
Abstract
Age-related macular degeneration (AMD) is the most common cause of blindness among the elderly in the developed world. While treatment is effective for the neovascular or “wet” form of AMD, no therapy is successful for the non-neovascular or “dry” form. Here we discuss the current knowledge on dry AMD pathobiology and propose future research directions that would expedite the development of new treatments. In our view, these should emphasize system biology approaches that integrate omic, pharmacological, and clinical data into mathematical models that can predict disease onset and progression, identify biomarkers, establish disease causing mechanisms, and monitor response to therapy.
Similar content being viewed by others


Introduction
Age-related macular degeneration (AMD) is the world’s leading cause of blindness among the elderly1. It is projected that the number of people with AMD worldwide will be 196 million in 2020, increasing to 288 million in 2040, with the pooled prevalence of early, late, and any AMD to be 8.01% (95% CI 3.98–15.49), 0.37% (0.18–0.77), and 8.69% (4.26–17.40), respectively1. While AMD is currently more prevalent in Europe and North America than Asia, given that Asia accounts for more than 60% of the world’s population, the largest projected number of AMD cases will occur in Asia1. In the US alone, approximately 11 million people have AMD, a prevalence that is similar to that of all invasive cancers combined, and more than double of that of Alzheimer’s disease2.
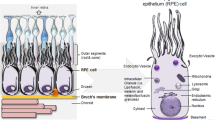
AMD is classified into two forms, a non-neovascular or “dry” form and a neovascular or “wet” form (Fig. 1). In the wet form, rapid, severe vision loss can occur due to the development of new blood vessels from the choroid into the subretinal space, within Bruch’s membrane, or in the subretinal pigmented epithelial (RPE) space that can leak fluid, hemorrhage, and with time, develop fibrosis around these neovascular tufts. In dry AMD, vision loss is typically gradual. Dry AMD is defined clinically by the presence of at least intermediate-size yellow sub-RPE deposits called drusen (63 µm or larger in diameter), RPE pigmentary abnormalities, and subretinal deposits called reticular pseudodrusen3. These pigmentary abnormalities are the clinical manifestation of RPE degeneration, which can ultimately culminate in death of the RPE and of the overlying photoreceptors. Multiple medium-sized drusen, large-sized drusen, RPE pigmentary changes, and AMD duration are independent risk factors for developing late AMD3. In late, dry AMD or geographic atrophy (GA), patches of RPE cell loss become confluent. When GA involves the fovea, vision loss is severe.

Spectrum of AMD. a Fundus photograph of a normal left macula. Arrowhead points to the fovea. b Right eye with intermediate, dry AMD. Arrowhead points to a typical large druse while arrow points to RPE hyperpigmentation. c Right eye with geographic atrophy. Arrowheads outline the area of GA. d Right eye with neovascular AMD. Arrowheads outline the area of choroidal neovascularization that is partially outlined by subretinal hemorrhage
While successful treatment using anti-vascular endothelial cell growth factor treatment is available for wet AMD4, no effective prevention or treatment is available for dry AMD. The Age-related Eye Disease Study (AREDS) trials demonstrated that antioxidant micronutrient supplements given to intermediate AMD patients modestly reduced the risk of developing advanced disease, and in particular, wet AMD5,6. Recently, lampalizumab, an inhibitor against complement Factor D, showed some efficacy in slowing GA progression in a Phase 2 study7, but this result was not confirmed in a Phase 3 study. A Phase 2 study conducted by Apellis using an inhibitor of complement factor 3 activation (APL-2) demonstrated a reduction in the rate of GA progression but was also associated with an increased risk of wet AMD in a subset of patients8. A number of other approaches for treating GA have failed in human trials (e.g., visual cycle inhibitors, emixustat, Acucela, sustained release of neurotrophic factors, NT501, Neurotech, and complement pathway inhibitors, eculizumab, Soliris)7,9.
The lack of preventive measures and treatment for dry AMD underscores the importance of gaining a better understanding of its pathobiology. Prior research has implicated strong roles for inflammation, and complement in particular, mitochondrial dysfunction, oxidative stress, lipid abnormalities, and cell death in dry AMD pathobiology, but their precise mechanisms are unclear. Furthermore, the relative magnitude and temporal contributions of these factors remain elusive. Due to the multifactorial etiology, effective management of dry AMD may require multiple targets that differ for prevention and therapy of early, intermediate, and late stage disease. These targets will likely result from the elucidation of the mechanistic pathways that are critically involved at each disease stage. The National Advisory Eye Council established a working group to evaluate the current knowledge on dry AMD pathobiology and propose future research directions that would expedite the development of new treatments and the purpose of this perspective is to report on the findings from this working group. The intention is to raise awareness of the impact of AMD on public health, review the current understanding of the pathobiology of this disease, offer future research directions that focus on unbiased systems approaches, encourage the continued efforts of dedicated vision scientists who focus on dry AMD, and encourage the broader scientific community to join in a collaborative effort to develop therapies for this complex and debilitating disease.
Hallmark pathological changes in dry AMD
The macula is the central area of the retina, 6 mm in diameter, which contains more than one layer of ganglion cell nuclei (Fig. 2). The central macula or fovea, 0.8 mm in diameter, is cone-dominated while the surrounding parafovea is rod-dominated. In the normal aging parafovea, the RPE become enlarged and often multinucleated10. In early, dry AMD, parafoveal rods die before either the RPE or cones11. With advancing dry AMD, the RPE degenerates with severe changes in cell shape, often becoming multilayered, and dissociated from Bruch’s membrane with migration into the retina or below Bruch’s membrane12. These changes suggest that RPE dysfunction has a central role in the development of photoreceptor loss in dry AMD.

Histological cross-section of a human macula. a Normal macula. b Hard drusen (*) within Bruch’s membrane. c A soft druse (*) within Bruch’s membrane. (b and c are courtesy of Philip Luthert, MBBS, UCL Institute of Ophthalmology.) d Subretinal drusenoid deposits (SDDs) between shortened photoreceptor outer segments and the RPE. Arrows highlight one of several SDDs (Courtesy of Christine Curcio, Ph.D., University of Alabama Birmingham and Project MACULA AMD Histopathology resource (http://projectmacula.cis.uab.edu/). HFL Henle fiber layer, NFL nerve fiber layer, GCL ganglion cell layer, IPL inner plexiform layer, INL inner nuclear layer, OPL outer plexiform layer, ONL outer nuclear layer, POS photoreceptor outer segment, RPE retinal pigment epithelium, BM Bruch’s membrane. Bar = 25 μm
Coincident with RPE changes, Bruch’s membrane develops basal deposits or accumulations of heterogeneous debris12. With aging, the inner Bruch’s membrane accumulates apolipoprotein B100-containing lipoproteins13, which stimulate inflammatory infiltration, accumulation of cellular debris, and the formation of basal deposits. Basal laminar deposits accumulate between the RPE and its basement membrane, and are associated with dry AMD when they become thick and composed of heterogeneous debris. Basal linear deposits, which form within Bruch’s membrane’s inner collagenous layer, are specific to dry AMD. Nodular-shaped basal linear deposits and focal basal laminar deposits accumulations are visualized as soft drusen on clinical exam.
The choriocapillaris is the sinusoidal capillary network of the choroidal circulation that is adjacent to Bruch’s membrane. Choriocapillaris endothelium has fenestrations that enable the bidirectional movement of fluid and macromolecules with the RPE and outer retina. Early dry AMD is characterized by loss of the choriocapillaris, which provides oxygen and nutrients for RPE and photoreceptor survival. Choriocapillaris loss precedes RPE atrophy and correlates with drusen size and density, thus implicating choriocapillaris dysfunction in RPE survival and drusenogenesis14. Finally, reticular pseudodrusen are extracellular deposits that accumulate in the subretinal space. While initially overlooked, improved imaging has shown that reticular pseudodrusen are part of a continuum of pathology that is referred to as subretinal drusenoid deposits (SDDs). The presence and progression of SDD can predict disease advancement to GA15.
