
Abstract
Besides high efficiency, the stability and reproducibility of perovskite solar cells (PSCs) are also key for their commercialization. Herein, we report a simple perovskite formation method to fabricate perovskite films with thickness over 1 μm in ambient condition on the basis of the fast gas−solid reaction of chlorine-incorporated hydrogen lead triiodide and methylamine gas. The resultant thick and smooth chlorine-incorporated perovskite films exhibit full coverage, improved crystallinity, low surface roughness and low thickness variation. The resultant PSCs achieve an average power conversion efficiency of 19.1 ± 0.4% with good reproducibility. Meanwhile, this method enables an active area efficiency of 15.3% for 5 cm × 5 cm solar modules. The un-encapsulated PSCs exhibit an excellent T80 lifetime exceeding 1600 h under continuous operation conditions in dry nitrogen environment.
Similar content being viewed by others


Introduction
Organic−inorganic lead halide perovskite solar cells (PSCs) have drawn a great deal of attention in the photovoltaic research community due to their high efficiency and simple manufacturing process1,2,3,4,5. The power conversion efficiency (PCE) of PSCs has reached over 22% 6. Similar to other kinds of thin film photovoltaics, film qualities of perovskite layers can heavily influence device performance of PSCs. Thus, a delicate control of film quality of perovskite layers is key to achieving both superior performance and high reproducibility. Many solution processable perovskite formation methods have been developed to fabricate high-quality perovskite films, such as one-step method2,7,8,9 and two-step method10. These methods usually involve a transformation of 2D lead halide inorganic framework to the 3D perovskite structure. This structure transformation is often accompanied with the formation of morphological defects and crystallographic structure dislocation within the grain and at grain boundaries, where defects are present as trap states causing serious charge recombination and limiting the charge carrier diffusion length (less than 1 μm)11,12. To ensure efficient charge collection in the presence of defects, perovskite films are usually made sufficiently thin. So far, high efficiency PSCs mainly adopt perovskite films with thickness ranging from 400 to 800 nm (Supplementary Table 1). The low thickness fluctuation tolerance and morphological defects of thin perovskite films decrease reproducibility of the fabrication process. Moreover, thin films are prone to thickness variations and pinhole defects, which increase significantly with the area.
It is obvious that thicker perovskite films are likely to reduce the risk of forming voids and pinholes due to the larger thickness fluctuation tolerance. A high-quality thick perovskite film is desirable to achieve a high device yield and reproducibility in making large-area solar modules. Besides, thicker perovskite films can not only improve the light harvesting13,14, but also broaden light response region by utilizing the below-band absorption15. As such, it is advantageous to construct a high-quality thick perovskite film with enough carrier diffusion length in PSCs to endorse both higher efficiency and manufacturing viability. However, thick perovskite films over 1 μm have been found to be generally less efficient than thin-films devices due to the poor mobility, limited carrier diffusion length and are seldom explored (Supplementary Table 1). Moreover, conventional perovskite formation methods are usually not suitable for fabricating large-area high-quality thick perovskite films. Thus, improving the quality of thick perovskite films is critical to balance the thickness and efficiency. Also, the development of new perovskite formation techniques is desirable to construct high-quality and thick perovskite films.
Recently, a perovskite formation process based on methylamine (CH3NH2) gas−solid reaction has been developed to prepare high-quality CH3NH3PbX3 (X = I, Br, Cl), mixed halides CH3NH3+-based perovskite and Cesium/CH3NH3+ mixed cations perovskite films16,17,18. This gas−solid reaction-based method introduces a liquid intermediate, which leads to induced morphology reconstruction, defects healing19,20. Meanwhile, CH3NH2 gas treatment can help reduce the defect density in perovskite films19,20 and also elimination of I213. Besides, CH3NH2-induced δ-NH2CH=NH2PbI3 in (CH3NH3)x(NH2CH=NH2)1−xPbI3 is also reported to passivate trap states to improve device performance21. Moreover, excess solvent is absent during this perovskite formation process, which avoids the possible detrimental effect-related residue solvent. Therefore, CH3NH2 gas-based processes are promising for depositing high-quality perovskite films across large area for PSC device fabrication22,23.
In terms of the synthetic techniques developed for perovskite, using chloride containing raw materials to prepare perovskite is a commonly used method3. Although the chlorine content in resultant perovskite films is usually much lower than the nominal ratio of chlorine added in the precursor solution and in some cases the chorine content in fully annealed perovskite films is below detection limit, both the optoelectronic properties and morphology can be tuned by appropriate chlorine incorporation24,25. The resultant carrier diffusion length improvement11,12, defects density reduction26, optimization of film growth27,28,29, and charge tranport30,31 make this method effective in obtaining high-quality perovskite films. In particular, CH3NH3PbI3−xClx (i.e., CH3NH3PbI3 incorporated with a small amount of Cl) has been reported to exhibit a longer carrier diffusion length over 1 μm compared with pure CH3NH3PbI3, and as a result device performance improved dramatically11,12.
These earlier works have inspired us to combine the CH3NH2 gas-based perovskite formation method and partial substitution of iodine ions by chorine ions to fabricate high quality over 1-μm-thick perovskite films with a sufficient charge diffusion length, which is not only beneficial for better light absorbing but also desirable for fabricating large-area solar modules with high yield and reproducibility via low-cost printing techniques13,32.
Here we report a simple perovskite formation method to fabricate over 1-μm-thick perovskite films based on the fast gas−solid reaction of chlorine-incorporated hydrogen lead triiodide (HPbI3(Cl)) and CH3NH2 gas. The comprehensive characterization results reveal that with the synergistic effect of CH3NH2 gas and partial substitution of iodine ions by chorine ions, the resultant CH3NH3PbI3(Cl) (MAPbI3(Cl)) films with a thickness over 1 μm and low thickness variation exhibit excellent film quality. The resultant PSCs gave an average PCE of 19.1% and low PCE standard deviation (±0.4%), which indicates the excellent reproducibility of this method. Meanwhile, this method enables an active area PCE of 15.3% for 5 cm × 5 cm solar modules. Besides, the un-encapsulated PSCs exhibit an excellent T80 lifetime exceeding 1600 h under continuous operation conditions in dry N2 environment. Our film stability study also offers the in-depth understanding for the underlying mechanisms responsible for device stability improvement.
