, were well tolerated (3aj–3aq). The polycyclic rings and heterocycles such as 2-naphenyl, 5-benzothiophyl, 5-benzofuranyl, 5-indyl, 6-quinolyl, and 1,3-benzodioxole substrates could be delivered to corresponding products (3ar–3aw) in 48–79% yields with up to 95.5:4.5 er. The coupling of ethyl benzene and 3,4-dimethyl benzyl bromide could give 3ax in 68% yield with 96:4 er.

Substrate scope. Standard conditions A: 1 (1.0 mL), 2 (0.2 mmol), Ir(dFCF3ppy)2(dtbbpy)Cl (2.2 mol%), NiCl2·DME (20 mol%), L1e (20 mol%), DMBP (25 mol%), and K2HPO4 (2.0 eq.) in dioxane (3 mL) under the irradiation of 8 W blue LEDs for 34 h. Standard conditions B: 1 (0.8 mmol), 2 (0.2 mmol), Ir(dFCF3ppy)2(dtbbpy)Cl (2.2 mol%), NiCl2·DME (20 mol%), L1e (20 mol%), DMBP (25 mol%), and K2HPO4 (2.0 eq.) in dioxane (3 mL) under the irradiation of 8 W blue LEDs for 96 h. Isolated yield, the er was determined by HPLC. aFor 48 h. bNMR yield using TMSPh as an internal standard
For the substituted benzenes, ethyl benzene with methoxyl, flouro, alkyl groups, and 2-ethylnaphthalene also serve as effective substrates in asymmetric benzylic C(sp3)−H arylation under standard conditions B to convert to 3ba–3ca in moderate yields with 86:14 to 89:11 ers. It should be noted that the chemoselective benzylic C–H arylation of the ethyl group rather than the isobutyl group on 1-ethyl-4-isobutylbenzene (1d) proceeded to deliver 3da in 35% yield and 94.5:5.5 er. The propyl and butyl benzenes were also used to give the corresponding arylation products (3fa, 3fd) in moderate yields with 90:10 er. The 1,2-diphenylethane, 1,3-diphenylpropane, and 1,4-diphenylbutane were mono-activated, providing 1,1,x (x = 2,3,4) triaryl alkanes in 42–46% yields with 92:8 to 94.5:5.5 ers. The asymmetric arylation of cyclic substrate 1j performed smoothly to afford 3ja in 37% yield with 90:10 er. Although low yields were observed in some cases under standard conditions B, the mass balances of alkyl benzenes were mostly quantitative.
The application of this protocol was also investigated by using readily available alkyl benzenes. A Menthol-derived substrate could be utilized to deliver 4 in 61% yield with good er. This strategy was also available in the synthesis of pharmaceutical active molecules such as compound 5, which was reported as a N1L protein (potent vaccinia and variola (smallpox) virulence factor) antagonists8.
Mechanistic studies
Several experiments were designed to figure out the reaction process. The observation of homocoupling byproduct is consistent with the existence of benzylic radical. The reaction of 1-(cyclopropylmethyl)-4-methoxybenzene 6 as a radical clock afforded 7 in 20% yield and 100% mass balance vs. 2a through a radical-ring-opening process followed by a irreversible capture by nickel species27, which strengthened the possibility on radical pathway (Fig. 3a). Yet, we cannot exclude the possibility of a β-carbon elimination pathway to afford the same product. The phenyl methyl ethyne 8 was used as a bromine atom-trapping agent under the standard conditions to afford a mixture of bromo-substituted alkenes 9 in 24% yield and 100% mass balance vs. 2a, which illustrated the existence of bromine free radical and aryl-nickel bromide species (Fig. 3b). The halide additive studies (Fig. 3c, also see Supplementary Table 3) with aryl chloride 2a-Cl or aryl iodide 2a-I could not afford 3aa under standard conditions A. 1.0 equivalent of KBr was added in the reaction of 2a-Cl to initiate nickel bromide by halide exchange affording 3aa with 58% yield. This is also an evidence for the bromine radical initiating HAT of benzylic C−H bond. The deuterium experiment (Fig. 3d) using a 1:1 mixture of 1e and D-1e was carried out and kinetic isotope effect (KIE) was 2.47, which indicated that H-atom abstraction might be the turnover limiting step. Kinetic experiments of ethylbenzene and 4-tBu-phenyl bromide (see Supplementary Figs. 145-147) illustrated zero order on the concentration of aryl bromide and first order on the concentration of ethyl benzene, which were alternative evidences of turnover-limiting HAT process.