Stressors and pathways implicated in dry AMD pathobiology
Oxidative stress
Photoreceptors and the RPE have high metabolic activity that results in reactive oxygen species (ROS) generation. The high metabolic demand requires a high oxygen partial pressure of 70–90 mmHg16, and the unique, photo-oxidative stress from light exposure make the macula a high oxidative stress microenvironment. In addition, several lifestyle choices such as cigarette smoking, and high fat or high glycemic index diets, add to the oxidative stress burden, and are associated with AMD risk17,18,19. The AREDS trials showed that, among patients with intermediate AMD, antioxidants lower the risk of developing advanced AMD5,6. Finally, genetic variants in oxidative stress-related genes, including MTND2*LHON-4917G, NADH subunits, SOD2, and PPARGC1A, are associated with AMD risk20,21,22.
Lipids
Because lipids can occupy more than 40% of drusen volume23, and polymorphisms in several lipid-related genes, including LIPC, CETP, ABCA1, and APOE, are associated with AMD risk24,25,26,27, lipids play a critical role in drusenogenesis. The RPE accumulates cholesterol either from phagocytosis of photoreceptor outer segments or from the ingestion of lipoproteins from the circulation. The RPE recycles cholesterol back to the photoreceptor or eliminates it through reverse cholesterol transport by effluxing cholesterol to ApoAI-I to form a high-density lipoprotein (HDL)28. With lipoproteinemia, lipoprotein ingestion is excessive, and the RPE becomes cholesterol-overloaded. Should the reverse cholesterol transport fail, the RPE will secrete apoB100 lipoproteins into Bruch’s membrane13,29. With aging, Bruch’s membrane accumulates advanced glycation endproducts, which induces lipoprotein lipase, causing the retention and oxidation of lipoproteins30,31. Hydroxyapatite surrounds the oxidized lipoproteins, which get coated with lipids and inflammatory proteins to promote drusen growth32. Since cones contain more cholesterol than rods, drusen tend to accumulate in the cone-rich fovea33.
SDDs contain unesterified cholesterol and apoE, which suggests that HDLs are involved in their formation34,35. Photoreceptors and the RPE have an active bidirectional cholesterol transport where HDLs accumulate cholesterol released into the subretinal space and cycles cholesterol between the RPE and photoreceptors28. With RPE dysfunction, cholesterol-laden HDLs accumulate in the subretinal space, triggering inflammation, and the accumulation of complement factors, vitronectin, and immune cells36. Figure 3 summarizes the role of lipids in AMD lesion formation.

Schema of the role of lipids in drusen and subretinal drusenoid deposit formation. a In the cone-rich fovea, the high cholesterol content of cones, along with lipids derived from the circulation due to a high cholesterol diet, induces apoB100 lipoprotein formation that is basally secreted. Due to age-related Bruch’s membrane changes, lipoproteins accumulate, forming the lipid wall. In the rod-rich parafovea, reverse cholesterol transport mediated through ABCA1 releases cholesterol to apoE and apoA1, forming high-density lipoproteins for the recycling of lipids including docohexanoic acid and cholesterol to rods. b Hydroxyapatite forms around retained lipids and lipoproteins, and combined with lipoprotein oxidation, induces an inflammatory response with the accumulation of inflammatory debris, leading to basal deposit and drusen formation. In the subretinal space, dysfunction of reverse cholesterol transport can lead to lipid accumulation, which induces an inflammatory response, forming subretinal drusenoid deposits. AGEs advanced glycation endproducts, BD basal deposit, BrM Bruch’s membrane, CC choriocapillaris, CEP carboxyethyllysine, Chol cholesterol, ER endoplasmic reticulum, HAP hydroxyapatite, HDL high-density lipoprotein, HFC high-fat cholesterol diet, HSPG heparan sulfate proteoglycan, Lp apoB100 containing lipoprotein, Ly lysosome, Mono/macro monocyte/macrophage, RCT reverse cholesterol transport, ROS reactive oxygen species, SDD subretinal drusenoid deposit; yellow circle surrounded by red, apoB100 containing lipoprotein with surrounding HAP
Inflammation and innate immunity
Altered immune responses that lead to destructive neuroinflammation are thought to contribute to the dry AMD phenotype. Parainflammation is a low-grade cytoprotective adaptation to local stress that is intermediate between immune-mediated homeostasis and chronic inflammation that maintains cellular and tissue function. Loss of parainflammation control contributes to dry AMD by invoking a chronic, heightened immune response that causes tissue destruction. The hallmarks of immune activation include drusen formation, subretinal and choroidal recruitment of microglia/macrophages, mast cell activation, and RPE immune activation37,38,39. This immune activation may involve close interplay between intracellular complement regulation and NLRP3 assembly in either immune cells or the RPE, although a recent study argues against NLRP3 inflammasome activation in the RPE40. At present, it is unclear at what point such immune activation converts from being protective to pathologic41,42. Figure 4 is a schema of the role of inflammation in AMD pathobiology.

Cartoon of inflammatory processes involved in AMD pathobiology. In the subretinal space, oxidized lipids and excess HDLs can elicit an inflammatory response that includes microglial and systemic monocyte recruitment. a Initially, immune responses are heightened, representing parainflammation to maintain cell health. This includes activation of RPE, microglia, and potentially choroidal macrophages, with heightened intracellular cytokine responses (IL-18, IL-33), upregulation of autophagy, and immunometabolic regulation to maintain mitochondrial health. b Conversion from parainflammation to chronic inflammation can induce inflammasome activation by microglia, monocytes and the RPE (including IL-1b production) that contributes to subretinal drusenoid deposit formation. In the RPE, complement and the inflammasome can be activated by a number of triggers. In Bruch’s membrane, the accumulation of lipoproteins and cellular debris elicits an inflammatory response from the RPE, mast cells, and monocytes/macrophages derived from the choroid or systemic circulation to activate both complement and the inflammasome. Inflammatory debris accumulates around lipoproteins/HAP particles during drusen formation in Bruch’s membrane. CEP carboxyethyllysine, CFH complement factor H, HSPG heparan sulfate proteoglycan, Ly lysosome, MDA malondialdehyde, Mit mitochondria, mono/macro monocyte/macrophage, PR photoreceptor, ROS reactive oxygen species, SDD subretinal drusenoid deposit, TLR Toll-like receptor
Complement system
Genetic studies have identified complement pathway gene variants with AMD risk, which strongly implicates the complement pathway in driving AMD progression. However, the assumption that these variants induce excessive complement activation that leads to tissue injury remains unvalidated. For example, the exact mechanism for how the complement factor H (CFH) 402H variant contributes to dry AMD has eluded researchers for over a decade. CFH acts as both a cofactor during Factor I-mediated C3b cleavage and as a decay accelerating agent against the alternative pathway C3-convertase. The 402H variant increases complement activity43. However, it is unclear whether this increased activity induces elements of the dry AMD phenotype because CFH 402H also has impaired interaction with C-reactive protein, malondialdehyde, and heparan sulfate proteoglycans that can increase inflammation and lipoprotein accumulation43,44,45. As a mechanistic understanding of the causal molecular pathways is incomplete, perhaps not surprisingly, clinical trials testing complement inhibitors have failed9.