Results
Perovskite formation
Conventional perovskite formation methods are usually not suitable for fabricating high-quality thick perovskite thin films (Supplementary Fig. 1). Here, we developed a fast gas−solid reaction of HPbI3(Cl) and CH3NH2 gas to prepare high-quality thick perovskite films. In this method, hydrogen lead triiodide (HPbI3) was used as the starting material aiming at taking advantage of its fully coordinated [PbI3]− structure to reduce iodide-vacancy and improve film quality (Supplementary Figs. 2, 3)33,34. Firstly, we tuned the thickness of perovskite films based on the HPbI3–CH3NH2 reaction20 by varying the substrate temperature (Supplementary Figs. 4, 5, 6, Supplementary Table 2 and Supplementary Note 1). As a result, perovskite films with thickness about 1.1 μm are used as our benchmark for further studies. Partial substitution of iodine ions by chorine ions step depicted in Fig. 1a was achieved by reacting CH3NH3Cl (MACl) with HPbI3 to form HPbI3(Cl). Considering that MACl exists in the solid state at room temperature but sublimes at elevated temperatures (higher than 100 °C), the introduction of MACl can not only achieve a small amount of substitution of iodine ions by chorine ions, but also tune the morphology of HPbI3(Cl) films27,35. Top set in Fig. 1b shows the top-view scanning electron microscope (SEM) images of HPbI3(Cl) films prepared using HPbI3/MACl precursor solution with different MACl contents. It is found that upon increasing the MACl content, the size of voids between islands-like crystals in the HPbI3(Cl) films gradually decreases, and the crystals gradually turn from the hexagonal shape to the square shape and then to crystals with round edges (Supplementary Fig. 7). The enhanced coverage of HPbI3(Cl) films is expected to be beneficial for the subsequent CH3NH2 gas-based perovskite formation process.

Perovskite film formation. a Schematic drawing showing the reaction process of partial substitution of iodine ions by chorine ions to form chlorine-incorporated perovskite MAPbI3(Cl) films: First, iodine ions in HPbI3 are partially substituted by chorine ions to form HPbI3(Cl). Then HPbI3(Cl) reacts with CH3NH2 gas to form MAPbI3(Cl) via the ultra-fast acid-base reaction. b Top-view SEM images of HPbI3(Cl) (top set, scale bar: 50 μm) and MAPbI3(Cl) (bottom set, scale bars: 5 μm) films prepared using different molar ratios of MACl versus HPbI3 in the HPbI3/MACl precursor solution
To further investigate the incorporation of chlorine into the HPbI3, we used X-ray diffraction (XRD) spectroscopy to characterize the obtained HPbI3(Cl) films. It is reported that the HPbI3 films show a hexagonal structure20,34,36. Upon increasing MACl content from 0 to 0.20, the low-angle peak around 11.7° shifts to a higher two theta value and the peak intensity increases significantly (Supplementary Fig. 8). This result suggests that the substitution of I– ion with smaller Cl– ion can shrink the crystal lattice, and HPbI3(Cl) still preserves the pseudo-hexagonal phase in the case of MACl content lower than 0.4. When the MACl content is increased to 0.40, the XRD peak intensity of the HPbI3(Cl) film at around 11.7° reduced substantially. This observation suggests that when the MACl content is too high, the crystal structure of HPbI3(Cl) cannot maintain the previous structure, which is also supported by the observed shape difference of crystal islands by SEM (Supplementary Fig. 7). This is because the large difference between the ionic radii of I– and Cl– makes the crystal structure unstable when the content of chlorine is too high. Such a phenomenon has also been observed in MAPbI3−xClx. When a small amount of Cl was incorporated, MAPbI3−xClx can still maintain the pseudo-cubic phase11,12. However, a continuous solid phase of MAPbI3−xClx theoretically cannot form at a high chlorine content due to the large size mismatch between chloride and iodide30. Besides, a slight splitting of the XRD peaks around 11.7° and the formation of MAPbI3 are observed for high MACl content of 0.70 and 1.00 cases. The former observation may be due to the incomplete removal of MACl or phase separation of HPbI3(Cl), and the latter observation suggests that the reaction of MACl with HPbI3 can form MAPbI3. The nonvisible perovskite peaks in HPbI3(Cl) films at low MACl content from 0.05 to 0.40 may be due to the low amount of MAPbI3 seeds and detection limit of XRD. Here, we can deduce that the possible reaction for the formation of HPbI3(Cl) at low MACl content up to 0.40 is as follows:
where the chlorine-incorporated HPbI3, i.e., H1−xPb1−xI3−4xClx is termed as HPbI3(Cl) hereafter; for MACl, x + y represents the initial amount of MACl in the HPbI3/MACl precusor solution, x represents the amount of MACl used for chlorine incorporation, y represents the amount of MACl that sublimes during thermal annealing.
The as-prepared HPbI3(Cl) films were then exposed to CH3NH2 gas atmosphere to form perovskite via the reaction, CH3NH2 + H+ → CH3NH3+ 20. As shown in Fig. 1b bottom set, the apparent grain size of the MAPbI3(Cl) films gradually increases upon the increase of MACl content. However, too much MACl is found to cause significant deterioration of the film morphology, especially for the MACl content of 1.00 case (Supplementary Fig. 9). The structural properties of MAPbI3(Cl) films were further examined by XRD (Fig. 2a, b and Supplementary Fig. 10). All the samples show a preferred orientation along the (110) crystallographic plane, and the corresponding peak intensity becomes stronger with consecutive substitution of iodine ions by chorine ions. This preferable (110) crystal orientation is induced by the CH3NH2-based perovskite formation process19,37. The gradually increased peak intensity is ascribed to the effect of chlorine on perovskite film growth27,35. Furthermore, it was found that the low-angle XRD peak around 14.1° shifts to a higher two theta value and the peak intensity also increases upon increasing the MACl content. The same as the aforementioned discussion about the HPbI3(Cl), substitution of I– by smaller-size Cl– can also lead to the shrinkage of the crystal lattice of perovskite, which is consistent with the cell parameters variation (Supplementary Fig. 11). A slight splitting of the XRD peaks is also observed in the case of the MACl content of 1.00, in which the large size mismatch between chloride and iodide precludes the formation of a continuous solid phase of MAPbI3−xClx, and results in phase segregation and large cell parameter variation (Fig. 2b and Supplementary Fig. 11). The observed additional peak around 28.2 ° is likely associated with the hydrate phase of perovskite38.

Partial substitution of iodine ions by chorine ions. a, b X-ray diffraction (XRD) patterns of MAPbI3 films prepared with different molar ratios of MACl versus HPbI3 in the HPbI3/MACl precursor solution. c UV−Vis spectra of MAPbI3 films prepared with different molar ratios of MACl versus HPbI3 in the HPbI3/MACl precursor solution. d SIMS profiles showing I (magenta), Pb (red), Cl (black), and Ti (blue) elements from the top to the bottom of the MAPbI3 film prepared with the molar ratio of MACl versus HPbI3 to be 0.10 in the HPbI3/MACl precursor solution
Although MACl is introduced into precursor with nominal ratios of MACl/HPbI3 from 0.1 to 1, the remaining contents of chlorine in HPbI3(Cl) and MAPbI3(Cl) can be significantly lower than the nominal content of chlorine in precursor solution. This is due to the fact that MACl tends to sublime at elevated temperatures. Furthermore, chlorine tends to form volatile chlorine-containing species, which also tends to gradually diffuse and sublimate from the film, especially in the presence of water25,27. Thus, after thermal annealing in open air (relative humidity of about 40−60%) the remaining chlorine content in the resultant MAPbI3(Cl) films is much lower than the chlorine content in the precursor solution. The low content of chlorine in the MAPbI3(Cl) films is proven by the tiny change in absorption edge (Fig. 2c)39. It should be noted that the fluctuation of absorption for the case of MACl content of 0.70 and 1.00 may be due to the observed slight phase segregation and large roughness (Supplementary Fig. 12)25,27,35,39. Similar to chorine inclusion, phase segregation is also observed in the bromine inclusion case with high content, i.e., (MAPb(IxBr1−x)3) (0.1 < x < 0.8)40, in which perovskite undergo phase separation into iodide-rich and bromide-rich regions under light illumination41. In the case of bromine inclusion with low content, the existence of bromine is found to assist grain growth, improve stability, and device performance40,41,42.