Mechanistic studies. a Radical-clock experiments consistent with the existence of benzylic radicals. b Compound 8 was added in the absence of ethylbenzene 1a for trapping bromine-free radical under conditions B. c Halide additive studies. d Kinetic isotopic effect was evaluated with 1e and deuterate 1e as substrate, indicating HAT process might be the turnover-limiting step
Based on mechanistic studies (also see Supplementary Discussion section in Supplementary Information) and previously reported literatures56,57,58,59,60, the proposed mechanism was shown in Fig. 4. The in-situ generated Ni(0) complex A could undergo oxidative addition with aryl bromide to generate aryl Ni(II) bromide species B, which could undergo visible-light-induced single-electron oxidation to give aryl Ni(II) species C and active bromine atom.

Proposed mechanism. Visible light induced HAT by in-situ-generated bromine radical followed by the asymmetric cross-coupling to 1,1-diaryl alkanes
Simultaneously, the photoexcited iridium complex was reduced to iridium(II) species. Yet, we cannot rule out the mechanism of ET process in the initiation of bromine-free radical from nickel-aryl adduct. The HAT process occurred between bromine-free radical (BDE (Bond Dissociation Energies) of H−Br is 366 kJ/mol)69 and alkyl benzene (BDE of benzylic C−H bond of ethylbenzene is 357 kJ/mol)69 rather than between bromine and dioxane (BDE of the C−H bond of dioxane is 406 kJ/mol)70 using DMBP as co-catalyst to deliver benzylic radical which was trapped by aryl Ni(II) species C to afford Ni(III) complex D. The reductive elimination of Ni(III) complex D could afford the chiral 1,1-diaryl alkanes and produce Ni(I) complex E, which could undergo single-electron reduction by iridium(II) species to regenerate Ni(0) species A and photocatalyst iridium(III) complex.
Conclusion
A direct enantioselective benzylic C−H arylation under photoredox/nickel dual catalysis was reported with a broad substrate scope and good level of enantioselectivity. This protocol provides an effective method for the asymmetric synthesis of 1,1-diaryl alkanes with preferable step- and atom economy. In addition, this protocol is redox neutral without using any additional single-electron oxidant or reductant. Furthermore, this method could be applied for the synthesis of pharmaceutical molecules and modification of complex compounds. A primary mechanism was proposed based on the previously reported literatures and mechanistic studies. Further studies on enantioselective C−H functionalization with photocatalysis are undergoing in our laboratory.
Methods
Materials
For NMR spectra of compounds in this manuscript, see Supplementary Figs. 1–107. For HPLC spectra of compounds in this manuscript, see Supplementary Figures 108-144. For the optimization of reaction conditions, see Supplementary Tables 1, 2. For control experiments, see Supplementary Table 3. For kinetic experiments, see Supplementary Figs. 145–147 and Supplementary Tables 4, 5. For radical-clock experiment, bromine radical-trapping experiment, KIE experiment, and catalytic active species experiment, see Supplementary Figs. 148–155. For the experimental procedures and analytic data of compounds synthesized, see Supplementary Methods.
Standard conditions A for chiral 1,1-diaryl alkanes
To a 20 mL vial with a stir bar was added L1e (0.04 mmol), NiCl2•DME (0.04 mmol) and 1 mL of dioxane in a N2-filled glovebox. The reaction was stirred at 50 °C for 30 min before cooling to room temperature. Dioxane (2 mL), 1 (1 mL), benzyl bromide 2 (0.2 mmol), Ir(dFCF3ppy)2(dtbbpy)Cl (0.0044 mmol), DMBP (0.05 mmol), and K2HPO4 (0.4 mmol) was added consistently. The vial was sealed with a Teflon cap and then allowed to remove from the glovebox. The reaction was stirred at 600 r.p.m. under the irradiation of 8 W blue LEDs in a distance of 5 cm at room temperature (25 °C) for 34 h. The reaction was quenched by adding Et2O, filtered through a short pad of silica, and eluted with Et2O. The solution was concentrated under reduced pressure to afford the crude residue, which was purified by flash column chromatography.