Mitochondrial dysfunction
While age-associated mitochondrial dysfunction reduces cytoprotective pathway function, mitochondrial impairment severe enough to cause tissue damage will herald disease. In dry AMD, RPE mitochondrial mass is reduced, mitochondria are morphologically abnormal, and mitochondrial DNA (mtDNA) damage increases with disease severity46,47. In early and dry AMD, before a patient loses vision, mtDNA damage in genes involved in electron transport complex I, II, IV, and V in the RPE accumulates more in the macula than in the periphery, and occurs in the RPE prior to the neurosensory retina47,48,49. With mitochondrial loss and injury, the RPE has reduced baseline oxygen consumption and reserve capacity, and elevated ROS, making them poorly responsive to stress50. This change results in decreased ATP production, compromised calcium- and ROS-mediated signaling, and altered nucleotide metabolism, amino acid, lipid, and heme biosynthesis, which subsequently impair essential cytoprotective or specialized functions51. In addition, mitochondrial damage can activate apoptosis, leading to RPE degeneration and death, two key features of AMD.
Cell death
Photoreceptor cell death is the basis for permanent visual decline in dry AMD. Therefore, identifying the mechanisms involved in photoreceptor death is critical for developing new treatments to prevent permanent visual loss. Photoreceptor cell death is principally caused by apoptosis and necrosis52. Preventing photoreceptor cell death by specifically blocking apoptosis has been unsuccessful when inhibiting caspase because photoreceptor death is also caused by receptor-interacting protein kinase (RIPK)-mediated necrosis53. Likewise, when RIPK is inhibited, photoreceptor loss is unaffected53. In contrast, inhibiting both RIPK and caspases impairs both necrosis and apoptosis and rescues photoreceptors53. The mechanisms for RPE cell death may differ from those of photoreceptors. Double stranded RNA (dsRNA), a component of drusen, is a ligand for Toll-like Receptor-3, which mediates the innate immune response and cell death. After injecting dsRNA into the eye, photoreceptors die by apoptosis and RPE cells die by necrosis54. Since cell death pathways are redundant and complementary, combination therapies that block both apoptosis and necrosis may be effective for dry AMD.
Strategies for understanding dry AMD etiology and pathogenesis
Genetics and genomics
Genetic approaches have contributed enormously to our understanding of AMD pathobiology. The association of APOE variants with AMD was the first indication that a specific gene affected disease risk55,56. The advent of high-throughput technologies and agnostic genome-wide searches using family-based linkage and genome-wide association study (GWAS) approaches facilitated the identification of genes on chromosomes 1 (CFH)57,58,59,60 and 10 (ARMS2/HTRA1)61,62,63,64. These discoveries are remarkable because they are among the few genetic associations for a common disease that increase the odds of disease by more than 2.5 among heterozygotes and 7.5 among homozygotes60,62. The association with CFH prompted focused investigation of complement system genes27 which uncovered associations with C3 65,66, BF/C2 67, and CFI 68.
Current research has focused on understanding the role of specific genes and related pathways that drive AMD pathobiology69,70,71. The genes identified in a recent GWAS highlight the importance of both previously recognized and novel mechanistic pathways including complement activation, collagen synthesis, lipid metabolism/cholesterol transport, receptor-mediated endocytosis, and extracellular matrix organization24. Variants located in seven extracellular matrix genes were associated with advanced AMD and not early/intermediate AMD, suggesting that extracellular remodeling pathway(s) become relevant in advanced disease72. Pathway analysis and systems biology approaches have provided clues that upstream regulators/modulators of AMD risk variants are potential therapeutic targets73,74. These approaches give insight into the relationship between genetic variation and disease mechanism that can be tested in appropriate in vivo and in vitro models.
Except for CFH and HTRA/ARMS2, AMD-associated common variants confer a very small effect on disease risk (i.e., odds ratios <1.2), with as much as 70% of AMD risk remaining unexplained24. Increasing the samples for GWAS will undoubtedly implicate more loci in addition to the 40+ already identified24,25,70,75,76,77, but the associated variants will account for an increasingly smaller amount of the genetic variance. A portion of the proverbial “missing heritability” may be the result of gene interactions and environmental factors78,79. Associations with highly penetrant rare variants in genes known to harbor common variants have helped to establish causation, but the extent that rare genetic variants with high impact will account for the remaining genetic causality of AMD is unknown24,80,81,82,83,84. It will be difficult to identify rare, highly penetrant alleles in novel genes using traditional case–control designs because the requisite sample size to detect rare variants is much greater than aggregated cohorts assembled for GWAS. Nonetheless, such discoveries will provide additional insight into disease mechanisms.
Studies of non-European ancestry populations have demonstrated variability in the strength of genetic associations for AMD across Hispanics, Asians, and African Americans77,85. Protective alleles may contribute to these population differences. For example, AMD is very rare (<1%) in the isolated Timor-Leste population, and the few affected individuals do not harbor risk alleles at the two most associated AMD loci, CFH and ARMS2/HTRA1 (ref. 86). However, the frequency of protective alleles at CFH, CFHR2, and C2 was increased in Timorese compared to outbred European ancestry populations, underscoring their potential importance. It would be valuable to study cohorts in which individuals with a high exposure to known AMD risk factors (e.g., smoking), advanced age, and elevated genetic risk for AMD do not develop AMD to gain insights into the protective mechanisms of specific genes or pathways that might serve as therapeutic targets.
Other types of genetic variants and mechanisms may have important roles in dry AMD. These include copy number variants or larger deletions and insertions, noncoding transcripts such as lncRNAs and microRNAs as modulators of gene expression and disease, and mitochondrial gene variants21,87. Somatic DNA modifications resulting from environmental exposures such as smoking and aging may also play a role in AMD pathogenesis88,89. Germline and somatic DNA changes may not directly impact pathogenic mechanisms, but rather influence expression or interact with other genes that are more proximate in pathways leading to disease. Thus, post-transcriptional DNA modifications that alter expression and their downstream consequences must be considered in studies that involve the transcriptome, post-translational modifications, proteome, and metabolome90. For example, using ATAC-Seq in the retina and RPE from AMD and control patients, Wang et al.91 found global decreases in chromatin accessibility in the RPE from early AMD globes and in the retina of advanced disease, which suggests that RPE dysfunction drives disease onset. Cigarette smoke treatment of RPE cells recapitulated the chromatin accessibility changes seen in AMD, providing a potential epigenetic link of a known AMD risk factor with AMD pathology.
In vitro dry AMD models
The ability to differentiate embryonic stem (ES) and induced pluripotent stem (iPS) cells into RPE cells and retinal organoids has enhanced the ability to uncover the role of specific pathways and genetic influences. The use of patient-specific iPS cells is especially attractive because they have both the specific gene mutation of interest and the underlying genotype that could influence the mutation’s contribution to the patient’s disease and phenotype. Galloway et al. studied iPS-derived RPE cells from patients with and without AMD and found that sub-RPE basal deposits were more abundant and contained a lipid- and protein-rich “drusen-like” composition than unaffected cells92. Saini et al.93 found that iPS-RPE cells derived from AMD patients with the ARMS2/HTRA1 AMD risk variant had increased complement and inflammatory proteins compared donors without AMD. Saini et al.94 also found a graded response to nicotinamide (NAM), with a marked inhibition of drusen-associated proteins, clusterin and vascular endothelial growth factor A, in iPS-RPE cells with the ARMS2/HTRA1 AMD risk variant. These experiments underscore the importance of targeting more than one disease pathway or mechanism at a time, while taking into consideration genotype risk95. Alternatively, the emergence of CRISPR/Cas9 gene editing has enabled researchers to further manipulate stem cell systems for mechanistic study. This approach can study the impact of a genetic variant by comparing its behavior with an isogenic control cell line without the mutation. With the possibility of personalized medicine, it may be possible to predict a drug response using iPS-derived cells or to differentiate iPS-derived retinal cells for transplantation into a diseased macula.
Improved culturing techniques have enabled the generation of retinal organoids. Using iPS cells, Zhong et al.96 recapitulated spatiotemporally each of the main steps of retinal development observed in vivo during the formation of three-dimensional retinal cups that contain all major retinal cell types arranged in their proper layers. Remarkably, the photoreceptors showed outer segment formation and were photosensitive. This system can enable the study of cellular interactions.