To further confirm the existence of chorine in MAPbI3(Cl) films, we employed secondary-ion mass spectrometry (SIMS) to characterize the depth profile of chlorine in the perovskite film with MACl content of 0.1 (which gives the best device performance as discussed in the device performance section). As shown in Fig. 2d, the Cl depth profile exhibits a continuous and gradual increase, and then shows a significant spatial overlap on the Ti profile. This observation is consistent with other reports revealing that the distribution of chlorine within MAPbI3(Cl) is often inhomogeneous25,43,44,45. One possible cause responsible for the inhomogeneous distribution of chlorine is due to the tendency of chlorine to form volatile chlorine-containing species gradually diffusing and sublimating from the top surface of the film, especially in ambient air, resulting in a gradient increase of the Cl content from the film surface to bottom25. Although the XRD results of the MAPbI3(Cl) films do not show obvious peaks of PbCl2 or MAPbCl3, it is worth noting that in addition to incorporation into perovskite, the chlorine may form some chloride compounds that are nondetectable by XRD due to the detection limit or the amorphous nature of these compounds. Here, we can complete the second step of MAPbI3(Cl) formation reaction depicted in Fig. 1a as follows:
where the chlorine-incorporated perovskite, i.e., MAPbI3−xClx is termed as MAPbI3(Cl) hereafter.
Device performance
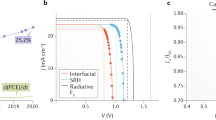
To study the device performance, we implemented our smooth 1.1-μm-thick MAPbI3(Cl) films in the device configuration of fluorine-doped tin oxide (FTO) /compact TiO2/meso-TiO2/perovskite/spiro-OMeTAD/Au. Supplementary Fig. 13 and Supplementary Table 3 show the device performance results. The MACl content of 0.10 case delivers the best device performance (Supplementary Note 2) with an average current density (JSC) of 22.2 ± 0.3 mA cm−2, open-circuit voltage (VOC) of 1.09 ± 0.02 V, fill factor (FF) of 0.793 ± 0.015, and PCE of 19.1 ± 0.4%. As shown in Fig. 3a, a best PCE of 20.0% was achieved. The integrated JSC from external quantum efficiency (EQE) is in excellent agreement with current density–voltage (J−V) measurements, with a discrepancy below 2% (Fig. 3b). The representative J–V curves under different scan directions indicate the negligible hysteresis effect (Supplementary Fig. 14). In addition, a stabilized PCE approaching 19% for device (with efficiency of 19.2% from J−V measurements) is also achieved by holding the voltage at the maximum power point for 500 s (Fig. 3c), which further confirms the negligible hysteresis effect. When compared with reports listed in Supplementary Table 1, the MAPbI3(Cl)-based devices in this work can also deliver a comparable high efficiency with perovskite film thickness over 1.1 μm as shown in Fig. 3d. This suggests that at least the 1.1-μm-thick MAPbI3(Cl) layer deposited here does not show a detrimental effect in carrier transport. To further inspect the devices, we performed the cross-sectional-view SEM (Fig. 4a). It is found that the grains extend across the entire absorber layer minimizing grain boundaries and providing facile pathways for efficient charge transport. All these results prove that we obtained high-quality thick perovskite films, and high efficiency PSCs can be fabricated based on these films.

Photovoltaic performance. a Current density–voltage (J−V) curve for the best performing PSC with AM 1.5 radiation under ambient condition. b External quantum efficiency (EQE) spectrum and integrated JSC for perovskite solar cells based on the 1.1-μm-thick MAPbI3(Cl) films. c Stabilized photocurrent density output by holding the voltage at the maximum power point (0.926 V) for PSCs based on 1.1-μm-thick MAPbI3(Cl) films. d Comparison of the PSCs and the corresponding perovskite film thickness in the representative reports and this work (Supplementary Table 1)

Solar cells and modules. a Cross-sectional-view SEM image of the PSC based on 1.1-μm-thick films. b, c Histograms of the thickness and root-mean-square (RMS) roughness of 16 1.1-μm-thick MAPbI3(Cl) samples, respectively. d Photograph of the 1.1-μm-thick MAPbI3(Cl) film on a 5 cm × 5 cm substrate with the mirror-like smooth surface. e Photograph of a 12.0 cm2 six-cell perovskite solar module fabricated using our new method. f J–V curve of the 5 cm × 5 cm perovskite module with an active area of 12.0 cm2 based on 1.1-μm-thick MAPbI3(Cl) films
Reproducibility is crucial for mass production of low-cost optoelectronic device applications. When compared with PSCs based on perovskite films fabricated using the antisolvent method, our 1.1-μm-thick MAPbI3(Cl) film-based devices show significantly improved reproducibility represented by a much smaller PCE standard deviation (decreasing from ±1.2% to ±0.4%, Supplementary Fig. 15 and Supplementary Table 4). The high reproducibility of device performance is attributed to the high reproducibility of 1.1-μm-thick MAPbI3(Cl) films. To confirm this point, we measured the thickness and roughness of 16 samples of the 1.1-μm-thick MAPbI3(Cl) films (Fig. 4b, c and Supplementary Figs. 16, 17). The average thickness and roughness together with their standard deviations are determined to be 1.13 ± 0.03 μm and 5.4 ± 0.4 nm, respectively. In addition, the device performance of 1.1-μm-thick MAPbI3(Cl) devices is higher than that of the devices based on 450-nm-thick perovskite film deposited by the antisolvent method. The higher efficiency for the former case is mainly due to the improved JSC and FF, which is also supported by the EQE results in Supplementary Fig. 15. This improvement is not only due to the thicker film but also its improved film quality. Moreover, a slightly wider spectral response is observed for the 1.1-μm-thick MAPbI3(Cl) film-based devices. These results suggest that high-quality, thick perovskite films can benefit the enhancement of light harvesting capability, especially at the near-infrared region, towards better device performance.
