
Abstract
Copper-based materials are promising electrocatalysts for CO2 reduction. Prior studies show that the mixture of copper (I) and copper (0) at the catalyst surface enhances multi-carbon products from CO2 reduction; however, the stable presence of copper (I) remains the subject of debate. Here we report a copper on copper (I) composite that stabilizes copper (I) during CO2 reduction through the use of copper nitride as an underlying copper (I) species. We synthesize a copper-on-nitride catalyst that exhibits a Faradaic efficiency of 64 ± 2% for C2+ products. We achieve a 40-fold enhancement in the ratio of C2+ to the competing CH4 compared to the case of pure copper. We further show that the copper-on-nitride catalyst performs stable CO2 reduction over 30 h. Mechanistic studies suggest that the use of copper nitride contributes to reducing the CO dimerization energy barrier—a rate-limiting step in CO2 reduction to multi-carbon products.
Similar content being viewed by others


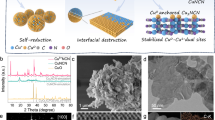
Introduction
Electrocatalytic CO2 reduction has been investigated extensively based on metals such as Au, Ag, Sn, Zn, In, Pd, Cu, and their associated compounds1,2,3,4. Among these materials, Cu-based catalysts are promising for olefin and oxygenate production thanks to their moderate CO binding energies5,6. Multi-carbon products such as ethylene (C2H4), ethanol (C2H5OH), and n-propanol (C3H7OH) are of great interest: C2H4, for example, is a valuable precursor in the manufacture of polymers7; C2H5OH can be directly used as fuel8; and C3H7OH has a higher mass energy density (30.94 kJ g−1)9,10 than does gasoline11. Furthermore, renewables-derived C2H5OH and C3H7OH can each be blended with gasoline to deliver a clean fuel12.
Polycrystalline Cu metal is known to produce CH4 with high selectivity4,13, whereas oxide-derived Cu favors C2+ products14,15,16,17, a fact attributed to the effects of grain boundaries18,19,20, high-local pH21,22, and residual oxygen14,23,24. Certain prior computational studies have suggested that the Cu+/Cu0 mixture synergistically promotes CO2 reduction to C2+ products due to CO2 activation and CO dimerization25,26. Experimentally, however, the stable presence of the active Cu+ species during CO2 reduction remains the subject of debate 27.
A Cu+–Cu0 core-shell structured catalyst offers an architecture wherein stable Cu0 deposited on top of a Cu+ support protects from further reduction. Recently, core-shell catalysts have been widely investigated in electrocatalysis and have achieved significantly improved activity and kinetics28,29,30,31,32,33,34,35. The core-support interactions modify the electronic structure of the surface catalyst, influencing the chemisorption of the intermediates in the electrocatalytic reaction31. Copper (I) oxide (Cu2O), which has been mostly used as a precursor to Cu-based CO2 reduction catalysts14,17,18,19,23,24, is a candidate as a Cu+ support; however, Cu+ from Cu2O is unstable under CO2 reduction conditions. Previous reports suggest that transition metal nitrides can be employed not only as a stable catalytic active species, but also as supports 36.
Here we sought therefore to investigate whether copper (I) nitride (Cu3N) could be used as Cu+ support during CO2 reduction. We hypothesize that the Cu3N support affects the electronic structure and oxidation state of the surface Cu, decreasing the energy barrier associated with CO dimerization during CO2 reduction. This, together with the prolonged presence of Cu+ over time, could allow for the realization of increased-stability C2+ electrosynthesis systems under CO2 reduction conditions.
Results
Synthesis and structural characterization
In order to challenge our hypothesis, we set out to synthesize Cu deposited on Cu3N (Cu-on-Cu3N) catalyst as depicted in Fig. 1a. We first synthesized Cu3N nanocrystals capped with long-chain octadecylamine (ODA) ligands37. We then performed a ligand exchange using short-chain azide (N3−) to replace the ODA. An outer oxide was formed at the surface of Cu3N nanocrystals by exposing samples to ambient air during the ligand exchange process. These nanocrystals then went through an initial electroreduction process: we swept the cyclic voltammetry (CV) curve from 0 to −1.75 V vs. RHE to obtain the active Cu-on-Cu3N catalyst.

