
Abstract
African swine fever virus (ASFV) is contagious and can cause highly lethal disease in pigs. ASFV DNA ligase (AsfvLIG) is one of the most error-prone ligases identified to date; it catalyzes DNA joining reaction during DNA repair process of ASFV and plays important roles in mutagenesis of the viral genome. Here, we report four AsfvLIG:DNA complex structures and demonstrate that AsfvLIG has a unique N-terminal domain (NTD) that plays critical roles in substrate binding and catalytic complex assembly. In combination with mutagenesis, in vitro binding and catalytic assays, our study reveals that four unique active site residues (Asn153 and Leu211 of the AD domain; Leu402 and Gln403 of the OB domain) are crucial for the catalytic efficiency of AsfvLIG. These unique structural features can serve as potential targets for small molecule design, which could impair genome repair in ASFV and help combat this virus in the future.
Similar content being viewed by others


Introduction
African swine fever virus (ASFV) is a large encapsulated double-stranded DNA virus. It belongs to the Asfivirus genus and is the only member of the Asfarviridae family. ASFV is highly contagious and can cause lethal disease in both domestic pigs and wild boars1. The disease caused by ASFV was first reported in Kenya in the 1920s and remained restricted to Africa till the mid 1950 s2. Since then, ASFV has spread into many countries in Europe, South America, the Caribbean region, as well as in Asia, especially Russian3. Very recently, the virus has been found in China4, the largest pork producer in the world. The virus is turning into a global threat; however, very unfortunately, no vaccine or other useful treatment against this virus has been developed5. The disease has caused very serious economic problems in many countries6. In 2011, more than 300,000 pigs in the Russian Federation region were killed by the disease with an estimated cost of 240 million US dollars.
The size of ASFV genome varies between 170 and 190 kb, and encodes more than 150 proteins that are involved in various stages of the ASFV life cycle, including gene expression, DNA replication, virion assembly, entry into host cells, and suppression of host immune response7. Though the DNA synthesis process starts in the nucleus, the replication and virion assembly of ASFV are completed in the cytoplasm of infected cells8, primarily swine macrophage cells9. Macrophages are very rich in free oxygen radicals10,11, which cause constant damages to the virus genome, such as strand breaks and spontaneous depurination/depyrimidation. To efficiently overcome these DNA damages, ASFV virus has evolved its own repair system. Interestingly, unlike in humans and many other species, the fidelities of the repair DNA polymerase (AsfvPolX)12 and ligase (AsfvLIG)13 of ASFV are very low; they can tolerate various base mismatches at the repair sites. Therefore, in addition to the maintenance of genome stability, these repair enzymes also play important roles in strategic mutagenesis and genotype formation of ASFV. By sequencing the C-terminal region of only one gene (B6464L), more than 20 ASFV genotypes have been identified14,15,16. As demonstrated in eastern and southern Africa, existence of various genotypes further complicates the epidemiology, diagnosis, and prevention of ASFV.
Surprisingly, though ASFV virus has been extensively studied over the past 90 years, the enzymes involved in the DNA repair pathway have not been well characterized;17,18 the structural information of ASFV repair enzymes has only begun to emerge in recent years19,20,21. The only crystal structures of AsfvPolX/substrate complex were reported by our group in 2017; these structures revealed a unique 5’-P binding pocket located at the finger domain of AsfvPolX22. AsfvLIG (Fig. 1a) is composed of 419 amino acids and is one of the smallest DNA ligases identified to date. AsfvLIG contains one adenylation domain (AD) in the center and one OB-fold domain (OB) at the C-terminus; both AD and OB domains are conserved in homologous proteins (Supplementary Fig. 1), including human DNA ligases (HsLig1-4)23,24,25, Sus scrofa DNA ligase 1 (SusLIG1), and archaeal ligases26,27. In addition to DNA repair, some DNA ligases also play a role in the ligation of Okazaki fragments occurring during replication28. Besides the AD and OB domains, many DNA ligases share one DNA-binding domain (DBD), which is critical for substrate binding and ligation processes. Interestingly, the N-terminal region (referred to as NTD hereafter, Supplementary Fig. 1a) of AsfvLIG has no similarity with any other ligase DBD domains. Owing to the lack of structural information, many fundamental questions, such as overall folding, substrate recognition, and the basis for mismatch tolerance by AsfvLIG, remain unanswered.

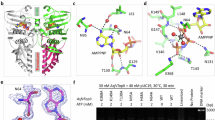
Domain architecture and ATP recognition pattern of AsfvLIG. a Schematic view comparing the domain compositions of AsfvLIG and HsLIG1. b In vitro DNA ligation catalyzed by wild-type AsfvLIG. Data represent the mean of three independent experiments, the standard deviation (SD) values are indicated by error bars. c Overall fold of the non-catalytic AsfvLIG:DNA complex. AsfvLIG is shown as surface with the NTD, AD, and OB domains colored in magenta, yellow, and green, respectively. DNA and ATP are shown as cartoons and spheres in white and atomic colors (C, light-blue; N, blue; O, red; P, orange), respectively. d Cartoon-and-stick view showing ATP recognition by the AsfvLIG AD domain. ATP is shown as sticks and outlined with simulated annealing 2Fo-Fc omit map, which is contoured at the 1.5 sigma level
Here we report the structural and functional studies of AsfvLIG. Four AsfvLIG crystal structures, including one in the non-catalytic form and three in the catalytic form, were determined. One unsealed C:G pair, one unsealed C:T pair, and one sealed C:T pair were, respectively, captured at the catalytic sites of the three catalytic form structures, referred to as AsfvLIG:CG, AsfvLIG:CT1, and AsfvLIG:CT2 hereafter. Though the molecular basis underlying the low fidelity of AsfvLIG remains elusive, our structural and biochemical studies clearly showed that both NTD domain and the unique active site residues of AsfvLIG play very important roles in substrate binding and catalysis. Similar to the novel 5′-P binding pocket of AsfvPolX22, the unique AsfvLIG NTD domain can also serve as a potential target for drug design to disrupt the DNA repair process of the ASFV genome.
Results
Low fidelity and ATP binding of AsfvLIG
To further verify the activity and fidelity of AsfvLIG, we carried out in vitro catalysis using wild type (WT) AsfvLIG and substrates containing either Watson–Crick paired or mismatched base pairs at the ligation sites (Fig. 1b and Supplementary Fig. 2). The substrates are named as DNA-XY (Supplementary Table 1), where X and Y denote the nucleotides at the template strand and at the 3′-end upstream of the nick site, respectively. We only calculated the simplified apparent rate constants (Kobs) in this study, the full enzymatic characterization leading to both Kcat and Kd values of AsfvLIG has been reported by Lamarche and co-worker previously13. AsfvLIG can efficiently catalyze the ligation reactions of the substrates with Watson–Crick base pairs; though not as efficiently as previously reported13, the ligating rate of DNA-CT is comparable to those of the Watson–Crick paired substrates (Supplementary Table 2). Interestingly, AsfvLIG can also efficiently catalyze the ligation reactions of several other substrates, such as DNA-TC, DNA-TG, and DNA-CA, which all possess a pyrimidine nucleotide on the template strands. Unlike the slow reaction previously reported13, we found that the catalytic efficiency of DNA-TC is comparable to the Watson–Crick paired substrates under our reaction conditions.
To clarify the functions of the individual domains and to reveal the basis for mismatch tolerance, we crystallized the AsfvLIG proteins with various combinations. Though no AsfvLIG structure in the absence of DNA was obtained, we successfully solved four AsfvLIG:DNA complex structures, including one in the non-catalytic form and three in the catalytic form. Crystals of the non-catalytic form structure were grown in the presence of ATP but without Mg2+, which is an essential cofactor of the adenylation reaction. We found that the non-catalytic structure adopts an extended fold (Fig. 1c) and the ATP is bound by the AD domain and stabilized by various interactions (Fig. 1d). Besides hydrophobic stacking with the side chains of Ile294 and Phe232, the nucleobase of ATP forms one hydrogen bond (H-bond) with Gln149, via the N6 and OE1 atoms. The sugar pucker 2′-OH group and the oxygen atoms of the phosphate groups form H-bonds with the side chains of Lys151, Glu203, and Lys316.