Development of scalable fabrication processes is key to further industrialization of the PSC technology. We demonstrate that our new perovskite formation method is scalable and can be used to fabricate large-area solar modules. Figure 4d shows the optical photograph of 1.1-μm-thick MAPbI3(Cl) film on 5 cm × 5 cm substrates with the mirror-like smooth surface. It is also found that the film is much more uniform over the entire 5 cm × 5 cm substrate than that of antisolvent case (Supplementary Figs. 18, 19). We then fabricated six-cell modules (active area = 12.0 cm2, geometric fill factor = 48%) as shown in Fig. 4e. A best active area PCE of 15.3% under reverse scan was achieved with a JSC of 3.66 mA cm−2, VOC of 6.65 V, and FF of 0.630 (Fig. 4f), which is among the top PCE values obtained for perovskite solar modules with an active area greater than 10 cm2 35,46,47,48,49,50,51,52,53,54,55,56. Furthermore, our new method shows good reproducibility in the large-area device fabrication with an average module PCE and standard deviation of 13.6 ± 0.8% (Supplementary Table 5), which is much better than that of the antisolvent case (8.6 ± 1.6%, Supplementary Fig. 20 and Supplementary Table 6). These results further confirm the excellent reproducibility of our 1.1-μm-thick MAPbI3(Cl) films on large scale, which is a key advantage for manufacturing in the realistic industrial large-scale setting. Note that small J–V hysteresis was observed for solar modules (Supplementary Fig. 21).
Charge carrier transport behavior
As shown in Fig. 3d, we obtained high efficiencies with 1.1-μm-thick perovskite films. The promising device performance inspired us to conduct systematical investigations of charge transport properties of our PSC devices to find out what factors lead to the high device performance. We first performed conductive atomic force microscopy (c-AFM) measurements on the 1.1-μm-thick MAPbI3(Cl) film and perovskite film prepared by the antisolvent method (denoted as MAPbI3(AS)). It was found that the former shows lower roughness (Supplementary Note 3) and rounded large grains with almost four times larger grain size, and its corresponding fewer grain boundaries are expected to reduce the defects density (Supplementary Fig. 22). Moreover, the 1.1-μm-thick MAPbI3(Cl) film also shows nearly an order of magnitude higher light-induced current (Fig. 5a), which indicates its good electrical conductivity. This observation can be ascribed to the improved crystallinity of the MAPbI3(Cl) films with preferential (110) orientation (Fig. 5b), as a result of synergistic effect of CH3NH2 gas19,57 and partial substitution of I− by Cl−11,12.

Charge carrier transport behavior. a The light-induced current extracted from the c-AFM results and b XRD spectra for 1.1-μm-thick MAPbI3(Cl) film prepared by the HPbI3(Cl)/CH3NH2 method and MAPbI3(AS) film prepared by the antisolvent method. c Time-resolved photoluminescence (TRPL) decay for 1.1-μm-thick MAPbI3(Cl) and MAPbI3 films prepared by the HPbI3(Cl)/CH3NH2 method. d–f Current density–voltage (J−V) characteristics of devices with FTO/perovskite/Au configuration and g–i corresponding cross-sectional-view SEM images utilized for estimating the trap density and mobility for 1.1-μm-thick MAPbI3(Cl) and MAPbI3 films prepared by the HPbI3(Cl)/CH3NH2 method, and MAPbI3(AS) film prepared by the antisolvent method. Scale bars: 1 μm
In addition to the improvement in terms of the film morphology and conductivity, the defect nature in perovskite films may also play a role in influencing charge carrier transport properties58. To investigate radiative and nonradiative charge carrier recombination channels within perovskite films, we carried out time-resolved photoluminescence (TRPL) measurements on glass/perovskite/polymethyl methacrylate (PMMA) samples (Supplementary Fig. 23). Figure 5c shows the TRPL decay curves obtained from both the glass side and PMMA side for the 1.1-μm-thick perovskite films with (MAPbI3(Cl)) and without (denoted as MAPbI3) chlorine incorporation. Considering the limited penetration depth (tens of nanometers) of the light source, the PL emission signal only contains the charge carrier information on the surface layer that is facing the light source59. The time constants (τ) are calculated to be 54 ns for the MAPbI3 film no matter whether the laser light incidents from the PMMA side or the glass side, which indicates that perovskite on the bottom and top of film has nearly the same charge carrier properties. In the MAPbI3(Cl) case, the PL lifetime obtained from the glass side is longer than that obtained from the PMMA side. This observation suggests that perovskite on the bottom has better charge carrier properties than the perovskite on the top in MAPbI3(Cl) film. This observation may be correlated with the inhomogeneous distribution of chlorine within MAPbI3(Cl) film, where chlorine within a certain range of concentrations can benefit charge carrier properties. Besides, the MAPbI3(Cl) film shows longer PL lifetimes than that of the MAPbI3 film. This improvement can be ascribed to the better crystallinity and a lower defect density of the MAPbI3(Cl) film60.
To quantitatively evaluate the defect density, we fabricated sandwich devices by inserting the perovskite films between FTO and gold, and characterized the evolution of the space-charge-limited current (SCLC) for different biases, as shown in Fig. 5d−i. In general, the presence of mobile species in perovskite can lead to complication in interpreting the SCLC measurement results, and often precludes a robust quantitative analysis based on such measurements. On the other hand, we observed low hysteresis on the basis of J–V measurements, which indicates a negligible effect of mobile species. Thus, in this case we can safely deduce defects density (Nt) within perovskite films based on the SCLC measurements according to the equation13,61:
where ε and ε0 are the dielectric constants of perovskite and the vacuum permittivity, respectively, VTFL is trap-filled limit voltage, L is the thickness of the perovskite films, and e is the elementary charge. We estimated the defect density to be 2.13×1015 and 2.51×1015 cm−3 for the 1.1-μm-thick MAPbI3(Cl) film and MAPbI3 film, respectively, prepared by the HPbI3(Cl)/CH3NH2 method (Table 1). These defect density values are significantly lower than antisolvent case (1.48×1017 cm−3). The similar magnitude in terms of defects densities derived from the SCLC measurements in contrast with the clear difference in terms of lifetime derived from PL measurements is possibly due to the fact that the former measurement gives the defect information in the entire films, and the latter measurements only provides the defect information for perovskite on the top or bottom of the film. When the bias voltage further increases in the SCLC curves, the current shows a linear relationship with the square of the voltage, where we can deduce the mobility (μ) from the Mott−Gurney law61:
The mobility are estimated to be 1.12 and 0.83 cm2 V−1 s−1 for the 1.1-μm-thick MAPbI3(Cl) film and MAPbI3 film, respectively. The much higher carrier conductivity is consistent with the higher device performance of the MAPbI3(Cl) case, which again verifies the positive effect of small content chlorine incorporation on the optoelectronic properties of perovskite films. In addition, these mobility values around 1 cm2 V−1 s−1 are two orders of magnitude higher than that in antisolvent case (0.014 cm2 V−1 s−1), which further confirms the excellent charge carrier transport properties of CH3NH2 gas-based perovskite films. The improved carrier properties of the 1.1-μm-thick MAPbI3(Cl) films when implemented into PSCs is also verified by impedance spectroscopy characterization of devices under light illumination (Table 1 Supplementary Fig. 24 and Supplementary Note 4).