The use of iPS cells also has limitations. Due to heterogeneity of iPS lines, a subclone could be selected that would misrepresent the cell’s behavior. The epigenetic memory could remain and maintain elements of the cell’s original phenotype. The relevance of iPS cells to an aging disease such as AMD is open to speculation since markers of cellular age, including mitochondrial function and telomere length, are reset to a youth-like state after old donor fibroblasts are reprogrammed to iPS cells97.
In vivo dry AMD models
To provide mechanistic evidence of AMD pathobiology, zebrafish, mice, rats, and non-human primates are a few of the animals used to model AMD. The only animal to possess a macula, non-human primates are prohibitively expensive to maintain, and difficult to both manipulate and follow longitudinally since their life span is long, outliving the duration of most grants. Mice are the most practical organisms for modeling AMD due to their short life span, genetic manipulability, relatively low maintenance cost, and retinal architecture that is similar to humans. The mouse does not possess a macula, but rather a rod-rich retina that is similar to the rod-rich human parafovea. While the lack of a macula has been used to question their relevance to human AMD, mice have greatly aided our understanding of AMD pathobiology. Despite this important limitation, mice offer a valuable means of investigating the interplay of aging, oxidative stress, immunity, lipid metabolism, nutrition, and the microbiome as potential modifiers of retinal/RPE disease43,98,99. Such studies cannot be done with in vitro models and are not feasible in humans. However, most dry AMD mouse models represent early disease stages. Advanced age is the strongest risk factor for AMD and should be considered in any model. In fact, the most robust AMD models incorporate advanced age and are chronic in duration. Many AMD pathological hallmarks have been recapitulated in different mouse models, including vision loss; photoreceptor degeneration; RPE lipofuscin accumulation, hypo- and hyperpigmentation, multinucleation, and atrophy; choriocapillaris atrophy; and subretinal immune cell infiltration43,100,101,102,103. However, few models faithfully display a combination of these changes. While mice do not develop drusen, they do accumulate basal laminar deposits, which share many of the same constituents found in drusen43,100,102. The models that most closely simulate human AMD are based on known AMD-associated mechanisms, including inflammation, lipid transport and metabolism, ECM remodeling, complement dysregulation, oxidative stress, and autophagy, which will enable study of how these factors interact with one another during lesion development, and how these lesions correlate with different stages of dry AMD43,100,101. However, because of differences in anatomy, metabolism, and the immune response between mice and humans, extrapolation to human disease should be made with caution.
Clinical imaging of dry AMD
Imaging is an essential component of the clinical assessment of AMD, and is used to diagnose, define disease severity and progression, assess treatment response, and complements mechanistic basic science analysis to understand AMD pathobiology. Currently, color fundus photographs are used to determine drusen size and total area, RPE pigmentary abnormalities, and GA, with the aim to categorize and predict AMD progression. Angiography with intravenously injected sodium fluorescein or indocyanine green (ICG) dyes provides detailed images of retinal and choroidal vasculature. Fundus autofluorescence identifies areas with excess fluorophores from RPE lipofuscin accumulation, loss of fluorophores from RPE loss, or fluorophore variation104. Reflectance-based structural optical coherence tomography (OCT), either spectral domain or the faster swept source OCT, provides micron-level resolution to enable detailed visualization of retinal layers, substructures, and AMD pathology105. OCT-angiography can provide detailed images of the choriocapillaris and choroidal abnormalities such as choriocapillaris dropout with GA106. By registering images, specific OCT features can be monitored in the same location over time to quantify morphologic progression107.
Several new imaging modalities may provide novel insight into AMD pathobiology due to their ability to visualize key cellular and subcellular structures during different disease phases. Adaptive optics compensates for wave front aberrations to improve resolution108. When used with a scanning laser ophthalmoscope and OCT, adaptive optics systems can visualize individual cones, changes in rods, RPE, and SDD in dry AMD. Polarization-sensitive OCT highlights tissue using polarized signals and birefringence so that the RPE or fibrosis may be detected109. Fluorescence lifetime and hyperspectral fluorescence imaging, which measure dynamic metabolic states of the retina, offer new opportunities for retinal biomarkers that may complement other functional methods such as psychophysical testing (such as dark adaptation thresholds and kinetics) and electrophysiology110. These diagnostic tools may identify preclinical risk factors or subtypes of AMD that may differ with respect to progression and/or response to different treatments.
However, validating these new imaging technologies either individually or in combination has been restricted to small cohorts over short or inconsistent follow-up intervals111,112. Furthermore, imaging data have been simplified for clinical use, which limits their utility to uncover a deep understanding of AMD pathobiology. To advance our understanding, we must identify imaging biomarkers of early changes that reflect AMD pathobiology, predict disease progression and/or treatment response, and correlate with molecular markers that are relevant in both animal models and humans.
Recommendations for an expedited, improved understanding, and clinical management of dry AMD
Cellular and animal dry AMD models enable direct interrogation of pathological mechanisms in controlled environments, an attribute obviously lacking in human studies. The question remains of how to best leverage these models to determine the role of AMD-associated mechanisms that will lead to effective treatment. Cellular models are excellent for manipulating treatments that alter intracellular interactions while animal models enable the study of interactions among tissues, and how aberrant pathways induce an AMD phenotype. Since many models are incompletely or variably phenotyped, which can confuse interpretation, we advocate developing a consensus on the phenotypic standards of AMD animal models. This goal could be accomplished by a careful literature review on animal models that forms the foundation for roundtable discussions by leading scientists who utilize AMD animal models and clinicians who have expertise in AMD imaging, epidemiology, and treatment. Importantly, we recommend that animal and cell-based models rely on multiple risk factors to mimic the events at different stages of dry AMD, and that multiple models should be utilized to deepen the rigor beyond what a single model will provide.
Systems biology approaches
So far, genetic and epidemiologic studies have pinpointed more than 40 genetic variants and environmental and lifestyle factors, such as smoking, sunlight exposure, and a high-fat diet, that define individual risk for AMD. Integrating such heterogeneous factors that influence AMD pathobiology and turning this knowledge into prevention, prediction, and treatment is a big challenge. The next step is to understand the functional consequences of these risk factors. Traditionally, interlinks between genetic and non-genetic factors have been studied in a reductionist fashion. This approach is insufficiently comprehensive to assign the relative contribution of these risk factors to disease, the interaction of these factors, and the disease stage that these factors are pathogenic. In contrast, linking multiple data sources has already led to identifying the alternative complement pathway, ECM turnover, and lipid metabolism in AMD pathobiology113. The integration of genes carrying risk alleles into the framework of complex endophenotypes suggests that AMD risk genes act in multiple molecular pathways and large networks, associate with different anatomical microenvironments in the macula, and affect diverse higher order physiological activities such as Bruch’s membrane homeostasis, protein and lipid turnover, energy metabolism, and complement regulation114. Thus, we recommend application of systems biology to address the complex interplay of pathogenic factors and provide the appropriate broad perspective needed to decipher the role of multiple factors in dry AMD pathogenesis. This approach would integrate basic, genomic, preclinical, medical, pharmacological, and clinical data into mathematical models of pathological processes at different stages of dry AMD in order to ask how relevant individual components act together within the living system. Research partnerships with companies that conduct dry AMD clinical trials could provide valuable phenotyping information that is required to assess disease progression and treatment response that could be integrated with genetics and AMD biomarkers. Furthermore, clinical trial participants should be encouraged to maintain clinical follow-up after the study’s completion and to consider donating their eyes after death for research. While the effect of diet and environmental exposures on the human microbiome and the implications of these changes on the host immune and inflammatory systems have been explored in AMD115, to gain a complete understanding of the complex interactions and their role in the pathogenesis and progression of dry AMD, all individual perturbations must be integrated into the entire system to establish a causal role.