Standard conditions B for chiral 1,1-diaryl alkanes
To a 20 mL vial with a stir bar was added L1e (0.04 mmol), NiCl2•DME (0.04 mmol), and 1 mL of dioxane in a N2-filled glovebox. The reaction was stirred at 50 °C for 30 min before cooled to room temperature. Dioxane (3 mL), 1 (0.8 mmol), benzyl bromide 2 (0.2 mmol), Ir(dFCF3ppy)2(dtbbpy)Cl (0.0044 mmol), DMBP (0.05 mmol), and K2HPO4 (0.4 mmol) was added consistently. The vial was sealed with a Teflon cap and then allowed to remove from the glovebox. The reaction was stirred at 600 r.p.m. under the irradiation of 8 W blue LEDs in a distance of 5 cm at room temperature (25 °C) for 96 h. The reaction was quenched by adding Et2O, filtered through a short pad of silica, and eluted with Et2O. The solution was concentrated under reduced pressure to afford the crude residue, which was purified by flash column chromatography.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information file, and from the corresponding authors upon reasonable request. The experimental procedures and characterization of all new compounds are provided in the Supplementary Information.
References
McRae, A. L. & Brady, K. T. Review of sertraline and its clinical applications in psychiatric disorders. Exp. Opin. Pharm. 2, 883–892 (2001).
Hills, C. J., Winter, S. A. & Balfour, J. A. Tolterodine. Drugs 55, 813–820 (1998).
Malhotra, B. et al. The design and development of fesoterodine as a prodrug of 5-hydroxymethyl tolterodine (5-HMT), the active metabolite of tolterodine. Curr. Med. Chem. 16, 4481–4489 (2009).
Abad, A. et al. Synthesis and antimitotic and tubulin interaction profiles of novel pinacol derivatives of podophyllotoxins. J. Med. Chem. 55, 6724–6737 (2012).
Silva, D. H. S. et al. Dihydrochalcones and flavonolignans from Iryanthera lancifolia. J. Nat. Prod. 62, 1475–1478 (1999).
Gu, X.-H. et al. Design, synthesis, and monoamine transporter binding site affinities of methoxy derivatives of indatraline. J. Med. Chem. 43, 4868–4876 (2000).
Schenk, S. Effects of GBR 12909, WIN 35,428 and indatraline on cocaine self-administration and cocaine seeking in rats. Psychopharmacology 160, 263–270 (2002).
Cheltsov, A. V. et al. Vaccinia virus virulence factor N1L is a novel promising target for antiviral therapeutic intervention. J. Med. Chem. 53, 3899–3906 (2010).
Cherney, A. H., Kadunce, N. T. & Reisman, S. E. Enantioselective and enantiospecific transition-metal-catalyzed cross-coupling reactions of organometallic reagents to construct C–C bonds. Chem. Rev. 115, 9587–9652 (2015).
Tollefson, E. J., Hanna, L. E. & Jarvo, E. R. Stereospecific nickel-catalyzed cross-coupling reactions of benzylic ethers and esters. Acc. Chem. Res. 48, 2344–2353 (2015).
Pathak, T. P. et al. Synthesis and preliminary biological studies of 3-substituted indoles accessed by a palladium-catalyzed enantioselective alkene difunctionalization reaction. J. Am. Chem. Soc. 132, 7870–7871 (2010).
Friis, S. D., Pirnot, M. T. & Buchwald, S. L. Asymmetric hydroarylation of vinylarenes using a synergistic combination of CuH and Pd catalysis. J. Am. Chem. Soc. 138, 8372–8375 (2016).
Chen, Y.-G. et al. Nickel-catalyzed enantioselective hydroarylation and hydroalkenylation of styrenes. J. Am. Chem. Soc. 141, 3395–3399 (2019).
Anthony, D. et al. Nickel-catalyzed asymmetric reductive diarylation of vinylarenes. Angew. Chem. Int. Ed. 58, 3198–3202 (2019).
Wang, Z. et al. Organocatalytic asymmetric synthesis of 1,1-diarylalkanes by transfer hydrogenation. J. Am. Chem. Soc. 137, 383–389 (2015).