Device stability and perovskite film stability
Operational stability is another key aspect of PSCs. We studied the stability of the small size devices under the operation conditions (Fig. 6a). It was found that the 1.1-μm-thick MAPbI3(Cl)-based devices showed much better stability than the MAPbI3(AS) devices. Especially, the MAPbI3(Cl) devices maintained over 90% of their initial performance for 800 h under continuous light illumination with maximum power point tracking (MPPT). While the PCEs of the MAPbI3(AS) devices decreased to 90% of their initial performance after approximately 300 h. An initial increase of PCEs for both samples is observed in the first 20 h of the stability test. A similar phenomenon was also observed previously55,62, and was ascribed to the effects such as elevated temperature (approximately 46 °C), light or field induced ion movement, light-induced traps formation, interfacial charge accumulation55, or spiro-OMeTAD conductivity variation63. In addition, both devices showed a fast, exponential decay region after reaching the highest performance points, which is followed by a slower linear decay64. By fitting the linear region, the MAPbI3(Cl) devices exhibit a linear slope of −0.0014% h−1, which is 30% slower than the antisolvent case with a linear slope of −0.0020% h−1. Using these slope values, it is expected to take 1660 and 870 h of operation to reach the T80 points for MAPbI3(Cl) devices and the MAPbI3(AS) devices, respectively. The nearly doubled T80 parameters for the MAPbI3(Cl) devices suggests the excellent long-term stability of the cells based on our Cl-incorporated thick films. Note that the stability measurements in this study were conducted under operation conditions, i.e., continuous illumination, MPPT, dry N2 gas environment simulating the case of encapsulation, and therefore the results obtained here can be used as a realistic indicator of the device operation lifetime of our PSCs. It should be noted that all of the components in PSCs play an important role in device stability. Considering that MAPbI3(Cl) devices and MAPbI3(AS) devices are assembled using the same device configuration, the impact of charge selective layers and electrode on device stability can be excluded out. Hence, the significant stability difference between MAPbI3(Cl) devices and MAPbI3(AS) devices is most likely caused by the quality of the perovskite films such as crystallinity, grain size and/or defects, etc. 13,22,34.

Device stability and film stability. a Operational stability of un-encapsulated PSCs based on 1.1 μm thick MAPbI3(Cl) films and perovskite films deposited via the antisolvent method examined under continuous full-sun illumination in dry nitrogen atmosphere. b Factors influence film stability of perovskites
To reveal the underlying mechanism responsible for the improved stability, we prepared four kinds of films, i.e., film A: 1.1 μm MAPbI3(Cl) film prepared by the HPbI3(Cl)/CH3NH2 method; film B: 1.1 μm MAPbI3 film by the HPbI3/CH3NH2 method; film C: 0.5 μm MAPbI3(Cl) films prepared by the HPbI3(Cl)/CH3NH2 method; film D: 0.45 μm MAPbI3(AS) films prepared by the antisolvent method. We tested the stability of these films under light illumination, a high relative humidity of about 100% and gentle thermal environment of 50 °C (denoted as LHT) as shown in Supplementary Fig. 25. It is found that that the stability of these four films is in the order film A > film B > film C > film D (Supplementary Figs. 25 to 28). The improved stability of film A is firstly attributed to its improved grain crystallinity and larger grain size, which can be clearly seen by comparing the XRD peak intensity and full width half maximum of fresh film A with fresh film D (Supplementary Fig. 28b, c and Supplementary Note 5). This is because the ions in highly crystalline perovskite films have higher chemical binding energies to prevent themselves from migrating, which benefits light and thermal stability65. This is confirmed by SEM results shown in Supplementary Fig. 29. We found that the decomposition of perovskite mainly started from grain boundaries for all films after 1 h of LTH testing, where H2O and O2 can easily react with perovskite films due to the facts that (1) chemical binding at grain boundaries with low crystallinity is much weaker than that within crystals and (2) the severe ion migration in low crystallinity region especially at grain boundaries facilitates degradation (Fig. 6b)65. When further comparing film A with film B, film A shows better crystallinity, larger grain size, and also better stability (Supplementary Figs. 26 to 30). In addition, better stability of films A and B than thinner films C and D also verifies that thicker films are more advantageous in achieving better stability because the extra top layer of the compact high-quality film in the thicker films naturally helps as a protection layer to protect the film underlying the top layer. Besides, previous reports have demonstrated that often a tiny amount of I2 existing in perovskite films can cause severe degradation of perovskite13,66. While, CH3NH2 used here provides an alkaline condition to suppress the formation of I2 during the perovskite formation process and/or reduce the I2 amount during the initial degradation of iodine containing perovskite films (Fig. 6b and Supplementary Fig. 28f), which may contribute to the better stability of films A, B and C than film D13,66.
Discussion
A simple perovskite formation method based on fast reaction of chlorine-incorporated HPbI3 and CH3NH2 has been developed to fabricate high-quality, over 1 μm thick and stable CH3NH3PbI3(Cl) perovskite films in ambient condition. The resultant films can not only enable high efficiencies of 19.1 ± 0.4% (champion PCE = 20.0%) for small size PSCs and 13.6 ± 0.8% (champion active area PCE = 15.3%) for 5 cm × 5 cm perovskite solar modules, but also deliver excellent device reproducibility. In addition, the resultant un-encapsulated small size PSCs exhibit an excellent T80 lifetime exceeding 1600 h under continuous light illumination with MPPT in dry N2 environment. The excellent device stability is mainly a result of the excellent films stability. Our study not only provides a highly reproducible method to fabricate PSCs and modules with enhanced efficiency and stability, but also offers the in-depth understanding for the underlying mechanisms responsible for device stability improvement. The encouraging device performance and excellent stability in this work points out to a promising direction, i.e., use of thick absorber films to realize PSCs and modules with high efficiency, reproducibility, and stability.
Methods
Materials
All reagents were used as received without further purification, including PbI2 (99.99%, TCI), hydroiodic acid (57 wt% in H2O, distilled, stabilized, 99.95%, Sigma-Aldrich), chlorobenzene (99.8%, Wako), methylamine (40% in methanol, TCI), 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-OMeTAD, Merck), N,N-dimethylformamide (DMF, 99.99%, Sigma-Aldrich), methylammonium chloride (Wako), ethanol (Wako), HI (57%, Alfa Aesar), 4-tert-butylpyridine (99.9%, Sigma-Aldrich), acetonitrile (99.9%, Sigma-Aldrich), titanium diisopropoxide bis(acetylacetonate) (75 wt% in isopropanol, Sigma-Aldrich), n-butyl alcohol (Sigma-Aldrich), FTO glasses (7−8 Ω sq−1, Opvtech.), TiO2 (DyeSol, 30 NR-D).