Overall folding of the catalytic AsfvLIG:DNA complex
Among the three catalytic structures, AsfvLIG:CT2 captured one sealed C:T pair, whereas AsfvLIG:CG and AsfvLIG:CT1 captured unsealed C:G and C:T pairs at the catalytic sites, respectively. The structures belong to two different space groups: P212121 for AsfvLIG:CT2 and P21 for both AsfvLIG:CG and AsfvLIG:CT1. The molecular packing of AsfvLIG:CT2 is very different from the other two structures in the crystal lattice. However, as indicated by the low root mean square deviation values (rmsd, about 0.75 Å based on the superposition of 404 pairs of Cα atoms) among them, the three complex structures are very similar to each other.
Owing to higher resolution (2.35 Å), the AsfvLIG:CT1 structure was used to demonstrate the overall folding of the catalytic form complexes (Fig. 2a and Supplementary Fig. 3). Unlike the extended non-catalytic structure (Fig. 1c), AsfvLIG undergoes large domain rearrangement and adopts a closed conformation in the catalytic AsfvLIG:DNA complex. AsfvLIG is assembled like a ring-shaped clamp and encircles the DNA duplex in its central channel. In the DNA, 19 out of 22 base pairs (bp) is covered by AsfvLIG. The AD domain of AsfvLIG mainly recognizes the broken strand at the nick site and the flanking region upstream, whereas the OB domain primarily interacts with the continuous template strand surrounding and downstream of the nick (Supplementary Fig. 4). In the non-catalytic form structure, the substrate DNA was bound by the NTD domains of two AsfvLIG molecules and adopts regular B-form conformation (Supplementary Fig. 5a); though the DNA adopts a regular B-form conformation at the two ends, it was significantly bent and unwound around the nick site in the catalytic form complex (Supplementary Fig. 5b). Similar conformational changes of the DNA have previously been observed in the HsLIG1:DNA complex structure23. As depicted in Fig. 2b and Supplementary Fig. 5c, the relative orientations of the DNA, AD, and OB domains are similar in the two structures.

The catalytic AsfvLIG:DNA complex structures. a The overall structure of AsfvLIG:CT1 complex demonstrating the arrangement of individual domains in the catalytic form complex. AsfvLIG and DNA are shown as surface and cartoon, respectively. b Comparison of the catalytic form complexes of AsfvLIG and HsLIG1 (PDB_ID: 1 × 9 N). DNAs are colored in white, whereas the ligase NTD (or DBD), AD and OB domains are colored in magenta, yellow and green, respectively. In the HsLIG1 structure, the AMP that pyrophosphate linked to the 5′-P downstream of the DNA is shown as red spheres
Unique fold of AsfvLIG NTD and its interaction with DNA
The NTD domain (residues 1–120) of AsfvLIG mimics the DBD domain (residues 262–538) of HsLIG1 in the catalytic complexes (Fig. 2b and Supplementary Fig. 5c). However, unlike the AD and OB domains, which share certain sequence similarities between the two proteins (Supplementary Figs. 1b, c), AsfvLIG NTD has no sequence similarity to the DBDs of HsLIG1 nor with any other DNA ligases. Interestingly, the overall folds of AsfvLIG NTD and HsLIG1 DBD are significantly different from each other. In fact, as revealed by the Dali search program, the overall fold of AsfvLIG NTD is completely novel. AsfvLIG NTD has a mixed α/β fold in nature (Fig. 3a and Supplementary Figs. 6a, b), which is composed of three α-helices (α1–α3) and seven β-strands (β1–β7). The β-strands form one flat β-sheet and α3 (residues 95–113) is packed against the β-sheet on one side.

The interaction between DNA and AsfvLIG NTD. a Cartoon-and-surface view showing the relative orientation between AsfvLIG NTD and DNA. b–f Detailed interactions between AsfvLIG NTD and DNA residues. AsfvLIG NTD residues are shown as sticks in atomic colors (C, green; N, blue; O, red); the DNAs are also shown as sticks, the C-atoms of the template strand, upstream and downstream of the broken strand are colored in white, yellow, and pink, respectively. g Nucleotide-residue contact map showing individual nucleotide-residues interactions for the preferred binding site. Small and large markers on each nucleotide represent the major and minor groove contacts, respectively. Filled-in pink markers highlight which nucleotides are contacted by at least one residue in the minor groove
As revealed by the AsfvLIG:CT1 structure, the NTD domain forms extensive interactions with the DNA substrate. The β3-β4 connecting loop (residues 23-27) points toward one end of the DNA duplex, forming three H-bonds (Fig. 3b). Lys27 forms one H-bond with the broken strand (2.9 Å, between its side chain Nz atom and the OP2 atom of G21) and the other two H-bonds are all formed by Lys24, including one (3.1 Å) between its Nz atom and the OP2 atom of template T5, and another (2.6 Å) between its main chain N atom and the OP1 atom of template C6. Unlike T5 and C6, both the OP1 and OP2 oxygen atoms of template G8 are involved in direct H-bond interactions with AsfvLIG NTD (Fig. 3c). OP1 interacts with the OG1 atom of Thr64 (of the β5 strand) and OP2 interacts with the NE1 atom of Trp31 (of the β4 strands).
α3 is the longest α-helix of AsfvLIG NTD; it lies along the major groove of the DNA duplex (Fig. 3a). α3 forms two H-bonds with the broken strand of the DNA: one (3.0 Å) between the NE2 atom of Gln98 and the OP1 atom of T8 and the other (2.9 Å) between the OG atom of Ser105 and the OP2 atom of G9 (Fig. 3d). T8 and G9 are located in the middle of the DNA duplex. Via direct or water-mediated H-bonding (Fig. 3e), the neighboring G6 and A7 of the upstream DNA interact with Lys85 and Asn86 of the β7-α3 connecting loop (residues 82-94); the H-bond distances are all within the range of 2.7–3.1 Å. The β7-α3 connecting loop points toward the end of the DNA duplex but at the opposite direction of the β3-β4 connecting loop (Fig. 3a). Though they do not form direct H-bonds, the shape of the three tip residues (Lys89, Lys90, and Asn91) matches with the major groove and the sugar puckers of the DNA template strand (Fig. 3f). As revealed by the DNAProDB program29, several other AsfvLIG NTD residues also interact with the DNA nucleotides (Fig. 3g).
NTD is critical for catalytic complex assembly and catalysis
In the catalytic AsfvLIG:DNA complex structures, the β6-β7 connecting linker of AsfvLIG NTD bends toward one short α-turn of the OB domain (Fig. 4a). Different from the HsLIG1:DNA structure that forms several direct H-bonds between the DBD and OB domains, the NTD and OB domains of AsfvLIG interact with each other via two water-mediated H-bonds. Compared to the OB domain, the AD domain forms many more interactions with the AsfvLIG NTD domain. As depicted in Fig. 4b, one α-turn of the AD domain resides next to the C-terminus of strand β3 and helix α3 of the NTD domain. The side chain of the α-turn residue Asn307 forms two H-bonds: one (2.9 Å) between its ND2 atom and the main chain O atom of Glu22 and the other (2.8 Å) between its OD1 atom and the side chain Nz atom of Lys114. Arg115 of α3 helix projects toward Tyr306, forming stacking interactions between their side chains (Fig. 4c). Side chains of two other residues of the NTD α3 helix are also involved in the interaction with the AD domain (Fig. 4d). The NH1 atom of Arg112 forms one H-bond (3.0 Å) with the side chain OG1 atom of Thr173; the NE2 atom of Gln113 also forms one H-bond (2.8 Å) with Thr173 but with the main chain O atom. Thr173 is located at the tip of the one loop, residing next to the nick site of the broken DNA strand.