Logan, K. M. & Brown, M. K. Catalytic enantioselective arylboration of alkenylarene. Angew. Chem. Int. Ed. 56, 851–855 (2017).
Rubial, B. et al. Enantiospecific synthesis of ortho-substituted 1,1-diarylalkanes by a 1,2-metalate rearrangement/anti-SN2′ elimination/rearomatizing allylic Suzuki-Miyaura reaction sequence. Angew. Chem. Int. Ed. 58, 1366–1370 (2019).
Do, H.-Q., Chandrashekar, E. R. R. & Fu, G. C. Nickel/Bis(oxazoline)-catalyzed asymmetric Negishi arylations of racemic secondary benzylic electrophiles to generate enantioenriched 1,1-diarylalkanes. J. Am. Chem. Soc. 135, 16288–16291 (2013).
Gutierrez, O. et al. Nickel-catalyzed cross-coupling of photoredox-generated radicals: uncovering a general manifold for stereoconvergence in nickel-catalyzed cross-couplings. J. Am. Chem. Soc. 137, 4896–4899 (2015).
Tellis, J. C. & Primer, D. N. & Molander, G. A. Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 345, 433–436 (2014).
Ohmura, T., Awano, T. & Suginome, M. Stereospecific Suzuki-Miyaura coupling of chiral α-(Acylamino)benzylboronic esters with inversion of configuration. J. Am. Chem. Soc. 132, 13191–13193 (2010).
Zhou, Q. et al. Nickel-catalyzed cross-couplings of benzylic privates with arylboroxines: stereospecific formation of diarylalkanes and triarylmethanes. J. Am. Chem. Soc. 135, 3307–3310 (2013).
Harris, M. R. et al. Retention or inversion in stereospecific nickel-catalyzed cross-coupling of benzylic carbamates with arylboronic esters: control of absolute stereochemistry with an achiral catalyst. J. Am. Chem. Soc. 135, 3303–3306 (2013).
Maity, P. et al. Nickel-catalyzed cross couplings of benzylic ammonium salts and boronic acids: stereospecific formation of diarylethanes via C−N bond activation. J. Am. Chem. Soc. 135, 280–285 (2013).
Yonova, I. M. et al. Stereospecific nickel-catalyzed cross-coupling reactions of alkyl Grignard reagents and identification of selective anti-breast-cancer agents. Angew. Chem. Int. Ed. 53, 2422–2427 (2014).
Ackerman, L. K. G. et al. Cobalt co-catalysis for cross-electrophile coupling: diarylmethanes from benzyl mesylates and aryl halides. Chem. Sci. 6, 1115–1119 (2015).
Poremba, K. E. et al. Nickel-catalyzed asymmetric reductive cross-coupling to access 1,1-diarylalkanes. J. Am. Chem. Soc. 139, 5684–5687 (2017).
Woods, B. P. et al. Nickel-catalyzed enantioselective reductive cross-coupling of styrenyl aziridines. J. Am. Chem. Soc. 139, 5688–5691 (2017).
Newton, C. G. et al. Catalytic enantioselective transformations involving C−H bond cleavage by transition-metal complexes. Chem. Rev. 117, 8908–8976 (2017).
Saint-Denis, T. G. et al. Enantioselective C(sp3)–H bond activation by chiral transition metal catalysts. Science 359, eaao4798 (2018).
Yan, S.-B., Zhang, S. & Duan, W.-L. Palladium-catalyzed asymmetric arylation of C(sp3)–H bonds of aliphatic amides: controlling enantioselectivity using chiral phosphoric amides/acids. Org. Lett. 17, 2458–2461 (2015).
Wang, H. et al. An enantioselective bidentate auxiliary directed palladium-catalyzed benzylic C–H arylation of amines using BINOL phosphate ligand. Angew. Chem. Int. Ed. 55, 15387–15391 (2016).
Wasa, M. et al. Ligand-enabled methylene C(sp3) –H bond activation with a Pd(II) catalyst. J. Am. Chem. Soc. 134, 18570–18572 (2012).
Chen, G. et al. Ligand-accelerated enantioselective methylene C(sp3)–H bond activation. Science 353, 1023–1027 (2016).
Zhang, F. L. et al. Functionalization of C(sp3)–H bonds using a transient directing group. Science 351, 252–256 (2016).