Synthesis of HPbI3: The HPbI3 crystals were prepared using an antisolvent vapor-assisted crystallization method as shown in Supplementary Fig. 2. Briefly, 11 g PbI2 and 7 g of HI were mixed and dissolved in 17 g of DMF to form an HPbI3 solution. The HPbI3 solution was heated at 100 °C in chlorobenzene vapor environment for 24 h. During this heat-treatment, chlorobenzene diffuses into the HPbI3/DMF solution and reduces the solubility of HPbI3. Light yellow needle-like HPbI3 crystals were formed, which were collected and washed with chlorobenzene/DMF (volume ratio = 3:1) for three times and then washed with ethanol for three times. Finally, the HPbI3 crystals were dried at 60 °C for 24 h under vacuum.
Fabrication of perovskite films and solar cells
To study the dependence of the film thickness on preparation conditions, the HPbI3 films are obtained by spin-coating HPbI3 precursor (60 wt% HPbI3 in DMF) at 5000 rpm for 30 s on substrates under different temperatures (room temperature, 60, 70, 80, 90, 100 °C) in ambient air condition. For chlorine-incorporated perovskite deposition, the precursor solutions containing different molar ratios of MACl/HPbI3 (0, 0.05, 0.10, 0.15, 0.20, 0.40, 0.70, 1.00) are deposited on substrates preheated at 90 °C at 5000 rpm for 30 s. After thermal annealing at 100 °C for 5 min to remove the solvent, part of the MACl and also part or all of the formed HI during this process, the obtained films react with CH3NH2 gas to form perovskite20,22. Then the obtained perovskite films are annealed at 100 °C for 5 min in ambient air condition before device fabrication or other characterization. A 30 μL of spiro-OMeTAD was spin coated on the perovskite film at 3000 rpm for 30 s, where a spiro-OMeTAD/chlorobenzene (72.3 mg mL−1) solution was employed with the addition of 17.5 μL Li-TFSI/acetonitrile (520 mg mL−1), and 28.8 μL 4-tert-butylpyridine. Finally, a gold layer with a thickness of 100 nm was deposited as the counter electrode on the top of spiro-OMeTAD layer through shadow masks via thermal evaporation under high vacuum (5×10−5 Torr).
Characterization
Current–voltage (J–V) characteristics of PSCs are measured under one sun illumination (AM 1.5 G, 100 mW cm−2, calibrated using a Newport reference Si-cell, Oriel Instruments Model Number 90026564, 2 × 2 cm2) using a solar simulator (Newport Oriel Sol 1A, Xenon-lamp, USHIO, UXL-150SO) and a Keithley 2400 source meter in ambient air at about 25 oC and a relative humidity of about 40−60%. The small size PSCs were measured using a 0.1 cm2 metal mask. No mask is used for measurement of perovskite modules. The active area of perovskite modules is 12.0 cm2, determined by the overlap areas of top and bottom electrodes described in our previous publication46. All the J−V curves are measured under reverse scan with a scan rate of 0.25 V s−1 without preconditioning unless otherwise specified. For stability measurements, the cells were subsequently loaded in our home-designed environmental chamber coupled with a solar simulator (Peccell PEC-L01, AM1.5G) and source meter (Keithley 2401) controlled by a LabView program allowing automatic sequential measurements on the devices with adjustable acquisition time intervals. To simulate continuous solar cell operation an active bias voltage was applied to the cells maintaining the solar cell operation at the maximum power point. The devices were kept at the maximum power output voltage during the intervals between consecutive measurements. No UV-filters were used, i.e., the UV component is included in illumination. The stability measurement was performed under nitrogen box with a relative humidity below 5%. EQE measurements were performed on an Oriel IQE 200 in DC mode.
SEM measurements was carried out in scanning electron microscope (Helios NanoLab G3 UC, FEI). XRD measurements were carried out in a Bruker D8 Discover instrument (Bruker AXS GmbH, Karlsruhe, Germany) equipped with Cu wavelength λ = 1.54 Å X-ray source operated at 1600 W and Goebel mirror. Data were collected from 5 to 60 two theta degrees with a 0.02 degrees step. Experimental data were fitted to obtain unit cell phase parameters using the Profex/BGMN software (v.3.12.0, http://profex.doebelin.org/). TRPL was acquired using the time-correlated, single-photon counting technique (Hamamatsu, C10627), and excitation was provided by a femtosecond mode-locked Ti:sapphire laser (Spectra-Physics, MAITAI XF-IMW) at 450 nm with an average power at 8 MHz of 0.74 mW. In TRPL, perovskite films were covered with PMMA on the top of the perovskite layer to exclude/minimize the influence of ambient air (especially the influence from H2O and O2)67,68,69,70. SIMS (Kratos Axis ULTRA) equipped with quadrupole mass spectrometer (HAL 7, Hiden Analytical) was used to collect the elemental signal in positive ion detection mode (PID). For sputtering in SIMS, 3 keV Ar+ primary beam with a 10 mA current and diameter of 100 µm were utilized. The beam was set at an angle of 45° with respect to the sample surface normal. The spectrometer was operated at a pressure of 10−8 Torr. The c-AFM measurement of the films was performed in contact-mode (MFP-3D series, Asylum Research) in ambient conditions (35% RH, 22 °C). Top side illumination was generated with a halogen lamp. Chromium/platinum-coated cantilevers with a nominal spring constant of 0.2 N m−1 were used to collect the photo-generated currents. The SCLC data were collected with a semiconductor characterization system in N2 (4200-SCS, Keithley). Absorbance was measured using a UV−Vis spectrometer (JASCO Inc., V-670). Impedance Spectroscopy measurements had been recorded with an Autolab PGSTAT204 potentiostat equipped with a frequency response analyzer module FRA32, AC perturbation was set to 10 mV and frequency ranged between 1 MHz to 0.5 Hz. The measurements were done in N2 environment with less than 0.1% relative humidity, and Oriel VeraSol-2class AAALED light source simulating AM 1.5 spectra.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
References
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Kim, H.-S. et al. Lead iodide perovskite sensitized all-solid-state submicron thin film mesoscopic solar cell with efficiency exceeding 9%. Sci. Rep. 2, 591 (2012).
Lee, M. M., Teuscher, J., Miyasaka, T., Murakami, T. N. & Snaith, H. J. Efficient hybrid solar cells based on meso-superstructured organometal halide perovskites. Science 338, 643–647 (2012).
Green, M. A., Ho-Baillie, A. & Snaith, H. J. The emergence of perovskite solar cells. Nat. Photon. 8, 506–514 (2014).
Gratzel, M. The light and shade of perovskite solar cells. Nat. Mater. 13, 838–842 (2014).
Yang, W. S. et al. Iodide management in formamidinium-lead-halide-based perovskite layers for efficient solar cells. Science 356, 1376–1379 (2017).
Jeon, N. J. et al. Solvent engineering for high-performance inorganic–organic hybrid perovskite solar cells. Nat. Mater. 13, 897–903 (2014).
Xiao, M. et al. A fast deposition-crystallization procedure for highly efficient lead iodide perovskite thin-film solar cells. Angew. Chem. 126, 10056–10061 (2014).
Ahn, N. et al. Highly reproducible perovskite solar cells with average efficiency of 18.3% and best efficiency of 19.7% fabricated via lewis base adduct of lead(II) iodide. J. Am. Chem. Soc. 137, 8696–8699 (2015).