Impacts of AsfvLIG NTD on DNA binding and ligation. a Relative orientation between the NTD and OB domains in the catalytic form AsfvLIG-DNA complex. b–d Relative orientation and detailed interactions between the NTD and AD domains in the catalytic form structure. e Comparison of nick and duplex DNA-CG binding by WT AsfvLIG and AsfvLIG NTD. All data points from three independent experiments are shown with the median expressed as bars. The standard deviation (±SD) values are indicated by error bars. f In vitro DNA-CG ligation catalyzed by WT AsfvLIG and AsfvLIG with NTD deletion (for AsfvLIG ΔN). The substrate and product bands are labeled as S and P, respectively. Uncropped gels are shown in Supplementary Fig. 7
Though AsfvLIG NTD has no sequence or structural similarity to HsLIG1 DBD, it mimics HsLIG1 DBD in the catalytic complexes (Fig. 2b and Supplementary Fig. 5c). The total numbers of DNA base pairs covered by NTD (19 bp) and DBD (18 bp) are similar in the two structures. DBD is important for DNA binding and catalysis of HsLIG1: deletion of DBD lowers the substrate binding affinity by > 75-fold and reduces the catalytic efficiency of HsLIG1 by > 4 × 105 fold23. To clarify the function of AsfvLIG NTD, we carried out an in vitro DNA-binding assay using nick and duplex DNA-CG. WT AsfvLIG can bind both nick and duplex DNA-CG (Fig. 4e and Supplementary Fig. 6c); within the concentration range of 0.2–0.8 μM, the nick DNA-binding affinity of WT AsfvLIG is slightly higher than that of duplex DNA. Compared to WT AsfvLIG, the DNA-binding affinity of NTD is much weaker. At a 1.6 μM concentration, NTD also showed weak preference for the nick DNAs. Similar nick DNA preference was also observed for HsLIG1 and its DBD domain previously23. Replacing residues 85–92 of the DNA-interacting β7-α3 loop with two Gly residues (for AsfvLIG LD, Supplementary Fig. 6d) lowered the DNA-binding affinity by 2-fold. Deletion of the NTD domain (for AsfvLIG ΔN) completely abolished the DNA-binding ability of the protein (Supplementary Fig. 6d).
In addition to binding activity, we also investigated the DNA ligation activity of WT AsfvLIG and mutants using nick DNA-CG. As depicted in Fig. 4f and Supplementary Fig. 7a, the WT AsfvLIG can efficiently catalyze the ligation reaction, the reaction rate (Kobs) is 134.83 ± 3.32 × 10−6 min−1; no ligation activity was observed for AsfvLIG ΔN (Supplementary Fig. 7b). AsfvLIG LD can support the ligation process (Supplementary Fig. 6e); consistent with its weak DNA-binding affinity, the reaction rate (1.48 ± 0.20 × 10−6 min_1) of AsfvLIG LD is significantly lower than that of the WT AsfvLIG. Together, these observations indicate that NTD plays important role in DNA binding; the cooperative interactions between NTD and other domains (especially the AD domain) can enhance the DNA-binding affinity and are critical for the catalytic complex assembly and catalysis of AsfvLIG.
Conformational comparison of the nick site base pairs
Consistent with a previous study13, our in vitro catalytic assays further confirmed that AsfvLIG is an error-prone DNA ligase (Fig. 1b and Supplementary Fig. 2). AsfvLIG can ligate various substrates with mismatched base pairs, as well as Watson–Crick paired DNA substrates (Supplementary Table 2). Our structures represent several different states of the reaction: the AsfvLIG:CG complex structure captured the Watson–Crick C:G pair prior to ligation (Supplementary Fig. 8a), whereas AsfvLIG:CT1 and AsfvLIG:CT2 captured the mismatched C:T pair prior to and after the ligation process (Supplementary Figs. 8b, c), respectively.
DNA-CT is one of the mismatched substrates that can be ligated by AsfvLIG as efficiently as the Watson–Crick paired substrates. Analysis and comparison of the three catalytic structures revealed some potential basis underlying the C:T mismatch tolerance by AsfvLIG. In the AsfvLIG:CT1 structure (Fig. 5a and Supplementary Fig. 8d), the nucleobases of the template C11 and the nick site T12 form one H-bond (3.1 Å) between their N4 and O4 atoms, respectively. Further, via the water-mediated H-bond, the O2 atom of the C11 nucleobase also interacts with the main chain N atom of Gln403 of the OB domain. Though not identical, the overall shapes and orientations of the C:T pair in AsfvLIG:CT1 and the C:G pair in AsfvLIG:CG are similar (Fig. 5b and Supplementary Fig. 8e). Structural superposition (Fig. 5c) further revealed that presence of mismatched C11:T12 pair does not cause obvious conformational difference on 3′-OH, 5′-P, or phosphodiester backbone at the active site, compared with the Watson–Crick C11:G12 pair.

Structural basis for C:T and C:G base pair recognition by AsfvLIG. a, b Local conformations of the C:T and C:G pairs captured in the AsfvLIG:CT1 and AsfvLIG:CG structures, respectively. c Superposition of the nick site base pairs in AsfvLIG:CT1 and AsfvLIG:CG. d Local conformations of the C:T pair captured in the AsfvLIG:CT2 structure. e Superposition of the nick site base pairs in AsfvLIG:CT1 and AsfvLIG:CT2. f Detailed conformations of the active site residues Leu402 and Gln403, based on the AsfvLIG:CT1 structure. g, h In vitro DNA ligation catalyzed by L402R and Q403F mutants, respectively. The data represent the mean of three independent experiments, the standard deviation (±SD) values are indicated by error bars. In panels (a–e), the C-atoms of protein and DNA residues are colored in green, yellow, and magenta for the AsfvLIG:CT1, AsfvLIG:CT2, and AsfvLIG:CG structures, respectively
In the AsfvLIG:CT2 structure (Fig. 5d), the template C11 and the sealed T12 form one H-bond (2.9 Å) between their nucleobases. However, though the pairing and overall shape of C11:T12 pairs are similar in the AsfvLIG:CT1 and AsfvLIG:CT2 structures, superposition reveals some subtle conformational changes at the local regions. Compared to the AsfvLIG:CT1 structure, the C11-T12 pair is shifted about 0.5 Å toward the main chain N atom of Gln403 in the AsfvLIG:CT2 structure, which may exclude the C11-interacting water molecule (Supplementary Fig. 8f). Meanwhile, the side chain of Gln403 rotates clock wise around the CG-CD bond for about 15° in the AsfvLIG:CT2 structure (Fig. 5e), leading to the formation of one H-bond (2.9 Å) between the NE2 atom of Gln403 and the O2 atom of the T12 nucleobase (Fig. 5d), thereby shifting the conformation to the ligated product.
Leu402 and Gln403 impact the ligation activity of AsfvLIG
Gln403 of AsfvLIG corresponds to Phe872 in HsLIG1; interestingly, the residues prior to Gln403 and Phe872 are also different in AsfvLIG and HsLIG1, which have Leu402 and Arg871, respectively. As depicted in Supplementary Fig. 1c, both Arg871 and Phe872 residues are highly conserved in HsLIG1 and other DNA ligases26,27,30. Though Leu402 does not form strong interactions with the C11:T12 pairs in the AsfvLIG:CT1 and AsfvLIG:CT2 structures, its side chain resides next to the neighboring C12:G11 pair from the minor groove side (Fig. 5f). Among AD and OB domains, six residues (Leu211, Ala215, and Asn219 of the AD domain; Tyr363, Leu402, and Gln403 of the OB domain) form contacts with DNA in the minor groove (Supplementary Fig. 4a); Leu402 and Gln403 are the only two residues next to the nick of the DNA.
To investigate the potential roles of Leu402 and Gln403, we first constructed one AsfvLIG protein with the whole OB domain deleted (referred to as ΔOB hereafter). As depicted in Supplementary Fig. 9a, AsfvLIG ΔOB can bind both duplex and nick DNAs. The DNA-binding affinity of AsfvLIG ΔOB is stronger than that of AsfvLIG NTD but is much weaker than the full-length WT AsfvLIG (Supplementary Fig. 6c). Unlike WT AsfvLIG, which mainly forms complex with DNA with a molar ratio of 1:1, ΔOB can form both 1:1 and 2:1 complex with DNA. However, as revealed by the in vitro ligation assay (Supplementary Fig. 9b), ΔOB could not support the ligation reaction, indicating that the observed ΔOB:DNA complexes are incompatible with the catalysis.