Kim, J. H. et al. Rh(I)/NHC*-catalyzed site- and enantioselective functionalization of C(sp3)–H bonds toward chiral triarylmethanes. ACS Catal. 6, 7652–7656 (2016).
Yan, S.-Y. et al. Pd(II)-catalyzed enantioselective arylation of unbiased methylene C(sp3)–H bonds enabled by 2-pyridinylisopropyl auxiliary and chiral phosphoric acids. Angew. Chem. Int. Ed. 57, 9093–9097 (2018).
Shi, H. et al. Enantioselective remote meta-C–H arylation and alkylation via a chiral transient mediator. Nature 558, 581–586 (2018).
Zhang, W. et al. Enantioselective arylation of benzylic C−H bonds via copper-catalyzed radical relay. Angew. Chem. Int. Ed. 58, 6425–6429 (2019) https://doi.org/10.1002/anie.201902191.
Vasilopoulos, A. & Zultanski, S. L. & Stahl, S. S. Feedstocks to pharmacophores: Cu-catalyzed oxidative arylation of inexpensive alkylarenes enabling direct access to diarylalkanes. J. Am. Chem. Soc. 139, 7705–7708 (2017).
Zuo, Z. et al. Merging photoredox with nickel catalysis: coupling of α-carboxyl sp3-carbons with aryl halides. Science 345, 437–440 (2014).
Brimioulle, R. et al. Enantioselective catalysis of photochemical reaction. Angew. Chem. Int. Ed. 54, 3872–3890 (2015).
Skubi, K. L., Blum, T. R. & Yoon, T. P. Dual catalysis strategies in photochemical synthesis. Chem. Rev. 116, 10035–10074 (2016).
Tellis, J. C. et al. Single-electron transmetalation via photoredox/nickel dual catalysis: unlocking a new paradigm for sp3−sp2 cross-coupling. Acc. Chem. Res. 49, 1429–1439 (2016).
Fabry, D. C. & Rueping, M. Merging visible light photoredox catalysis with metal catalyzed C–H activations: on the role of oxygen and superoxide ions as oxidants. Acc. Chem. Res. 49, 1969–1979 (2016).
Lu, Q. & Glorius, F. Radical enantioselective C(sp3)−H functionalization. Angew. Chem. Int. Ed. 56, 49–51 (2017).
Ackerman, L. K. G., Martinez Alvarado, J. I. & Doyle, A. G. Direct C−C bond formation from alkanes using Ni-photoredox catalysis. J. Am. Chem. Soc. 140, 14059–14063 (2018).
Perry, I. B. et al. Direct arylation of strong aliphatic C–H bonds. Nature 560, 70–75 (2018).
Twilton, J. et al. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 1, 52 (2017).
Zuo, Z. et al. Enantioselective decarboxylative arylation of α-amino acids via the merger of photoredox and nickel catalysis. J. Am. Chem. Soc. 138, 1832–1835 (2016).
Stache, E. E., Rovis, T. & Doyle, A. G. Dual nickel- and photoredox-catalyzed enantioselective desymmetrization of cyclic meso-anhydrides. Angew. Chem. Int. Ed. 56, 3679–3683 (2017).
Ding, W. et al. Bifunctional photocatalysts for enantioselective aerobic oxidation of β-ketoesters. J. Am. Chem. Soc. 139, 63–66 (2017).
Wang, D. et al. Enantioselective decarboxylative cyanation employing cooperative photoredox catalysis and copper catalysis. J. Am. Chem. Soc. 139, 15632–15635 (2017).
Sha, W. et al. Merging photoredox and copper catalysis: enantioselective radical cyanoalkylation of styrenes. ACS Catal. 8, 7489–7494 (2018).
Lu, F.-D. et al. Asymmetric propargylic radical cyanation enabled by dual organophotoredox and copper catalysis. J. Am. Chem. Soc. 14, 6167–6172 (2019) https://doi.org/10.1021/jacs.9b02338.
Zhang, H.-H., Zhao, J.-J. & Yu, S. Enantioselective allylic alkylation with 4-alkyl-1,4-dihydro-pyridines enabled by photoredox/palladium cocatalysis. J. Am. Chem. Soc. 140, 16914–16919 (2018).