Imaging data provide the foundation for a systems biology approach because they can identify the major pathological events in chronological sequence. These images should provide objective and reproducible longitudinal data that can be integrated with large multisystems data from large cohorts. The imaging data must accurately co-localize morphologic changes in each of the multiple imaging modalities, and individual patient data must be cross-correlated before they can be incorporated into the overall analysis. Deep learning will expand the capability of analyzing these complex data sets116. To identify novel biomarkers, we recommend using agnostic, artificial intelligence methods that are independent of known biomarkers and/or prior categorizations to both confirm known mechanisms and identify novel associations. Because of the cost and scientific complexity of imaging modalities, we acknowledge that the analysis of large cohorts of imaging data may be limited to a few sites that are linked to a wide net of collaborating centers.
The imaging data must be integrated with content-rich clinical, epidemiological, pharmacological, and genetic data, as well as multi-omic approaches including genomic, transcriptomic, epigenomic, lipidomic, metabolomic, microbiome, and proteomic studies on fresh blood, donor eye tissue, or aqueous and vitreous biopsies from patients with dry AMD. Advances in multi-scale data integration of large throughput experiments and analysis have opened new avenues for discovery and have bridged genetic risk variants with expression and protein level information117,118. We recommend leveraging large data sets, whether public or proprietary, that are now available including those containing common and rare genetic variants for AMD, transcriptomic data derived from the mouse and eventually human retina, and proteomic data for developing a systems biology model of dry AMD119. The emergence of single-cell RNA and ATAC sequencing offers the possibility of defining the relative contributions of individual cell types to AMD pathogenesis, and will enrich the models that are developed. Finally, pathogenic molecular pathways that have been interrogated in cell and animal models must be integrated with these data, which must be quality controlled and consistently curated. These efforts require centralized and shared resources to disseminate the information, and could provide an unprecedented resource to understand AMD pathobiology, re-define stratified clinical phenotypes that are based on molecular and dynamic parameters, develop combinatorial biomarkers or biomarker signatures, and initiate novel diagnostic, prevention, prediction, and therapeutic approaches where reductionist approaches have failed.
The need for a personalized approach to treat dry AMD
Since dry AMD is a multifactorial disease resulting from perturbations in multiple pathways, the pathogenic signals are likely to vary among individuals and at different disease stages. Thus, we advocate designing computer models that are tailored to the individual so that physicians can accurately predict dry AMD risk, disease progression, and response to treatment. We believe that this precision medicine approach for AMD, which was unimaginable just a few years ago, is realistic and similar to what is being achieved in other complex diseases such as cancer120 or diabetes121. These individualized risk prediction models may potentially enable targeting patients with specific dry AMD subtypes for tailor-made therapies. Genomic and proteomic biomarkers, and other risk factors identified from a systems approach could monitor the effectiveness of preventive therapies to modify dry AMD risk before the onset of vision-threatening complications. Once single markers or marker signatures are linked to specific stages and molecular mechanisms of AMD, they can be employed as predictive or companion biomarkers and biomarker signatures that hallmark specific pathogenic features or pathological mechanisms of AMD. Having a set of multimodal markers at hand to group and stratify patients according to their individual risk and expected response pattern, therapeutic trials tailored to specific patient groups could be designed. Individuals within high-risk groups could be better motivated to comply with an early intervention on the basis of a predicted cumulative risk and a likely positive expected response to therapy prior to losing vision. The prerequisite for this rationale, however, depends upon the successful advancement of promising, preclinical drugs into clinical trials.
Benefits and challenges of a systems biology approach for dry AMD
This approach would be a departure from the current research paradigm. Such an approach would require the integrated collaboration of leading clinicians, imaging experts, a wide variety of basic scientists, bioinformaticians, and biostatisticians among other necessary expertise. Besides vision scientists, this research will benefit by recruiting scientists outside of dry AMD research who will provide not only additional technical expertise, but potentially a “Medici effect” (i.e., the most important innovations occur when concepts from diverse disciplines are conjoined) where scientists without AMD expertise could provide disruptive innovation that would complement the conventional approach. Due to the perceived cost, the clinical and basic science may be conducted at only a few centers that have the appropriate infrastructure to implement this large-scale approach, or require coordinated efforts of investigators at multiple institutions. However, implementing large-scale team systems biology research should be possible. We can now link heterologous large data sets to acquire novel information on which to generate new hypotheses, calculate risks on an individual basis, identify drivers of disease manifestation and progression, and turn knowledge into risk assessment and clinical recommendations. The EYE-RISK consortium is an example of how this goal can be achieved. A consortium of participants from different European countries, EYE-RISK (www.eye-risk.eu) funded by the EU Horizon 2020 program, uses a multidisciplinary approach to examine comprehensive epidemiologic data and biosamples from large European epidemiologic eye cohorts and biobanks (total study population N = 100,000). Collaborative studies conducted by EYE-RISK, the International AMD Genomics Consortium, and the Three Continent Consortium have been instrumental in identifying genetic risk variants and assessing environmental risk factors.
Currently, there is no organized, cost-effective system for obtaining human donor globes that have short death-to-dissection times with high-quality RNA, metabolite, DNA, and protein that is adequate to meet the needs of the AMD research community122. Likewise, the few existing repositories of blood or ocular fluid samples from well-phenotyped patients for study are not sufficiently resourced to distribute specimens on a wide scale. To enable this systems approach, we recommend developing mechanisms to create these libraries of AMD eyes, blood and ocular fluid samples, an array of omics data (e.g., genomic, transcriptomic, methylomic, proteomic, etc.) derived from these samples, and a program that ensures their effective use for dry AMD research. The National Cell Repository for Alzheimer disease which banks blood and DNA specimens, brain tissue, and associated data for Alzheimer disease research (https://www.nia.nih.gov/research/resource/national-cell-repository-alzheimers-disease-ncrad) is one example of a coordinated approach to address this need.
The current National Eye Institute and foundation-based funding mechanisms for AMD research are largely confined to grants awarded to individual researchers for a defined amount of money. This approach, which has been the cornerstone of our current understanding of AMD pathobiology to date, enables researchers to address specific, focused mechanistic questions, but in relative isolation. A systems biology approach using sophisticated high-throughput “-omics” assays is not feasible with a typical individual R01 NIH grant. We recommend that the NIH and other funding agencies examine how they allocate precious resources and consider including large programs that would enable the integration of basic discovery research, mechanistic investigation using well-phenotyped human populations, and suitable animal and cell-based models that are needed to gain a detailed understanding of dry AMD pathobiology. Mechanisms that foster access and coordination of multiple investigators to resources that would not be achievable on an individual basis would address this next step in dry AMD research. The implementation of this approach could be a template for tackling other complex diseases.
Conclusion
Future directions in dry AMD research should emphasize systems biology approaches that integrate omic, pharmacological, and clinical data into mathematical models that can predict disease onset and progression, identify biomarkers, establish disease causing mechanisms, and monitor response to therapy. Success in these areas will likely be achieved most expediently and effectively by promoting collaborative efforts of multidisciplinary investigator teams and developing centralized resources including clinical, imaging, omic, and other types of data as well as carefully phenotyped eye tissue from large cohorts of patients with and without AMD.
References
Wong, W. L. et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob. Health 2, e106–e116 (2014).
Pennington, K. L. & DeAngelis, M. M. Epidemiology of age-related macular degeneration (AMD): associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. (Lond.) 3, 34 (2016).
Chew, E. Y. et al. Ten-year follow-up of age-related macular degeneration in the age-related eye disease study: AREDS report no. 36. JAMA Ophthalmol. 132, 272–277 (2014).
Rosenfeld, P. J. et al. Ranibizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 355, 1419–1431 (2006).