Besides AsfvLIG ΔOB, we also constructed two AsfvLIG mutants (L402R and Q403F) and carried out in vitro DNA ligation assays using DNA-CT and DNA-TC, the two most tolerable mismatched substrates of AsfvLIG. Compared to WT AsfvLIG (Fig. 1b), replacing Leu402 with Arg402 (for L402R mutant) lowered the DNA-CT and DNA-TC ligation rates of the protein by about 20-fold (Fig. 5g and Supplementary Fig. 10a), to 10.06 ± 0.36 × 10−6 min−1 and 8.03 ± 0.26 × 10−6 min−1, respectively; replacing Gln403 with Phe403 (for Q403F mutant) caused more than 600-fold reduction on the DNA-CT and DNA-TC ligation rates of the protein (Fig. 5h and Supplementary Fig. 10b), to only 0.31 ± 0.02 × 10−6 min−1 and 0.25 ± 0.03 × 10−6 min−1, respectively.
As noted previously, mismatched C:T pair and Watson–Crick C:G pair have similar conformations in the AsfvLIG:CT1 and AsfvLIG:CG structures (Supplementary Fig. 8e). To test whether Leu402 and Gln403 impact the ligation efficiency of Watson–Crick paired substrates, we performed in vitro ligation assays using DNA-CG, DNA-GC, DNA-AT, and DNA-TA (Fig. 5g, h). Compared to the WT AsfvLIG, the L402R and Q403F mutants had much lower ligation rates, varied from ~40–75-fold for DNA-AT and DNA-TA substrates to ~100–200-fold for DNA-GC and DNA-CG substrates. In general, the impact of the Q403F mutation was greater than that of L402R mutation toward these DNA substrates.
Comparison with homologous proteins
The AD and OB domains are conserved in AsfvLIG and all HsLIG1-4 proteins. As depicted in Supplementary Fig. 1b, AsfvLIG AD shares 38%, 36%, and 29% sequence similarity with the AD domains of HsLIG1, HsLIG3, and HsLIG4, respectively. Compared to the AD domains, the sequence similarity between the OB domains (Supplementary Fig. 1c) are lower between AsfvLIG and HsLIG1, HsLIG3, and HsLIG4, which are 18%, 17%, and 20%, respectively. In addition to HsLIG1:DNA complex, one close catalytic form HsLIG3:DNA complex (PDB_ID: 3L2P)24 and several open form HsLIG4 structures31 have been reported. The overall structures of HsLIG1:DNA and HsLIG3:DNA complexes are very similar. Superposition between AsfvLIG1:CT1 and HsLIG1:DNA (Supplementary Fig. 5c) or HsLIG3:DNA (Supplementary Fig. 11a) resulted in rmsd values of 1.9 Å in their AD domains, based on the superposition of 166 and 173 pairs of Cα atoms. Possibly due to the lower sequence similarity, AsfvLIG1 OB domain exhibits more obvious conformational differences with HsLIG1 and HsLIG3, especially for the α-helix located at the edge. Based on the superposition of 82 and 74 pairs of Cα atoms, the rmsd values between the OB domains of AsfvLIG1 and HsLIG1 and HsLIG3 are 2.9 ÅÅ and 2.8 Å, respectively.
All the HsLIG4 structures adopt an open conformation (Supplementary Fig. 11b). The relative orientations between the AD and OB domains of HsLIG4 structures are different from our open form AsfvLIG:DNA complex (Fig. 1c and Supplementary Fig. 5a). However, structural comparison showed that the overall conformations of the AD domains of HsLIG4 and AsfvLIG are similar (Supplementary Fig. 11c); the rmsd value between them is 1.8 Å, based on the superposition of 159 pairs of Cα atoms. Compared to HsLIG1 and HsLIG3, the OB domain of AsfvLIG showed more significant conformational difference to HsLIG4 (Supplementary Fig. 11d). Superposition of the central β-sheet core resulted in a rmsd value great than 3.0 Å; all α-helice took significant different locations in the two structures.
In addition to Leu402 and Gln403 (or Arg871 and Phe872) of the OB domain, the above comparison revealed that sequences and conformations of several AD domain residues surrounding the DNA nick sites are different in AsfvLIG and HsLIG1 (Fig. 6a–c). Instead of the neighboring base pair, Arg871 of HsLIG1 is located directly under the nick site base pair and forms one salt bridge with Asp570. The Arg871-Asp570 pair is flanked by Phe872 on one side and by another Phe residue (Phe536) on the opposite side. Besides DNA distortion, previous studies suggested that the Asp570-Arg871 salt bridge and packing interactions between the Phe residues (Phe872 and Phe635) and the ribose sugars of nucleotides on the 5′ and 3′ ends of the nick all contribute to the fidelity and efficiency of HsLIG123. Both Asp570 and Phe635 of the HsLIG1 AD domain are highly conserved in many DNA ligases, whereas they are replaced by Asn153 and Leu211 in AsfvLIG, respectively (Supplementary Fig. 1b).

Unique ligase residues in proximity of the nick in the substrate DNA of AsfvLIG. a, b Cartoon representation showing the DNA-binding surface formed by the AD and OB domains of AsfvLIG and HsLIG1, respectively. c Superposition of the ligase residues in proximity of the nick in the substrate DNA. In (a–c), all the ligase residues are shown as sticks. Identical colors are utilized for the N-atoms (blue) and O-atoms (red) in both structures, but the C-atoms are colored in yellow and green in AsfvLIG and HsLIG1, respectively. d Comparison of the in vitro DNA-CT binding by WT AsfvLIG, and L402R and Q403F mutants. In the right panel, WT AsfvLIG, and L402R and Q403F mutants are colored in red, green, and blue, respectively. The complexes with protein:DNA molecular ratio of 2:1 are colored in light-green or light-blue. All data points from three independent experiments are shown with the median expressed as bars. The standard deviation (±SD) values are indicated by error bars. Uncropped gels are shown in Supplementary Fig. 13. e In vitro DNA ligation catalyzed by WT HsLIG1. f In vitro DNA ligation catalyzed by HsLIG1-d mutant. HsLIG1-d and HsLIG1-q stand for R871L/F872Q double mutant and D570N/F635L/R871L/F872Q quadruple mutant, respectively. The data represent the mean of three independent experiments, and the standard deviation (±SD) values are indicated by error bars
The conformations of Asn153 and Asp570 are similar in the two structures, whereas the conformations of Leu211, Leu402, and Gln403 of AsfvLIG are different from the corresponding residues of HsLIG1. To further investigate the functions of these unique residues, we carried out in vitro ligation assays using L402R/Q403F double mutant, N153D/L402R/Q403F triple mutant, and N153D/L211F/L402R/Q403F quadruple mutant (Supplementary Fig. 12 and Supplementary Table 3). Interestingly, none of these AsfvLIG mutants showed enhanced catalytic efficiency toward any substrates, including Watson–Crick paired and mismatched DNAs. In contrast, the activities of the three mutants were much lower than the WT AsfvLIG; and, for most of the substrates, their ligation activities were even lower than the L402R and Q403F mutants.