Burschka, J. et al. Sequential deposition as a route to high-performance perovskite-sensitized solar cells. Nature 499, 316–319 (2013).
Stranks, S. D. et al. Electron-hole diffusion lengths exceeding 1 micrometer in an organometal trihalide perovskite absorber. Science 342, 341–344 (2013).
Xing, G. et al. Long-range balanced electron- and hole-transport lengths in organic-inorganic CH3NH3PbI3. Science 342, 344–347 (2013).
Liu, Z. et al. Chemical reduction of intrinsic defects in thicker heterojunction planar perovskite solar cells. Adv. Mater. 29, 1606774 (2017).
Yuan, Z. et al. Approximately 800-nm-thick pinhole-free perovskite films via facile solvent retarding process for efficient planar solar cells. ACS Appl. Mater. Interfaces 8, 34446–34454 (2016).
Chen, Z. et al. Thin single crystal perovskite solar cells to harvest below-bandgap light absorption. Nat. Commun. 8, 1890 (2017).
Li, C., Pang, S., Xu, H. & Cui, G. Methylamine gas based synthesis and healing process toward upscaling of perovskite solar cells: progress and perspective. Sol. RRL 1, 1700076 (2017).
Zhou, Y. & Padture, N. P. Gas-induced formation/transformation of organic–inorganic halide perovskites. ACS Energy Lett. 2, 2166–2176 (2017).
Raga, S. R., Jiang, Y., Ono, L. K. & Qi, Y. B. Application of methylamine gas in fabricating organic–inorganic hybrid perovskite solar cells. Energy Tech. 5, 1750–1761 (2017).
Zhou, Z. et al. Methylamine-gas-induced defect-healing behavior of CH3NH3PbI3 thin films for perovskite solar cells. Angew. Chem. Int. Ed. 54, 9705–9709 (2015).
Pang, S. et al. Transformative evolution of organolead triiodide perovskite thin films from strong room-temperature solid-gas interaction between HPbI3-CH3NH2 precursor pair. J. Am. Chem. Soc. 138, 750–753 (2016).
Zhang, Y. et al. Trash into treasure: δ‐FAPbI3 polymorph stabilized MAPbI3 perovskite with power conversion efficiency beyond 21%. Adv. Mater. 30, 1707143 (2018).
Long, M. et al. Textured CH3NH3PbI3 thin film with enhanced stability for high performance perovskite solar cells. Nano Energy 33, 485–496 (2017).
Noel, N. K. et al. A low viscosity, low boiling point, clean solvent system for the rapid crystallisation of highly specular perovskite films. Energy Environ. Sci. 10, 145–152 (2017).
Nenon, D. P. et al. Structural and chemical evolution of methylammonium lead halide perovskites during thermal processing from solution. Energy Environ. Sci. 9, 2072–2082 (2016).
Aguiar, J. A. et al. In situ investigation of halide incorporation into perovskite solar cells. MRS Commun. 7, 575–582 (2017).
Xie, F. X., Su, H., Mao, J., Wong, K. S. & Choy, W. C. H. Evolution of diffusion length and trap state induced by chloride in perovskite solar cell. J. Phys. Chem. C 120, 21248–21253 (2016).
Zhao, Y. & Zhu, K. CH3NH3Cl-assisted one-step solution growth of CH3NH3PbI3: structure, charge-carrier dynamics, and photovoltaic properties of perovskite solar cells. J. Phys. Chem. C 118, 9412–9418 (2014).
Williams, S. T. et al. Role of chloride in the morphological evolution of organo-lead halide perovskite thin films. ACS Nano 8, 10640–10654 (2014).
Zhou, H. et al. Interface engineering of highly efficient perovskite solar cells. Science 345, 542–546 (2014).
Chen, Q. et al. The optoelectronic role of chlorine in CH3NH3PbI3(Cl)-based perovskite solar cells. Nat. Commun. 6, 7269 (2015).
Colella, S. et al. MAPbI3-xClx mixed halide perovskite for hybrid solar cells: the role of chloride as dopant on the transport and structural properties. Chem. Mater. 25, 4613–4618 (2013).
Deng, Y. et al. Scalable fabrication of efficient organolead trihalide perovskite solar cells with doctor-bladed active layers. Energy Environ. Sci. 8, 1544–1550 (2015).
Wang, F., Yu, H., Xu, H. & Zhao, N. HPbI3: a new precursor compound for highly efficient solution-processed perovskite solar cells. Adv. Funct. Mater. 25, 1120–1126 (2015).
Long, M. et al. Nonstoichiometric acid–base reaction as reliable synthetic route to highly stable CH3NH3PbI3 perovskite film. Nat. Commun. 7, 13503 (2016).
Yang, M. et al. Perovskite ink with wide processing window for scalable high-efficiency solar cells. Nat. Energy 2, 17038 (2017).
Zhao, L. et al. Redox chemistry dominates the degradation and decomposition of metal halide perovskite optoelectronic devices. ACS Energy Lett. 1, 595–602 (2016).
Chang, Y. et al. CH3NH2 gas induced (110) preferred cesium-containing perovskite film with reduced PbI6 octahedron distortion and enhanced moisture stability. J. Mater. Chem. A 5, 4803–4808 (2017).
Shirayama, M. et al. Degradation mechanism of CH3NH3PbI3 perovskite materials upon exposure to humid air. J. Appl. Phys. 119, 115501 (2016).
Li, Y. et al. H High-performance planar solar cells based on CH3NH3PbI3-xClx perovskites with determined chlorine mole fraction. Adv. Funct. Mater. 25, 4867–4873 (2015).
Hoke, E. T. et al. Reversible photo-induced trap formation in mixed-halide hybrid perovskites for photovoltaics. Chem. Sci. 6, 613–617 (2015).
Slotcavage, D. J., Karunadasa, H. I. & McGehee, M. D. Light-induced phase segregation in halide-perovskite absorbers. ACS Energy Lett. 1, 1199–1205 (2016).
Yang, M. et al. Facile fabrication of large-grain CH3NH3PbI3-xBrx films for high-efficiency solar cells via CH3NH3Br-selective Ostwald ripening. Nat. Commun. 7, 12305 (2016).
Starr, D. E. et al. Direct observation of an inhomogeneous chlorine distribution in CH3NH3PbI3-xClx layers: surface depletion and interface enrichment. Energy Environ. Sci. 8, 1609–1615 (2015).
Cao, C. et al. Iodine and chlorine element evolution in CH3NH3PbI3–xClx thin films for highly efficient planar heterojunction perovskite solar cells. Chem. Mater. 28, 2742–2749 (2016).
Luo, Y. et al. Spatially heterogeneous chlorine incorporation in organic–inorganic perovskite solar cells. Chem. Mater. 28, 6536–6654 (2016).
Jiang, Y. et al. Combination of hybrid CVD and cation exchange for upscaling Cs-substituted mixed cation perovskite solar cells with high efficiency and stability. Adv. Funct. Mater. 28, 1703835 (2017).