Age-Related Eye Disease Study Research, G. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch. Ophthalmol. 119, 1417–1436 (2001).
Age-Related Eye Disease Study 2 Research Group. Lutein+zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA 309, 2005–2015 (2013).
Rhoades, W., Dickson, D. & Do, D. V. Potential role of lampalizumab for treatment of geographic atrophy. Clin. Ophthalmol. 9, 1049–1056 (2015).
Taskintuna, I., Elsayed, M. E. & Schatz, P. Update on clinical trials in dry age-related macular degeneration. Middle East Afr. J. Ophthalmol. 23, 13–26 (2016).
Ach, T. et al. Quantitative autofluorescence and cell density maps of the human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 55, 4832–4841 (2014).
Starnes, A. C. et al. Multi-nucleate retinal pigment epithelium cells of the human macula exhibit a characteristic and highly specific distribution. Vis. Neurosci. 33, e001 (2016).
Sarks, S. H. Ageing and degeneration in the macular region: a clinico-pathological study. Br. J. Ophthalmol. 60, 324–341 (1976).
Malek, G., Li, C. M., Guidry, C., Medeiros, N. E. & Curcio, C. A. Apolipoprotein B in cholesterol-containing drusen and basal deposits of human eyes with age-related maculopathy. Am. J. Pathol. 162, 413–425 (2003).
Mullins, R. F., Johnson, M. N., Faidley, E. A., Skeie, J. M. & Huang, J. Choriocapillaris vascular dropout related to density of drusen in human eyes with early age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 52, 1606–1612 (2011).
Lee, M. Y., Yoon, J. & Ham, D. I. Clinical characteristics of reticular pseudodrusen in Korean patients. Am. J. Ophthalmol. 153, 530–535 (2012).
Khaliq, A., Jarvis-Evans, J., McLeod, D. & Boulton, M. Oxygen modulates the response of the retinal pigment epithelium to basic fibroblast growth factor and epidermal growth factor by receptor regulation. Invest. Ophthalmol. Vis. Sci. 37, 436–443 (1996).
Smith, W. et al. Risk factors for age-related macular degeneration: pooled findings from three continents. Ophthalmology 108, 697–704 (2001).
Mares-Perlman, J. A. et al. Dietary fat and age-related maculopathy. Arch. Ophthalmol. 113, 743–748 (1995).
Chiu, C. J., Milton, R. C., Gensler, G. & Taylor, A. Association between dietary glycemic index and age-related macular degeneration in nondiabetic participants in the Age-Related Eye Disease Study. Am. J. Clin. Nutr. 86, 180–188 (2007).
SanGiovanni, J. P. et al. DNA sequence variants in PPARGC1A, a gene encoding a coactivator of the omega-3 LCPUFA sensing PPAR-RXR transcription complex, are associated with NV AMD and AMD-associated loci in genes of complement and VEGF signaling pathways. PLoS ONE 8, e53155 (2013).
SanGiovanni, J. P. et al. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS ONE 4, e5508 (2009).
Canter, J. A. et al. Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS ONE 3, e2091 (2008).
Curcio, C. A., Millican, C. L., Bailey, T. & Kruth, H. S. Accumulation of cholesterol with age in human Bruch’s membrane. Invest Ophthalmol. Vis. Sci. 42, 265–274 (2001).
Fritsche, L. G. et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 48, 134–143 (2016).
Neale, B. M. et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc. Natl Acad. Sci. USA 107, 7395–7400 (2010).
Chen, W. et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc. Natl Acad. Sci. USA 107, 7401–7406 (2010).
Conley, Y. P. et al. Candidate gene analysis suggests a role for fatty acid biosynthesis and regulation of the complement system in the etiology of age-related maculopathy. Hum. Mol. Genet. 14, 1991–2002 (2005).
Tserentsoodol, N. et al. Intraretinal lipid transport is dependent on high density lipoprotein-like particles and class B scavenger receptors. Mol. Vis. 12, 1319–1333 (2006).
Fujihara, M., Cano, M. & Handa, J. T. Mice that produce ApoB100 lipoproteins in the RPE do not develop drusen yet are still a valuable experimental system. Invest. Ophthalmol. Vis. Sci. 55, 7285–7295 (2014).
Cano, M., Fijalkowski, N., Kondo, N., Dike, S. & Handa, J. Advanced glycation endproduct changes to Bruch’s membrane promotes lipoprotein retention by lipoprotein lipase. Am. J. Pathol. 179, 850–859 (2011).
Yamada, Y. et al. Oxidized low density lipoproteins induce a pathologic response by retinal pigmented epithelial cells. J. Neurochem. 105, 1187–1197 (2008).
Thompson, R. B. et al. Identification of hydroxyapatite spherules provides new insight into subretinal pigment epithelial deposit formation in the aging eye. Proc. Natl Acad. Sci. USA 112, 1565–1570 (2015).
Pikuleva, I. A. & Curcio, C. A. Cholesterol in the retina: the best is yet to come. Prog. Retin. Eye Res. 41, 64–89 (2014).
Rudolf, M. et al. Sub-retinal drusenoid deposits in human retina: organization and composition. Exp. Eye Res. 87, 402–408 (2008).
Ebrahimi, K. B., Fijalkowski, N., Cano, M. & Handa, J. T. Decreased membrane complement regulators in the retinal pigmented epithelium contributes to age-related macular degeneration. J. Pathol. 229, 729–742 (2012).
Greferath, U., Guymer, R. H., Vessey, K. A., Brassington, K. & Fletcher, E. L. Correlation of histologic features with in vivo imaging of reticular pseudodrusen. Ophthalmology 123, 1320–1331 (2016).
Bhutto, I. A. et al. Increased choroidal mast cells and their degranulation in age-related macular degeneration. Br. J. Ophthalmol. 100, 720–726 (2016).
Camelo, S., Lavelette, S., Guillonneau, X., Raoul, W. & Sennlaub, F. Association of choroidal interleukin-17-producing T lymphocytes and macrophages with geographic atrophy. Ophthalmologica 236, 53–58 (2016).
Cherepanoff, S., McMenamin, P., Gillies, M. C., Kettle, E. & Sarks, S. H. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br. J. Ophthalmol. 94, 918–925 (2010).
Kosmidou, C. et al. Issues with the specificity of immunological reagents for NLRP3: implications for age-related macular degeneration. Sci. Rep. 8, 461 (2018).
Theodoropoulou, S. et al. Interleukin-33 regulates tissue remodelling and inhibits angiogenesis in the eye. J. Pathol. 241, 45–56 (2017).
Tarallo, V. et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149, 847–859 (2012).
Toomey, C. B., Kelly, U., Saban, D. R. & Bowes Rickman, C. Regulation of age-related macular degeneration-like pathology by complement factor H. Proc. Natl Acad. Sci. USA 112, E3040–E3049 (2015).
Weismann, D. et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 478, 76–81 (2011).
Molins, B. et al. Complement factor H binding of monomeric C-reactive protein downregulates proinflammatory activity and is impaired with at risk polymorphic CFH variants. Sci. Rep. 6, 22889 (2016).
Feher, J. et al. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 27, 983–993 (2006).
Karunadharma, P. P., Nordgaard, C. L., Olsen, T. W. & Ferrington, D. A. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 51, 5470–5479 (2010).
Nordgaard, C. L., Karunadharma, P. P., Feng, X., Olsen, T. W. & Ferrington, D. A. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 49, 2848–2855 (2008).
Terluk, M. R. et al. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 35, 7304–7311 (2015).
Rohrer, B., Bandyopadhyay, M. & Beeson, C. Reduced metabolic capacity in aged primary retinal pigment epithelium (RPE) is correlated with increased susceptibility to oxidative stress. Adv. Exp. Med. Biol. 854, 793–798 (2016).