Together with the mutants with a single mutation, the ligation assay results of the double, triple, and quadruple mutants indicated that the four nick site residues play important roles in the catalytic efficiency of AsfvLIG. Compared to the Asp570-Arg871 salt bridge and the aromatic rings of Phe635 and Phe872 observed in the HsLIG1 structure, the local conformation formed by Asn153, Leu211, Leu402, and Gln403 were more flexible in the AsfvLIG structure; instead of discrimination between Watson–Crick paired and mismatched base pairs, our ligation results indicate that these residues may play a more important role in the assembly and stabilization of the catalytic complex. To further support this hypothesis, we carried out in vitro DNA-binding assays (Fig. 6d and Supplementary Fig. 13). Compared with WT AsfvLIG, replacing either Leu402 with Arg402 or Gln403 with Phe403 at low concentrations (ranging from 0.1 to 0.8 μM) has no clear impacts on DNA binding. At high concentration (1.6 μM), the overall DNA-CT binding abilities of L402R and Q403F were comparable to that of WT AsfvLIG; however, in contrast to WT AsfvLIG, a significant number of slower moving bands were observed on the native gel in the presence of mutants, especially Q403F. We speculate that the slower moving band may correspond to a complex with a protein:DNA molecular ratio of 2:1; similar assembly has been observed in the non-catalytic form AsfvLIG:DNA structure (Supplementary Fig. 5a).
Switching of nick site residues between AsfvLIG and HsLIG1
The four HsLIG1 residues (Asp570, Phe635, Arg871, and Phe872) in the proximity of the nick in the DNA substrate are highly conserved in many DNA ligases. However, our in vitro ligation assays clearly indicated that these residues can not replace the corresponding residues of AsfvLIG in catalysis. To further investigate the functional role of these nick site residues, we also constructed and purified the WT HsLIG1, one R871L/F872Q double mutant (HsLIG1-d), and one D570N/F635L/R871L/F872Q quadruple mutant (HsLIG1-q) of HsLIG1. During in vitro ligation assays, we found that the overall ligation activity of WT HsLIG1 is higher than that of AsfvLIG. The ligation rates of Watson–Crick paired DNAs are comparable in the presence of 0.01 μM HsLIG1 (Fig. 6e, Supplementary Fig. 14, and Supplementary Table 4) or 0.05 μM AsfvLIG (Fig. 1b and Supplementary Fig. 2). Among the non-Watson–Crick paired substrates, HsLIG1 can catalyze the ligation of DNA-TG most efficiently, but the DNA-TG ligation rate (Kobs) is 2–3 fold lower than those of Watson–Crick paired DNAs. A similar phenomenon has been observed previous32, maybe due to the T:G wobble-pair formation in the nick site. Unlike WT HsLIG1, HsLIG1-d could not catalyze the ligation of any non-Watson–Crick paired DNA substrate (Supplementary Fig. 15). Though HsLIG1-d can still catalyze the ligation of Watson–Crick paired DNA substrates (Fig. 6f, Supplementary Fig. 15, and Supplementary Table 4), the reaction rate is much lower than those of WT HsLIG1. HsLIG1-q could not catalyze the ligation of any Watson–Crick or non-Watson–Crick paired DNA substrates (Supplementary Fig. 16 and Supplementary Table 4). Though we are not sure about their exact role in the fidelity of HsLIG1, our in vitro ligation assay results clearly indicated that these nick site residues play important role in the catalytic efficiency of HsLIG1.
Catalytic mechanism and in vitro nick DNA binding
Despite their differences in sequence and size, all the characterized ATP-dependent DNA ligases catalyze phosphodiester bond formation between adjacent 3′-OH and 5′-phosphate in DNA duplex through a similar three-step mechanism (Fig. 7a). In the first step, the NH2 group of the catalytic Lys residue attacks the α-phosphate of ATP, forming an enzyme-AMP intermediate and inorganic pyrophosphate (PPi). In the second step, nick DNA binds to the enzyme-AMP intermediate and the 5′-P of the downstream DNA attacks Lys-AMP to form a pyrophosphate linked AppDNA intermediate. In the third step, the 3′-OH of the upstream DNA attacks the pyrophosphate group of AppDNA, covalently joining the DNA strands and liberating AMP. The ATP was captured in the active site of the non-catalytic form AsfvLIG:DNA structure (Fig. 1c, d). The catalytic Lys residue (Lys151) and ATP-interacting residues of AsfvLIG are highly conserved in the homologous proteins (Supplementary Fig. 1). Though the domain arrangement of the non-catalytic form AsfvLIG:DNA complex is very different from that of HsLig1 complexed with adenylated DNA23, the interactions between ATP and the interacting residues are very similar in the two structures (Fig. 7b), indicating that AsfvLIG follows a conserved mechanism in ATP binding and catalysis.

Comparison of ATP and DNA binding by AsfvLIG and HsLIG1. a The conserved catalytic mechanism of ATP-dependent DNA ligation. b Superposition showing the similar ATP binding observed in AsfvLIG and HsLIG1 structures. AsfvLIG of the non-catalytic AsfvLIG:DNA complex is shown as cartoon in yellow, and ATP and ATP-interacting residues are shown as sticks in atomic colors (C, yellow; N, blue; O, red; P, orange). HsLIG1 in the complex with adenylated DNA (PDB_ID: 1 × 9 N) is shown as cyan cartoon. DNA, AMP, and AMP-interacting residues of HsLIG1 are shown as sticks in atomic colors (C, cyan; N, blue; O, red; P, orange). c In vitro DNA binding by AsfvLIG1. d In vitro DNA binding by HsLIG1. All data points from three independent experiments are shown with the median expressed as bars. The standard deviation (±SD) values are indicated by error bars
Conceptually, differentiation between matched and mismatched base pairs first takes place in step 2 of the reaction. To verify whether AsfvLIG discriminates between match and mismatch during step 2, we carried out in vitro DNA-binding assay (Fig. 7c and Supplementary Fig. 17). Similar to the four Watson–Crick paired DNAs, DNAs with mismatched base pairs at the 3’-end of the nick can all be efficiently bound by AsfvLIG; the binding affinities between AsfvLIG and all DNAs are very similar. Besides AsfvLIG, we also carried out in vitro DNA-binding assay using HsLig1 (Fig. 7d and Supplementary Fig. 18). Compared to AsfvLIG, HsLig1 has similar binding affinity to mismatched DNAs with pyrimidines (C or T) on the template strand; however, when the protein concentrations are within the range of 0.2–0.8 μM, the binding affinities between HsLig1 and mismatched DNAs with purines (A or G) on the template strands are significantly weaker than those of AsfvLIG. Very surprising, compared to the mismatched DNAs, the binding affinities between HsLig1 and the four matched DNAs are much weaker.
Discussion
ASFV is contagious and can cause lethal diseases in both domestic pigs and wild boars. AsfvLIG catalyzes the DNA ligation reaction during the base excision repair (BER) process of ASFV genome. However, unlike the homologous proteins (such as HsLIG1 and SusLIG1), the fidelity of AsfvLIG is very low. By solving four AsfvLIG:DNA complex structures, including the non-catalytic open form (Fig. 1c) and the catalytic closed forms (Fig. 2a), we showed that the NTD, AD, and OB domains of AsfvLIG undergo a large conformational change during the catalytic assembly. A bound ATP molecule is present in the non-catalytic structure (Fig. 1c and Supplementary Fig. 5a), demonstrating that AsfvLIG has ATP binding and catalytic mechanism commonly shared by many other ligases (Supplementary Fig. 5b)33,34,35,36.
In the catalytic form AsfvLIG:DNA complex structures, AsfvLIG NTD domain mimics the DBD domains of the homologous ligases; however, AsfvLIG NTD has a complete novel DNA-binding fold. Deletion of NTD (for AsfvLIG ΔN) completely abolished the DNA binding (Supplementary Fig. 6d) and ligation (Fig. 4f) activities of AsfvLIG. Clearly, the extensive interactions between AsfvLIG NTD and nick DNAs contribute to the strong DNA-binding affinity of AsfvLIG, which are equal for matched and mismatched DNAs (Fig. 7c). HsLig1 is a high fidelity DNA ligase (Fig. 6e). Based on the HsLig1:DNA complex structure (PDB_ID: 1 × 9 N), it has been previously proposed that HsLig1 can cause some local distortion on the duplex; this distortion is important for the alignments of the 3′-OH and the adenylated 5′-P for nick sealing23. The overall matched DNA ligation activity of HsLig1 is much higher than that of AsfvLIG. However, very surprisingly, HsLig1 has much weaker binding affinities towards the matched nick DNAs, compared to the mismatched nick DNAs (Fig. 7d). These observations suggested that HsLig1 may be able to sense the structural perturbation at the position of the mismatch and stay bound to the DNA, leading to ligation inhibition.