Electrocatalyst design and the corresponding XPS characterization. a Schematic of preparing the Cu-on-Cu3N catalyst. b XPS spectra of Cu 2p, N 1 s, and Auger Cu LMM of the Cu3N nanocrystals with long organic ODA (i), the Cu3N nanocrystals with an oxide layer after N3- ligand exchange (ii), and the Cu-on-Cu3N composite after initial electroreduction (iii)
To investigate surface electronic properties, we conducted X-ray photoelectron spectroscopy (XPS) measurements of the samples (Fig. 1b). In the case of the Cu3N nanocrystals capped with ODA (Fig. 1b–i), the spectra of Cu 2p and Auger Cu LMM confirm a preponderance of Cu+38. The sharp peak of N at a binding energy of 399 eV is consistent with that of the metal nitride37,39. Furthermore, X-ray diffraction (XRD) attests to the formation of Cu3N nanocrystals (Supplementary Fig. 1)37.
Implementing the ligand exchange (Fig. 1b–ii and Supplementary Fig. 2) led to a different Cu composition compared to that before ligand exchange. A mixture of Cu2+ and Cu+ are present as observed in Cu 2p and LMM spectra38, which suggests that copper (II) oxide (CuO) is formed in ambient air during the Cu3N ligand exchange. The new peak in the N 1 s spectrum located at 403.1 eV aligns with that of the N3− group in the ligand at the nanocrystal surface40. When taken together with Fourier-transform infrared (FTIR) spectra (Supplementary Fig. 3), these findings reveal that the ODA organic ligands are completely replaced by the N3− short ligands. In addition, the Cu 2p peak areas indicate that the content of Cu2+ is significantly higher relative to the Cu+: we propose that CuO exists at the surface and substantially encompasses the Cu3N. As shown in the O 1 s spectrum (Supplementary Fig. 4), the dominant peak at 513.3 eV was assigned to O species in the surface CuO on the sample.
After initial reduction (Fig. 1b–iii), the Cu spectra show the presence of both Cu+ and Cu0, which indicates that the surface of the catalyst was reduced to Cu. The N 1 s peak at 399 eV remains after reduction, indicating that the Cu3N phase is intact. The disappearance of the N peak at 403.1 eV, which is the characteristic of the N3− ligands, can be ascribed to the weak electrostatic interaction between the ligands and the surface of the Cu3N nanocrystals when a potential was applied.
We used transmission electron microscopy (TEM) to investigate further the structure of the catalyst (Supplementary Fig. 5). Before ligand exchange, Cu3N nanocrystals have an average diameter of 30 nm. After ligand exchange, a reduced spacing between the nanocrystals is observed, similar to the case of quantum dot ligand exchanges 41.
The local atomic-scale elemental composition on individual Cu-on-Cu3N nanoparticle was further examined (Fig. 2a, b). From high-resolution transmission electron microscopy with electron energy loss spectroscopy (HRTEM-EELS, Fig. 2c), we observed that Cu was distributed across the volume of each nanoparticle; while N was concentrated in the core and was notably lower at the surface. The catalyst surface exhibited indications of surface reconstruction following operation under reducing conditions42. Our analysis of HRTEM-EELS data indicates a <= 3 nm surface Cu layer on top of Cu3N (Supplementary Fig. 6).

TEM characterization of the Cu-on-Cu3N catalyst. a, b HADDF-STEM images. c STEM-EELS Cu and N Element mapping of one individual particle in b, d, EEL spectra of element N K-edge circled as point A and point B in b. The scale bars are 50 nm in a, and 10 nm in b and c
We further investigated the distribution of N using EELS spectra. In a given nanoparticle, looking at two different positions (Fig. 2d), we found that for point A (inner), a strong absorption feature starting from 401 eV was obtained, consistent with the N K-shell absorption edge43. No obvious absorption signal was observed for point B (surface), indicating no N at the surface. These observations indicate a Cu-on-Cu3N structure.