He, Y. & Tombran-Tink, J. Mitochondrial decay and impairment of antioxidant defenses in aging RPE cells. Adv. Exp. Med. Biol. 664, 165–183 (2010).
Schweichel, J. U. & Merker, H. J. The morphology of various types of cell death in prenatal tissues. Teratology 7, 253–266 (1973).
Trichonas, G. et al. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc. Natl Acad. Sci. USA 107, 21695–21700 (2010).
Murakami, Y. et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 21, 270–277 (2014).
Souied, E. H. et al. The epsilon4 allele of the apolipoprotein E gene as a potential protective factor for exudative age-related macular degeneration. Am. J. Ophthalmol. 125, 353–359 (1998).
Klaver, C. C. et al. Genetic association of apolipoprotein E with age-related macular degeneration. Am. J. Hum. Genet. 63, 200–206 (1998).
Klein, M. L. et al. Age-related macular degeneration. Clinical features in a large family and linkage to chromosome 1q. Arch. Ophthalmol. 116, 1082–1088 (1998).
Klein, R. J., et al. Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 (2005).
Haines, J. L., et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 308, 419–421 (2005).
Edwards, A. O. et al. Complement factor H polymorphism and age-related macular degeneration. Science 308, 421–424 (2005).
Weeks, D. E. et al. A full genome scan for age-related maculopathy. Hum. Mol. Genet. 9, 1329–1349 (2000).
Rivera, A. et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum. Mol. Genet. 14, 3227–3236 (2005).
Dewan, A., et al. HTRA1 Promoter polymorphism in wet age-related macular degeneration. Science 314, 989–992 (2006).
Yang, Z., et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science 314, 992–993 (2006).
Yates, J. R., et al. Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 357, 553–561 (2007).
Maller, J. B. et al. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat. Genet. 39, 1200–1201 (2007).
Gold, B. et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 38, 458–462 (2006).
Fagerness, J. A. et al. Variation near complement factor I is associated with risk of advanced AMD. Eur. J. Hum. Genet. 17, 100–104 (2009).
Jun, G. et al. Influence of ROBO1 and RORA on risk of age-related macular degeneration reveals genetically distinct phenotypes in disease pathophysiology. PLoS ONE 6, e25775 (2011).
Logue, M. W. et al. Search for age-related macular degeneration risk variants in Alzheimer disease genes and pathways. Neurobiol. Aging 35, 1510.e1517–1510.e1518 (2014).
Hall, J. B. et al. Estimating cumulative pathway effects on risk for age-related macular degeneration using mixed linear models. BMC Bioinformatics 16, 329 (2015).
DeAngelis, M. M. et al. Genetics of age-related macular degeneration (AMD). Hum. Mol. Genet. 26, R246 (2017).
Silveira, A. C. et al. Convergence of linkage, gene expression and association data demonstrates the influence of the RAR-related orphan receptor alpha (RORA) gene on neovascular AMD: a systems biology based approach. Vis. Res. 50, 698–715 (2010).
Hecker, L. A. et al. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum. Mol. Genet. 19, 209–215 (2010).
Yu, Y. et al. Common variants near FRK/COL10A1 and VEGFA are associated with advanced age-related macular degeneration. Hum. Mol. Genet. 20, 3699–3709 (2011).
Fritsche, L. G. et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 45, 439e431–432 (2013).
Cheng, C. Y. et al. New loci and coding variants confer risk for age-related macular degeneration in East Asians. Nat. Commun. 6, 6063 (2015).
Schmidt, S. et al. Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. Am. J. Hum. Genet. 78, 852–864 (2006).
Seddon, J. M., George, S., Rosner, B. & Klein, M. L. CFH gene variant, Y402H, and smoking, body mass index, environmental associations with advanced age-related macular degeneration. Hum. Hered. 61, 157–165 (2006).
Helgason, H. et al. A rare nonsynonymous sequence variant in C3 is associated with high risk of age-related macular degeneration. Nat. Genet. 45, 1371–1374 (2013).
Seddon, J. M. et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet. 45, 1366–1370 (2013).
van de Ven, J. P. et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat. Genet. 45, 813–817 (2013).
Zhan, X. et al. Identification of a rare coding variant in complement 3 associated with age-related macular degeneration. Nat. Genet. 45, 1375–1379 (2013).
Corominas, J. et al. Whole-exome sequencing in age-related macular degeneration identifies rare variants in COL8A1, a component of Bruch’s membrane. Ophthalmology 125, 1433–1443 (2018).
Restrepo, N. A. et al. Genetic determinants of age-related macular degeneration in diverse populations from the PAGE study. Invest. Ophthalmol. Vis. Sci. 55, 6839–6850 (2014).
Morrison, M. A. et al. Ancestry of the Timorese: age-related macular degeneration associated genotype and allele sharing among human populations from throughout the world. Front. Genet. 6, 238 (2015).
Persad, P. J. et al. Joint analysis of nuclear and mitochondrial variants in age-related macular degeneration identifies novel loci TRPM1 and ABHD2/RLBP1. Invest. Ophthalmol. Vis. Sci. 58, 4027–4038 (2017).
Oliver, V. F. et al. Differential DNA methylation identified in the blood and retina of AMD patients. Epigenetics 10, 698–707 (2015).
Seddon, J. M., Reynolds, R., Shah, H. R. & Rosner, B. Smoking, dietary betaine, methionine, and vitamin D in monozygotic twins with discordant macular degeneration: epigenetic implications. Ophthalmology 118, 1386–1394 (2011).
Morgan, D. J. & DeAngelis, M. M. Differential gene expression in age-related macular degeneration. Cold Spring Harb. Perspect. Med. 5, a017210 (2014).
Wang, J. et al. ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat. Commun. 9, 1364 (2018).
Galloway, C. A. et al. Drusen in patient-derived hiPSC-RPE models of macular dystrophies. Proc. Natl Acad. Sci. USA 114, E8214–E8223 (2017).
Saini, J. S. et al. Nicotinamide ameliorates disease phenotypes in a human iPSC model of age-related macular degeneration. Cell Stem Cell 20, 635–647 e637 (2017).
Maiese, K., Chong, Z. Z., Hou, J. & Shang, Y. C. The vitamin nicotinamide: translating nutrition into clinical care. Molecules 14, 3446–3485 (2009).
Farrer, L. A. & DeAngelis, M. M. Human induced pluripotent stem cells illuminate pathways and novel treatment targets for age-related macular degeneration. Stem Cell Invest. 4, 92 (2017).
Zhong, X. et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 5, 4047 (2014).
Mahmoudi, S. & Brunet, A. Aging and reprogramming: a two-way street. Curr. Opin. Cell Biol. 24, 744–756 (2012).
Fujihara, M., Bartels, E., Nielsen, L. B. & Handa, J. T. A human apoB100 transgenic mouse expresses human apoB100 in the RPE and develops features of early AMD. Exp. Eye Res. 88, 1115–1123 (2009).
Rowan, S. et al. Involvement of a gut-retina axis in protection against dietary glycemia-induced age-related macular degeneration. Proc. Natl Acad. Sci. USA 114, E4472–E4481 (2017).
Hollyfield, J. G. et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat. Med. 14, 194–198 (2008).
Kaneko, H. et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 471, 325–330 (2011).
Malek, G. et al. Apolipoprotein E allele-dependent pathogenesis: a model for age-related retinal degeneration. Proc. Natl Acad. Sci. USA 102, 11900–11905 (2005).
Nakayama, M. et al. Overexpression of HtrA1 and exposure to mainstream cigarette smoke leads to choroidal neovascularization and subretinal deposits in aged mice. Invest Ophthalmol. Vis. Sci. 55, 6514–6523 (2014).
Pfau, M. et al. Green-light autofluorescence versus combined blue-light autofluorescence and near-infrared reflectance imaging in geographic atrophy secondary to age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 58, BIO121–BIO130 (2017).