Interestingly, though the overall folds of the AD and OB domains are conserved in AsfvLIG and the homologous proteins, their sequence identities are very low (Supplementary Figs. 1b, c). At the active site, AsfvLIG contains four unique residues, including Asn153 and Leu211 of the AD domain and Leu402 and Gln403 of the OB domain. Compared to AsfvLIG, the local conformation formed by the corresponding nick site residues (including Asp570, Phe635, Arg871, and Phe872) of HsLig1 is much more rigid. The nick site residues could not be switched between AsfvLIG and HsLig1, suggested that these residues might work in a cooperative manner with their corresponding NTD or DBD domain only. Though they are not involved in the direct catalytic process, our in vitro DNA binding and ligation assay results (Figs. 5g, h, 6d, 7b, and Supplementary Figs. 10, 12, 14-16) suggested that these nick site residues are important for nick recognition and thus catalytic efficiency of AsfvLIG and HsLig1.
Consistent with a previous study37, the obvious different DNA-binding behavior, unique fold of the NTD domain, and unique residues at the nick site all confirm that AsfvLIG is one atypical DNA ligase. Before any AsfvLIG structure was reported, Showalter and co-workers already demonstrated that AsfvLIG has very low adenylation activity towards DNAs with 3′-dideoxy- or 3′-amino-terminated nicks, compared to regular nick DNAs;13 these observations indicated that 3′-OH of the nick is a critical component of the active site architecture during 5′-P adenylation. The interaction between the NTD domain and the four nick site residues will help the reorientation of DNA 3′-OH and facilitate the ligation reaction. Compared to the other three nick site residues, Gln403 appears to be more important for the catalytic efficiency of AsfvLIG, maybe due to its H-bond interaction with the nick site base pairs (Fig. 5b). At present, the exact molecular basis underlying the low fidelity of AsfvLIG still remains elusive. More biochemical and structural data, especially the structures of AsfvLIG complexed with adenylated DNA, are required to better understand the tolerance of mismatched DNAs during the catalytic process.
Recent discovery of the poly (ADP-ribose) polymerase inhibitor (which can selectively target the DNA repair defect in hereditary breast cancer) has stimulated the inhibitor development of other repair enzymes38. As the enzyme required during almost all the repair events, ligase has been most extensively targeted39,40,41. Obvious repair inhibitory effects have been observed for many compounds targeting the DBD domains of human ligases (including HsLIG1-4) in vitro and/or in vivo42. In combination with their subtoxicities, some compounds can preferentially sensitize human cancer cell lines to the cytotoxic effects of other DNA agents. However, due to the conservation of DBD domains, many inhibitors can simultaneously target various human ligases, which limited their therapeutic potential.
ASFV is replicated and assembled in swine macrophage cells, which are oxidative and cause continuous damage to the virus genome. Though it is error-prone and may contribute to quick genotype formation, AsfvLIG is the sole ligase involved in the repair pathway. Therefore, inhibiting its activity will certainly disrupt the repair process and impair the genome stability of ASFV. AsfvLIG NTD has a mixed α/β fold in nature and represents a complete novel DNA-binding motif; though it may be not important for the fidelity, AsfvLIG NTD plays critical role in substrate binding and catalytic complex assembly of AsfvLIG. Although the structures of pig DNA ligases, including SusLIG1-4, have not been characterized, they should be very similar to those of human HsLIG1-4 evidenced by the high sequence similarities (all around 90%). Since AsfvLIG NTD has no sequence or folding similarity with any DNA ligases in pigs, it can serve as an excellent target for the development of small molecule inhibitor, and the AsfvLIG NTD targeting inhibitors will not interfere with the normal DNA joining functions of the pig ligases. Besides NTD, the nick site residues (Asn153, Leu211, Leu402, and Gln403) of AsfvLIG are very unique. As revealed by our mutagenesis and in vitro catalytic studies, these nick site residues cannot be switched between AsfvLIG and the homologous proteins. Small molecules targeting the nick site residues of AsfvLIG and other unique features of ASFV repair enzymes, such as the 5′-P binding pocket in the finger domain of AsfvPolX, can all impair ASFV repair process and help provide tools to combat this deadly virus.
Methods
Plasmid construction
The gene containing the codon-optimized cDNA sequence of full-length WT AsfvLIG (Supplementary Table 5) was purchased from Shanghai Generay Biotech Co., Ltd, China. The gene was cleaved with BamHI and XhoI and visualized using an agarose gel. The target fragment was recovered and recombined into the pET28-Sumo vector. The recombinant vector (coding for His-Sumo-AsfvLIG) was then transformed into Escherichia coli BL21 DE3 competent cells for protein expression. The recombinant His-Sumo-AsfvLIG coding vector was utilized as the template during the plasmid constructions of all AsfvLIG NTD, AsfvLIG ΔN, AsfvLIG LD (in which the residues 85–92 of WT AsfvLIG were replaced by two Gly residues), L402R, and Q403F mutants, via overlap polymerase chain reactions (PCR) or site direct mutagenesis according to the manufacturer’s protocols. The resulting His-Sumo-AsfvLIG L402R plasmid DNA was then used as the template for constructing double (L402R/Q403F), triple (L402R/Q403F/N153D), and quadruple (L402R/Q403F/N153D/L211F) mutants. Detailed sequences of the primers used in the constructions are listed in Supplementary Table 5.
The region coding for residues 262 to 919 of HsLIG1 was amplified by PCR using human genomic cDNA as template and sub-cloned into the BamHI/XhoI restriction sites of pET28a- Sumo vector. The resulting recombinant vector was transformed into the Escherichia coli BL21 DE3 competent cells and the plasmid DNA was extracted and used as template for the R871L/F872Q double mutant construction with site direct mutagenesis kit. The R871L/F872Q plasmid DNA was then used in the preparation of the HsLIG1 D570N/F635L/R871L/F872Q quadruple mutant. Detailed sequences of the primers used in WT and mutant HsLIG1 constructions are listed in Supplementary Table 6. Sequences of all WT and mutant of His-Sumo-AsfvLIG and His-Sumo-HsLIG1 plasmids were confirmed by DNA sequencing. All recombinant strains were preserved using 30% glycerol and stored in a −80 °C freezer prior to use.
Protein expression and purification
All His-Sumo-AsfvLIG and His-Sumo-HsLIG1 proteins were expressed using the same procedures. Briefly, the frozen recombinant strains were revived in Lysogeny broth (LB) medium supplemented with 50 µg/mL kanamycin at 37 °C overnight. Every 20 mL revived bacterium suspension was inoculated into 1 L LB medium supplemented with kanamycin (50 µg/mL) and cultured at 37 °C with continuous shaking. Protein expression was induced at OD600 ≈ 0.6 by adding of isopropyl β-D-1-thiogalacto-pyranoside (IPTG) at a final concentration of 0.1 mM. The induced cultures were then grown at 18 °C for an additional 18 h. The cells were harvested by centrifugation.
For overproduction of the Se-Met substituted AsfvLIG, the revived recombinant strains from 20 mL overnight cultures were inoculated into 1 L LB medium supplemented with 50 µg/mL kanamycin and grown at 37 °C. When OD600 reached 0.4, the cells were harvested by centrifugation and resuspended in 100 mL M9 medium (47.7 mM Na2HPO4, 22 mM KH2PO4, 8.6 mM NaCl, and 28.2 mM NH4Cl). The resuspended cells were centrifuged and transferred into 900 mL fresh M9 medium supplemented with 50 µg/mL kanamycin and 30 mg/L Se-Met (J&K). After growing at 37 °C for 1 h, the temperature was lowered to 18 °C and the protein expression was induced by addition of IPTG at a final concentration of 0.1 mM. The induced cultures were then grown at 18 °C for an additional 18 h and the cells were harvested by centrifugation.