Heitz, D. R., Tellis, J. C. & Molander, G. A. Photochemical nickel-catalyzed C−H arylation: synthetic scope and mechanistic investigations. J. Am. Chem. Soc. 138, 12715–12718 (2016).
Shields, B. J. & Doyle, A. G. Direct C(sp3)−H cross coupling enabled by catalytic generation of chlorine radicals. J. Am. Chem. Soc. 138, 12719–12722 (2016).
Shaw, M. H. et al. Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science 352, 1304–1308 (2016).
Shen, Y., Gu, Y. & Martin, R. sp3 C−H arylation and alkylation enabled by the synergy of triplet excited ketones and nickel catalysts. J. Am. Chem. Soc. 140, 12200–12209 (2018).
Chen, J. et al. Iron-catalyzed asymmetric hydrosilylation of 1,1-disubstituted alkenes. Angew. Chem. Int. Ed. 54, 4661–4664 (2015).
Guo, J., Shen, X. & Lu, Z. Regio- and enantioselective cobalt-catalyzed sequential hydrosilylation/hydrogenation of terminal alkynes. Angew. Chem. Int. Ed. 56, 615–618 (2017).
Cheng, B. et al. Highly enantioselective cobalt-catalyzed hydrosilylation of alkenes. J. Am. Chem. Soc. 139, 9439–9442 (2017).
Guo, J. et al. Cobalt-catalyzed asymmetric sequential hydroboration/hydrogenation of internal alkynes. J. Am. Chem. Soc. 139, 15316–15319 (2017).
Cheng, B., Liu, W. & Lu, Z. Iron-catalyzed highly enantioselective hydrosilylation of unactivated terminal alkenes. J. Am. Chem. Soc. 140, 5014–5017 (2018).
Chen, X. et al. Asymmetric remote C−H borylation of internal alkenes via alkene isomerization. Nat. Commun. 9, 3939 (2018).
Hao, X.-Q. et al. Biimidzoline ligands for palladium-catalyzed asymmetric allylic alkylation. Tetrahedron Asymmetry 26, 1360–1368 (2015).
Clark, J. R. et al. Manganese-catalysed benzylic C(sp3)–H amination for late-stage functionalization. Nat. Chem. 10, 583–591 (2018).
Luo, Y. -R. & Cheng, J. -P. Bond dissociation energies. In: CRC Handbook of Chemistry and Physics (Rumble, J. R., ed.) 99 edn (CRC Press/Taylor & Francis, Boca Raton, FL, 2018).
Kranenburg, M. et al. Carbon−oxygen bond dissociation enthalpies in peroxyl radicals. J. Phys. Chem. A 104, 915–921 (2000).
Acknowledgements
Financial support was provided by NSFC (21772171), National Basic Research Program of China (2015CB856600), Zhejiang Provincial Natural Science Foundation of China (LR19B020001), Zhejiang University K. P. Chao’s High Technology Development Foundation, ZJU-NHU United Research and Development Center, and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
X.C. and H.L. performed the experiments. X.C. prepared the Supplementary Information. Z.L. and X.C. designed the experiments. Z.L. and X.C. prepared the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Nature Communications thanks Daniel A. Di Rocco and other anonymous reviewer(s) for their contribution to the peer review of this work
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cheng, X., Lu, H. & Lu, Z. Enantioselective benzylic C–H arylation via photoredox and nickel dual catalysis. Nat Commun 10, 3549 (2019). https://doi.org/10.1038/s41467-019-11392-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-11392-6
This article is cited by
-
Ni-catalyzed enantioconvergent deoxygenative reductive cross-coupling of unactivated alkyl alcohols and aryl bromides
Nature Communications (2024)
-
Site- and enantioselective cross-coupling of saturated N-heterocycles with carboxylic acids by cooperative Ni/photoredox catalysis
Nature Communications (2023)
-
Modulating stereoselectivity in allylic C(sp3)-H bond arylations via nickel and photoredox catalysis
Nature Communications (2023)
-
Nickel/biimidazole-catalyzed electrochemical enantioselective reductive cross-coupling of aryl aziridines with aryl iodides
Nature Communications (2023)
-
Highly selective synthesis of all-carbon tetrasubstituted alkenes by deoxygenative alkenylation of carboxylic acids
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