Bu, T. et al. A novel quadruple-cation absorber for universal hysteresis elimination for high efficiency and stable perovskite solar cells. Energy Environ. Sci. 10, 2509–2515 (2017).
Chen, H. et al. A solvent- and vacuum-free route to large-area perovskite films for efficient solar modules. Nature 550, 92–95 (2017).
Chiang, C.-H., Nazeeruddin, M. K., Gratzel, M. & Wu, C.-G. The synergistic effect of H2O and DMF towards stable and 20% efficiency inverted perovskite solar cells. Energy Environ. Sci. 10, 808–817 (2017).
Qiu, W. et al. Pinhole-free perovskite films for efficient solar modules. Energy Environ. Sci. 9, 484–489 (2016).
Leyden, M. R., Jiang, Y. & Qi, Y. B. Chemical vapor deposition grown formamidinium perovskite solar modules with high steady state power and thermal stability. J. Mater. Chem. A 4, 13125–13132 (2016).
Liao, H. C. et al. Enhanced efficiency of hot-cast large-area planar perovskite solar cells/modules having controlled chloride incorporation. Adv. Energy Mater. 7, 1601660 (2017).
Yeo, J.-S. et al. Reduced graphene oxide-assisted crystallization of perovskite via solution-process for efficient and stable planar solar cells with module-scales. Nano Energy 30, 667–676 (2016).
Heo, J. H., Han, H. J., Kim, D., Ahn, T. K. & Im, S. H. Hysteresis-less inverted CH3NH3PbI3 planar perovskite hybrid solar cells with 18.1% power conversion efficiency. Energy Environ. Sci. 8, 1602–1608 (2015).
Grancini, G. et al. One-year stable perovskite solar cells by 2D/3D interface engineering. Nat. Commun. 8, 15684 (2017).
Li, Z. et al. Scalable fabrication of perovskite solar cells. Nat. Rev. Mater. 3, 18017 (2018).
Zhang, M.-J. et al. Carrier transport improvement of CH3NH3PbI3 film by methylamine gas treatment. Acs Appl. Mater. Interfaces 8, 31413–31418 (2016).
Ball, J. M. & Petrozza, A. Defects in perovskite-halides and their effects in solar cells. Nat. Energy 1, 16149 (2016).
Ding, J., Lian, Z., Li, Y., Wang, S. & Yan, Q. The role of surface defects in photoluminescence and decay dynamics of high-quality perovskite MAPbI3 single crystals. J. Phys. Chem. Lett. 9, 4221–4226 (2018).
Wu, X. et al. Trap states in lead iodide perovskites. J. Am. Chem. Soc. 137, 2089–2096 (2015).
Dong, Q. et al. Electron-hole diffusion lengths>175 μm in solution-grown CH3NH3PbI3 single crystals. Science 347, 967–970 (2015).
Hou, Y. et al. A generic interface to reduce the efficiency-stability-cost gap of perovskite solar cells. Science 358, 1192–1197 (2017).
Hawash, Z., Ono, L. K. & Qi, Y. B. Moisture and oxygen enhance conductivity of LiTFSI-doped spiro-MeOTAD hole transport layer in perovskite solar cells. Adv. Mater. Interfaces 3, 1600117 (2016).
Domanski, K., Alharbi, E. A., Hagfeldt, A., Grätzel, M. & Tress, W. Systematic investigation of the impact of operation conditions on the degradation behaviour of perovskite solar cells. Nat. Energy 3, 61–67 (2018).
Yuan, Y. & Huang, J. Ion migration in organometal trihalide perovskite and its impact on photovoltaic efficiency and stability. Acc. Chem. Res. 49, 286–293 (2016).
Wang, S., Jiang, Y., Juarez-Perez, EmilioJ., Ono, LuisK. & Qi, Y. B. Accelerated degradation of methylammonium lead iodide perovskites induced by exposure to iodine vapour. Nat. Energy 2, 16195 (2016).
Galisteo-López, J. F., Anaya, M., Calvo, M. E. & Míguez, H. Environmental effects on the photophysics of organic–inorganic halide perovskites. J. Phys. Chem. Lett. 6, 2200–2205 (2015).
Fang, H.-H. et al. Ultrahigh sensitivity of methylammonium lead tribromide perovskite single crystals to environmental gases. Sci. Adv. 2, e1600534 (2016).
Brenes, R. et al. Metal halide perovskite polycrystalline films exhibiting properties of single crystals. Joule 1, 155–167 (2017).
Tian, Y. et al. Mechanistic insights into perovskite photoluminescence enhancement: light curing with oxygen can boost yield thousandfold. Phys. Chem. Chem. Phys. 17, 24978–24987 (2015).
Acknowledgements
This work was supported by funding from the Energy Materials and Surface Sciences Unit of the Okinawa Institute of Science and Technology Graduate University, the OIST Proof of Concept (POC) Program, the OIST R&D Cluster Research Program, and JSPS KAKENHI Grant Number JP18K05266. S.L. acknowledges the funding support from the 111 Project (B14041). The authors thank Dr. Mikas Remeika for writing the software for steady-state power measurements, Mr. Toshio Sasaki, Mr. Laszlo Szikszai and the Nanofab members for SEM characterization, Dr. Zhanhao Hu and Dr. Qijing Wang for insightful discussion for SCLC characterization.
Author information
Authors and Affiliations
Contributions
Y.Q. supervised the project. Y.Q. and Z.L. conceived the ideas and designed the experiments. Z.L. conducted the corresponding device fabrication and basic characterization. L.Q. helped with TiO2 fabrication and stability study. E.J.J.-P. helped with EIS characterization, performed XRD cell parameter calibration, and TRPL fitting. Z.H. performed the conductive AFM characterization. T.K. and Z.H. performed SIMS characterization. Y.J. helped with module fabrication. Z.W. helped with mesoporous TiO2 fabrication. S.R.R. and L.K.O. helped with CH3NH2 gas reaction system setup. S.L. provided valuable suggestions for this work. Z.L. and Y.Q. participated in all the data analysis. All authors contributed to writing the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Z., Qiu, L., Juarez-Perez, E.J. et al. Gas-solid reaction based over one-micrometer thick stable perovskite films for efficient solar cells and modules. Nat Commun 9, 3880 (2018). https://doi.org/10.1038/s41467-018-06317-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-06317-8
This article is cited by
-
Efficient micrometer-scale thick-film perovskite solar cells with superior stability
Rare Metals (2024)
-
Unveiling the surface-interface properties of perovskite crystals and pivotal regulation strategies
Nano Research (2024)
-
Ammonia for post-healing of formamidinium-based Perovskite films
Nature Communications (2022)
-
Vapor phosphorus-coated cobalt vanadate as a high-performance anode for a lithium-ion battery
Journal of Solid State Electrochemistry (2022)
-
A holistic approach to interface stabilization for efficient perovskite solar modules with over 2,000-hour operational stability
Nature Energy (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