All AsfvLIG proteins were purified using the same procedures. The cell pellets were resuspended in Buffer A (20 mM Tris pH 8.0, 500 mM NaCl, 25 mM imidazole pH 8.0) and lysed under high pressure via a JN-02C cell crusher. The homogenate was clarified by centrifugation and the supernatant was loaded onto a HisTrapTM HP column equilibrated with Buffer A. The fusion protein was eluted from the column using Buffer B (20 mM Tris pH 8.0, 500 mM NaCl, 500 mM imidazole pH 8.0) with a gradient. The fractions containing the desired fusion proteins were pooled and dialyzed against Buffer S (20 mM Tris pH 8.0, 500 mM NaCl, 25 mM imidazole pH 8.0) at 4 °C for 3 h; Ulp1 protease was also added to the sample during the dialysis process. The sample was again loaded onto the HisTrapTM HP column, the target protein was collected, and mixed with Tris buffer (20 mM, pH 8.0). The diluted sample was applied to a HiTrap SP HP column equilibrated with S binding buffer (20 mM Tris pH 8.0, 100 mM NaCl) and eluted using S elution buffer (20 mM Tris pH 8.0, 1 M NaCl) with a continuous gradient. The target protein was concentrated and loaded onto a Hi 16/60 Superdex G200 column equilibrated with gel filtration buffer (20 mM Tris pH 8.0, 100 mM NaCl). All HsLIG1 proteins were purified using the similar procedures without the HiTrap SP HP column purification step. A volume of 1 mM DTT was present in all buffers, to prevent the oxidation of the proteins. The purity of the proteins was analyzed using an SDS-PAGE gel and the samples were stored at −80 °C until use.
In vitro DNA binding and catalysis assays
All DNAs used (Supplementary Table 1) in the binding and catalytic assays were purchased from Shanghai Generay Biotech Co., Ltd. The sixteen DNA substrates containing either Watson–Crick paired or mismatched base pairs were assembled by mixing the continuous template strand with upstream and downstream oligos in a molar ratio of 1:1:1 in gel filtration buffer. For catalysis assays, a 10-μL reaction system (composed of 6 µL gel filtration buffer, 1 µL 100 mM MgCl2, 1 µL 10 mM ATP, 1 µL 8 µM DNA, and 1 µL protein) was established. The final AsfvLIG and HsLIG1 protein concentrations are 0.05 µM and 0.01 µM, respectively. The reactions were carried out at 37 °C and quenched by the addition of 10 µL termination buffer (90% formamide, 20 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene blue) at various time points. A total of 8 µL samples were loaded onto pre-warmed 18% urea sequencing gels and run at 18–20 W and 40–45 °C for 60 min. The gel was visualized using Typhoon FLA 9000, and intensities of the substrate and product bands were quantified by ImageQuantTL. Data were then fitted to the exponential Y = Ymax[1-e(-Kobst)] using non-linear regression in GraphPad Prism 5. The observed rate constant (Kobs) and maximum ligation yield (Ymax) were determined from the regression curve.
Substrates DNA-CT or DNA-CG were diluted to 4 µM with gel filtration buffer and used in electrophoretic mobility shift assays (EMSA). A 10-µL reaction system, composed of 7 µL gel filtration buffer, 1 µL DNA (4 µM), and 2 µL proteins (either WT or mutants at a concentration of 8 µM, 4 µM, 2 µM, 1 µM, or 0.5 µM) was established. The samples were incubated at 0 °C for 90 mins and mixed with 10 µL loading buffer (20% glycerol, 0.05% bromophenol blue, and 0.05% xylene blue). A total of 8 µL samples were loaded onto pre-cooled 10% native gels and run at 120 V for 60 min. The gel was imaged using Typhoon FLA 9000. The intensities of the bands were quantified by ImageQuantTL and the data were compared using GraphPad Prism.
Crystallization and data collection
All DNAs used in the structural studies were dissolved in ddH2O; detailed sequences of the DNAs are listed in Supplementary Table 7. The crystallization samples were prepared by mixing AsfvLIG, DNA, and MgCl2 and/or ATP (if present) at room temperature. The initial crystallization conditions for all crystals were identified at 18 °C using the crystallization robot system and commercial crystallization kits. During the initial screening, the sitting-drop vapor diffusion method was used, whereas all the crystal optimization procedures were performed using the hanging-drop vapor diffusion method. The compositions of the final crystallization conditions are also listed in Supplementary Table 7.
All crystals were cryoprotected using their mother liquor supplemented with 25% glycerol and snap-frozen in liquid nitrogen. X-ray diffraction data were collected on beamline BL17U and BL19U at the Shanghai Synchrotron Radiation Facility (SSRF). One crystal was used for each structure; data processing was carried out using the HKL2000 or HKL3000 programs43. The data collection and processing statistics are summarized in Supplementary Table 8.
Structure determination and refinement
The Se-AsfvLIG:DNA structure was solved using the single-wavelength anomalous diffraction (SAD) method44 with the AutoSol program45 embedded in the Phenix suite46. The Figure of Merit (FOM) value was 0.36. The initial model, which covers ~70% of protein residues in the asymmetric unit, was built using the Autobuild program and was refined against the diffraction data using the Refmac5 program47 of CCP4i. During refinement, 5% of randomly selected data was set aside for free R-factor cross validation calculations. The 2Fo-Fc and Fo-Fc electron density maps were regularly calculated and used as guides for building the missing amino acids, DNA, and solvent molecules using COOT48. All AsfvLIG:CG, AsfvLIG:CT1, and AsfvLIG:CT2 structures were solved using the MR method with the Phaser program of the CCP4i suite using the individual domains of Se-AsfvLIG:DNA structure as the search model. The final refinement of all structures was done using the phenix.refine program49 of Phenix. The structural refinement statistics are summarized in Supplementary Table 8.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Structural factors and coordinates have been deposited in the Protein Data Bank under accession codes 6IMJ, 6IMK, 6IML, and 6IMN for Se-AsfvLIG:DNA, AsfvLIG:CG, AsfvLIG:CT1, and AsfvLIG:CT2, respectively. All other data are available from the corresponding author upon reasonable request.
References
Tulman, E. R., Delhon, G. A., Ku, B. K. & Rock, D. L. African swine fever virus. Curr. Top. Microbiol. Immunol. 328, 43–87 (2009).
Arzt, J., White, W. R., Thomsen, B. V. & Brown, C. C. Agricultural diseases on the move early in the third millennium. Vet. Pathol. 47, 15–27 (2010).
Gogin, A., Gerasimov, V., Malogolovkin, A. & Kolbasov, D. African swine fever in the North Caucasus region and the Russian Federation in years 2007-2012. Virus Res. 173, 198–203 (2013).
Zhou, X. et al. Emergence of African Swine Fever in China, 2018. Transbound Emerg Dis. 65, 1482–1484 (2018).
Sanchez-Vizcaino, J. M., Mur, L. & Martinez-Lopez, B. African swine fever: an epidemiological update. Transbound. Emerg. Dis. 59(Suppl 1), 27–35 (2012).
Vinuela, E. African swine fever virus. Curr. Top. Microbiol. Immunol. 116, 151–170 (1985).
Yanez, R. J. et al. Analysis of the complete nucleotide sequence of African swine fever virus. Virology 208, 249–278 (1995).
Rojo, G., Garcia-Beato, R., Vinuela, E., Salas, M. L. & Salas, J. Replication of African swine fever virus DNA in infected cells. Virology 257, 524–536 (1999).
Alcami, A., Carrascosa, A. L. & Vinuela, E. Interaction of African swine fever virus with macrophages. Virus Res. 17, 93–104 (1990).
Akaike, T. Role of free radicals in viral pathogenesis and mutation. Rev. Med. Virol. 11, 87–101 (2001).
Forman, H. J. & Torres, M. Redox signaling in macrophages. Mol. Asp. Med. 22, 189–216 (2001).