We synthesized Cu deposited on Cu2O (Cu-on-Cu2O) and pure Cu catalysts as control samples using a process similar to the synthesis of the Cu-on-Cu3N catalyst. XRD patterns confirm the formation of Cu-on-Cu2O and pure Cu after electroreduction (Supplementary Fig. 7a–c). Their morphology and size are similar to those of the Cu-on-Cu3N catalyst (Supplementary Fig. 7b–d). Double-layer capacitance measurements yielded electrochemical roughness factors of 9.7, 8.0, and 9.3 for the Cu-on-Cu3N, Cu-on-Cu2O, and Cu catalysts, respectively, indicating similar surface: geometric area ratios (Supplementary Fig. 8 and Supplementary Table 1).
We also obtained valence band spectra (VBS) to examine differences in the valence electronic structure between the Cu and the composite (Supplementary Fig. 9). Comparing with the case of pure Cu, we found that the Fermi-level (EF) shifted toward the VBm by 0.33 eV for Cu-on-Cu3N and 0.08 eV for Cu-on-Cu2O, respectively, indicating that the core-level Cu3N and Cu2O supports have an effect on the electronic structure of the surface Cu.
Spectroscopic characterization
To investigate the structure and chemical state of the active catalysts with time evolution under CO2 reduction, we obtained in situ X-ray absorption spectra (XAS) of the three catalysts at −0.95 V vs. RHE during CO2 reduction.
As depicted in Fig. 3a, b, the Cu K-edge XAS spectrum of the as-prepared Cu-on-Cu3N catalyst presents an absorption edge between Cu (8979 eV) and Cu3N (8980.5 eV)—and in particular exhibits a prominent shoulder at 8980.0 eV. Over the course of CO2 reduction, both Cu and Cu3N features are still present, with a shoulder energy at 8979.4 eV after 2 h. In contrast, the Cu-on-Cu2O catalyst shows a prominent metallic Cu feature after 1 h (Supplementary Fig. 10a). Pure Cu presents a metallic Cu feature under CO2 reduction (Supplementary Fig. 10b).

In situ characterization of the structure and chemical state for the catalysts during CO2 reduction. a Cu K-edge XAS spectra of the Cu-on-Cu3N catalyst as function of reaction time at −0.95 V vs RHE. b The first derivatives of the spectra in a. c In situ Cu K-edge spectra during the initial 30 min on the catalysts: Cu-on-Cu3N (green) and Cu-on-Cu2O (orange). Spectra of Cu (red) and Cu3N (yellow) are also listed as references. d Ratio of Cu+ relative to the reaction time at −0.95 V vs RHE
To gain more insight into the role of the Cu+ support, we acquired in situ Cu K-edge spectra of Cu-on-Cu3N and Cu-on-Cu2O catalysts following 30 min under CO2 reduction (Fig. 3c). We found that the absorption edges of the two catalysts are between Cu+ and Cu0, indicating the presence of a mixture during the reaction. However, the absorption edge of Cu-on-Cu2O was at a lower energy than that of Cu-on-Cu3N, with energies at 8979.4 eV and 8979.8 eV, respectively. We also calculated the ratio of Cu oxidation states as function of the reaction time (Fig. 3d). The Cu-on-Cu3N catalyst shows that the structure becomes stable with Cu3N and Cu after the initial 60 min, while Cu-on-Cu2O only presents the Cu component after 1 h. This observation indicates suppression of the partial reduction of the catalyst when we use the Cu3N support.