Ouyang, Y., Heussen, F. M., Hariri, A., Keane, P. A. & Sadda, S. R. Optical coherence tomography-based observation of the natural history of drusenoid lesion in eyes with dry age-related macular degeneration. Ophthalmology 120, 2656–2665 (2013).
Schwartz, D. M. et al. Phase-variance optical coherence tomography: a technique for noninvasive angiography. Ophthalmology 121, 180–187 (2014).
Sadda, S. R. et al. Consensus definition for atrophy associated with age-related macular degeneration on OCT: classification of atrophy report 3. Ophthalmology 125, 537–548 (2018).
Hofer, H. et al. Improvement in retinal image quality with dynamic correction of the eye’s aberrations. Opt. Express 8, 631–643 (2001).
Elsner, A. E., Weber, A., Cheney, M. C., VanNasdale, D. A. & Miura, M. Imaging polarimetry in patients with neovascular age-related macular degeneration. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 24, 1468–1480 (2007).
Dysli, C. et al. Fluorescence lifetime imaging ophthalmoscopy. Prog. Retin Eye Res. 60, 120–143 (2017).
Boretsky, A. et al. In vivo imaging of photoreceptor disruption associated with age-related macular degeneration: a pilot study. Lasers Surg. Med. 44, 603–610 (2012).
Dubra, A. et al. Noninvasive imaging of the human rod photoreceptor mosaic using a confocal adaptive optics scanning ophthalmoscope. Biomed. Opt. Express 2, 1864–1876 (2011).
Fritsche, L. G. et al. Age-related macular degeneration: genetics and biology coming together. Annu Rev. Genom. Hum. Genet. 15, 151–171 (2014).
Kortvely, E. & Ueffing, M. Common mechanisms for separate maculopathies? Adv. Exp. Med. Biol. 723, 61–66 (2012).
Zinkernagel, M. S. et al. Association of the intestinal microbiome with the development of neovascular age-related macular degeneration. Sci. Rep. 7, 40826 (2017).
Bogunovic, H. et al. Machine learning of the progression of intermediate age-related macular degeneration based on OCT imaging. Invest. Ophthalmol. Vis. Sci. 58, BIO141–BIO150 (2017).
Bouhaddani, S. E. et al. Integrating omics datasets with the OmicsPLS package. BMC Bioinformatics 19, 371 (2018).
Grapov, D., Fahrmann, J., Wanichthanarak, K. & Khoomrung, S. Rise of deep learning for genomic, proteomic, and metabolomic data integration in precision medicine. OMICS 22, 630–636 (2018).
Budak, G., Dash, S., Srivastava, R., Lachke, S. A. & Janga, S. C. Express: a database of transcriptome profiles encompassing known and novel transcripts across multiple development stages in eye tissues. Exp. Eye Res. 168, 57–68 (2018).
Friedman, A. A., Letai, A., Fisher, D. E. & Flaherty, K. T. Precision medicine for cancer with next-generation functional diagnostics. Nat. Rev. Cancer 15, 747–756 (2015).
Merino, J. & Florez, J. C. Precision medicine in diabetes: an opportunity for clinical translation. Ann. NY Acad. Sci. 1411, 140–152 (2018).
Williams, A. M., Stamer, W. D. & Allingham, R. R. Increasing the availability and quality of donor eyes for research. JAMA Ophthalmol. 134, 351–352 (2016).
Acknowledgements
We thank Paul Sieving, MD, Ph.D., Director of the NEI and the National Eye Institute for organizing the AMD Pathobiology group. We also thank Anna E. Mazzucco, Ph.D. for her contributions to the group. J.T.H.: NIH EY027691; NIH R42 EY029625-01; BrightFocus Foundation; Macular Degeneration Foundation; Wilmer-Bayer Alliance Grant, Bayer Pharmaceuticals, Inc.; Unrestricted Grant from Research to Prevent Blindness (Wilmer Eye Institute), Robert Bond Welch Professorship. C.B.R.: NIH R01 EY026161; P30 EY005722 to Duke University, a Research to Prevent Blindness (RPB)/International Retinal Research Foundation (IRRF) Catalyst Award for Innovative Research Approaches for AMD, an unrestricted grant from RPB (to the Duke Eye Center), and a Fighting Blindness Individual Investigator Award. A.D.D.: NIHR Biomedical Research Centre Moorfields Eye Hospital and UCL-Institute of Ophthalmology, National Eye Research Centre UK, Macular Society UK, Medical Research Council UK, Rosetrees Trust UK. M.B.G.: Harold and Pauline Price Foundation, Research to Prevent Blindness, NY, NY, NIH/NEI R01 EY09859 Gorin (PI). J.W.M.: NEI Core Facility Grant EY014104, Yeatts Retina Fund, Research to Prevent Blindness, Retina Research Fund, Champalimaud Vision Award. C.A.T.: an unrestricted grant from RPB (to the Duke Eye Center). M.U.: EYE-RISK is funded by the Horizon 2020 program of the European Union. Funding is provided to Marius Ueffing in the period of 2015–2019 under Grant Agreement number 634479. M.Z.: Joseph J. and Marguerite DiSepio Retina Research Fund; Eng Family Foundation; New Jersey Lions Eye Research Foundation; Paid Consultant for: California Institute of Regenerative Medicine, Cell Cure, Chengdu Kanghong Biotechnology Co., Coherus Biosciences, Inc., Daiichi Sankyo, Frequency Therapeutics, Foundation Fighting Blindness, Genentech/Roche, Healios KK, Inc., Iridex, Isarna Therapeutics, Makindus, Novartis Pharma AG, Ophthotech Corp., Percept Corp. L.A.F.: NIH U01-AG032984, NIH UF1-AG046198, NIH R01-AG048927, NIH RF1-AG057519.
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions to the conception and design of the work, and have contributed to the manuscript writing and revisions.
Corresponding authors
Ethics declarations
Competing interests
J.T.H.: Grant funding from Bayer Pharmaceuticals, Inc. J.W.M.: Consultant/Advisor for Genentech/Roche, Bausch+Lomb, Kalvista Pharmaceuticals, ONL Therapeutics; Grant Support from Lowy Medical Research Institute; Equity in ONL Therapeutics; Patents/Royalties from ONL Therapeutics/Mass. Eye and Ear, Valeant Pharmaceuticals/Mass. Eye and Ear. C.A.T.: Alcon royalties for surgical technologies. M.Z.: Cell Cure, Chengdu Kanghong Biotech, Coherus Biosciences, Daiichi Sankyo, Frequency Therapeutics, Healios KK, Iridex, Isarna Therapeutics, Genentech/Roche, Makindus, Novartis Pharma AG, Ophthotech, Percept Corp., Rutgers University (patent). The remaining authors declare no competing interests.
Additional information
Peer review information: Nature Communications thanks Paul Baird and the other anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Handa, J.T., Bowes Rickman, C., Dick, A.D. et al. A systems biology approach towards understanding and treating non-neovascular age-related macular degeneration. Nat Commun 10, 3347 (2019). https://doi.org/10.1038/s41467-019-11262-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-11262-1
This article is cited by
-
Fairer AI in ophthalmology via implicit fairness learning for mitigating sexism and ageism
Nature Communications (2024)
-
The AKT2/SIRT5/TFEB pathway as a potential therapeutic target in non-neovascular AMD
Nature Communications (2024)
-
Large scale plasma proteomics identifies novel proteins and protein networks associated with heart failure development
Nature Communications (2024)
-
Qihuang Granule protects the retinal pigment epithelium from oxidative stress via regulation of the alternative complement pathway
BMC Complementary Medicine and Therapies (2023)
-
The role of the retinal vasculature in age-related macular degeneration: a spotlight on OCTA
Eye (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