Oliveros, M. et al. Characterization of an African swine fever virus 20-kDa DNA polymerase involved in DNA repair. J. Biol. Chem. 272, 30899–30910 (1997).
Lamarche, B. J., Showalter, A. K. & Tsai, M. D. An error-prone viral DNA ligase. Biochemistry 44, 8408–8417 (2005).
Boshoff, C. I., Bastos, A. D., Gerber, L. J. & Vosloo, W. Genetic characterisation of African swine fever viruses from outbreaks in southern Africa (1973-1999). Vet. Microbiol. 121, 45–55 (2007).
Bastos, A. D. et al. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 148, 693–706 (2003).
Achenbach, J. E. et al. Identification of a new genotype of African swine fever virus in domestic pigs from Ethiopia. Transbound. Emerg. Dis. 64, 1393–1404 (2017).
Redrejo-Rodriguez, M., Garcia-Escudero, R., Yanez-Munoz, R. J., Salas, M. L. & Salas, J. African swine fever virus protein pE296R is a DNA repair apurinic/apyrimidinic endonuclease required for virus growth in swine macrophages. J. Virol. 80, 4847–4857 (2006).
Garcia-Escudero, R., Garcia-Diaz, M., Salas, M. L., Blanco, L. & Salas, J. DNA polymerase X of African swine fever virus: insertion fidelity on gapped DNA substrates and AP lyase activity support a role in base excision repair of viral DNA. J. Mol. Biol. 326, 1403–1412 (2003).
Wu, W. J. et al. How a low-fidelity DNA polymerase chooses non-Watson-Crick from Watson-Crick incorporation. J. Am. Chem. Soc. 136, 4927–4937 (2014).
Maciejewski, M. W. et al. Solution structure of a viral DNA repair polymerase. Nat. Struct. Biol. 8, 936–941 (2001).
Showalter, A. K., Byeon, I. J., Su, M. I. & Tsai, M. D. Solution structure of a viral DNA polymerase X and evidence for a mutagenic function. Nat. Struct. Biol. 8, 942–946 (2001).
Chen, Y. et al. Unique 5’-P recognition and basis for dG:dGTP misincorporation of ASFV DNA polymerase X. PLoS Biol. 15, e1002599 (2017).
Pascal, J. M., O’Brien, P. J., Tomkinson, A. E. & Ellenberger, T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature 432, 473–478 (2004).
Cotner-Gohara, E. et al. Human DNA ligase III recognizes DNA ends by dynamic switching between two DNA-bound states. Biochemistry 49, 6165–6176 (2010).
De Ioannes, P., Malu, S., Cortes, P. & Aggarwal, A. K. Structural basis of DNA ligase IV-Artemis interaction in nonhomologous end-joining. Cell Rep. 2, 1505–1512 (2012).
Nishida, H., Kiyonari, S., Ishino, Y. & Morikawa, K. The closed structure of an archaeal DNA ligase from Pyrococcus furiosus. J. Mol. Biol. 360, 956–967 (2006).
Petrova, T. et al. ATP-dependent DNA ligase from Thermococcus sp. 1519 displays a new arrangement of the OB-fold domain. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 68(Pt 12), 1440–1447 (2012).
MacNeill, S. A. DNA replication: partners in the Okazaki two-step. Curr. Biol. 11, R842–R844 (2001).
Sagendorf, J. M., Berman, H. M. & Rohs, R. DNAproDB: an interactive tool for structural analysis of DNA-protein complexes. Nucleic Acids Res. 45(W1), W89–W97 (2017).
Pascal, J. M. et al. A flexible interface between DNA ligase and PCNA supports conformational switching and efficient ligation of DNA. Mol. Cell 24, 279–291 (2006).
Ochi, T., Gu, X. & Blundell, T. L. Structure of the catalytic region of DNA ligase IV in complex with an Artemis fragment sheds light on double-strand break repair. Structure 21, 672–679 (2013).
Bhagwat, A. S., Sanderson, R. J. & Lindahl, T. Delayed DNA joining at 3 ‘ mismatches by human DNA ligases. Nucleic Acids Res. 27, 4028–4033 (1999).
Unciuleac, M. C., Goldgur, Y. & Shuman, S. Structure and two-metal mechanism of a eukaryal nick-sealing RNA ligase. Proc. Natl Acad. Sci. USA 112, 13868–13873 (2015).
Nair, P. A. et al. Structural basis for nick recognition by a minimal pluripotent DNA ligase. Nat. Struct. Mol. Biol. 14, 770–778 (2007).
Nandakumar, J., Nair, P. A. & Shuman, S. Last stop on the road to repair: structure of E. coli DNA ligase bound to nicked DNA-adenylate. Mol. Cell 26, 257–271 (2007).
Odell, M., Sriskanda, V., Shuman, S. & Nikolov, D. B. Crystal structure of eukaryotic DNA ligase-adenylate illuminates the mechanism of nick sensing and strand joining. Mol. Cell 6, 1183–1193 (2000).
Showalter, A. K. et al. Mechanistic comparison of high-fidelity and error-prone DNA polymerases and ligases involved in DNA repair. Chem. Rev. 106, 340–360 (2006).
Lord, C. J. & Ashworth, A. Targeted therapy for cancer using PARP inhibitors. Curr. Opin. Pharmacol. 8, 363–369 (2008).
Tseng, H. M. et al. A high-throughput scintillation proximity-based assay for human DNA ligase IV. Assay. Drug Dev. Technol. 10, 235–249 (2012).
Sun, D. & Urrabaz, R. Development of non-electrophoretic assay method for DNA ligases and its application to screening of chemical inhibitors of DNA ligase I. J. Biochem. Biophys. Methods 59, 49–59 (2004).
Zhong, S. et al. Identification and validation of human DNA ligase inhibitors using computer-aided drug design. J. Med. Chem. 51, 4553–4562 (2008).
Chen, X. et al. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 68, 3169–3177 (2008).
Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D. Biol. Crystallogr. 62(Pt 8), 859–866 (2006).
Giacovazzo, C. & Siliqi, D. Phasing via SAD/MAD data: the method of the joint probability distribution functions. Acta Crystallogr. D. Biol. Crystallogr. 60(Pt 1), 73–82 (2004).
Terwilliger, T. C. et al. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D. Biol. Crystallogr. 65(Pt 6), 582–601 (2009).
Adams, P. D. et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D. Biol. Crystallogr. 58(Pt 11), 1948–1954 (2002).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr. 67(Pt 4), 355–367 (2011).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60(Pt 12 Pt 1), 2126–2132 (2004).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr. 68(Pt 4), 352–367 (2012).
Acknowledgements
We thank the BL17U and BL19U beamline staff at the Shanghai Synchrotron Radiation Facility (SSRF) for help during data collection. This work was supported by the National Natural Science Foundation of China (31870721, 31470724, 31670878, 21572146, 21761132029) and the Key Research and Development Project of China (2016YFA0500600), and Key R&D Program of Sichuan (2018NZ0151).
Author information
Authors and Affiliations
Contributions
Y.C. produced and purified the proteins, and performed the biochemical assays. Y.C., H.L., and L.Z. grew the crystals. Y.C., C.Y., Y.G., X.Y., and X.C. collected the X-ray data. Y.C., H.L., and J.G. determined the structures. Y.C., H.L., R.C., S.L., X.L., J.M., Z.H., J.L., and J.G. analyzed the data. Y.C., H.L., Z.H., J.L., and J.G. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Liu, H., Yang, C. et al. Structure of the error-prone DNA ligase of African swine fever virus identifies critical active site residues. Nat Commun 10, 387 (2019). https://doi.org/10.1038/s41467-019-08296-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-08296-w
This article is cited by
-
Massive expansion of the pig gut virome based on global metagenomic mining
npj Biofilms and Microbiomes (2024)
-
Structures of African swine fever virus topoisomerase complex and their implications
Nature Communications (2024)
-
Structures of LIG1 that engage with mutagenic mismatches inserted by polβ in base excision repair
Nature Communications (2022)
-
A unique DNA-binding mode of African swine fever virus AP endonuclease
Cell Discovery (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