We sought a method to probe with greater surface-specificity catalyst as function of reaction time. We acquired angle-resolved XPS (ARXPS) at a 20° emission angle relative to the sample normal (Supplementary Fig. 11a). The detection depth is below 2 nm44. Cu LMM spectra (Supplementary Fig. 11b–f, left column) indicate the presence of Cu+ and Cu0, and the N 1 s spectra (Supplementary Fig. 11 b–f, right column) are consistent with the spectrum of metal nitride, indicating the presence of Cu3N in the first ~2 nm of the surface over the course of CO2 reduction. In the initial 60 min, Cu0 content increased and Cu3N content decreased; thereafter, such as following a 2-h reaction, the catalyst gradually reached a stable surface composition. This result agrees with the observed in situ XAS data (Fig. 3d).
Both in situ XAS and ex situ ARXPS indicate the presence of Cu+ following CO2 reduction. Further, the N signal suggests the presence of Cu3N, and STEM-EELS mapping (Supplementary Fig. 6) shows evidence of Cu3N in the subsurface layer following CO2 reduction. Nevertheless, we point out also that XAS has a bulk penetration depth; and that air-sensitive Cu complicates the interpretation of the ARXPS studies herein. For these reasons, direct and unambiguous confirmation of the presence of Cu+ at the surface of the catalyst remains an ongoing opportunity for further advances in the field of Cu-based electrocatalysis and model development.
CO2 electroreduction performance
To verify the effect of the Cu+ support on the surface catalyst, we carried out CO2 reduction using the Cu-on-Cu3N, Cu-on-Cu2O, and pure Cu catalysts, respectively. To analyze the selectivity toward various products with different applied potentials, we performed stepped-potential electrolysis between −0.55 and −1.45 V vs RHE (with iR correction in Supplementary Fig. 12).
Cu-on-Cu3N gives the highest C2+ production among the three catalysts (Fig. 4a, b). When the applied potential is less negative than −0.65 V vs RHE, CH4, and HCOOH are the main products; whereas, when we sweep toward more strongly negative potentials, we obtain production of reduced C2+ species, such as C2H4, C2H5OH, and C3H7OH. This indicates CO dimerization beyond the potential of −0.65 V vs. RHE (Supplementary Figs. 13a, 14 and Supplementary Table 2). The highest FE for total C2+ reaches 64 ± 2% at −0.95 V vs. RHE, with C2H4, C2H5OH, and C3H7OH accounting for 39 ± 2%, 19 ± 1%, and 6 ± 1%, respectively.

CO2 electroreduction performance of the designed catalysts. a Faradaic efficiency of the C2+ distribution on Cu-on-Cu3N at different potentials. b Comparison of faradaic efficiency for C2+ and the ratio of C2+/CH4 at −0.95 V vs RHE on Cu, Cu-on-Cu2O, and Cu-on-Cu3N. c C2+ partial current density at different potentials on the three catalysts. d Stability test of C2+ selectivity on the three catalysts. Experiments from a to d were performed in triplicates and the results are shown as mean ± standard deviation
The Cu-on-Cu3N catalyst achieves a 6.3-fold enhancement in the ratio of C2+ to CH4 compared to Cu-on-Cu2O; and a 40-fold enhancement over pure Cu (Fig. 4b). CH4 production is thus strongly suppressed for the catalysts that use Cu2O and Cu3N support compared with pure Cu (Supplementary Fig. 13b–c and Supplementary Tables 3−4).
To further compare the CO2 reduction activity of the three samples, we obtained C2+ partial current densities at a variety of potentials between −0.75 and −1.45 V vs RHE (Fig. 4c). The Cu-on-Cu3N catalyst exhibits a higher C2+ partial current density relative to Cu-on-Cu2O and pure Cu across the entire potential window, with a maximum 14 mA cm−2 at −0.95 V vs RHE, 2.2x and 4.4x higher than Cu-on-Cu2O and pure Cu catalysts, respectively.
To test the operational stability of the catalysts, we carried out CO2 reduction over an extended period of time. The Cu-on-Cu3N catalyst exhibits relatively stable Faradaic efficiencies toward C2H4, C2H5OH, and C3H7OH, with a relative 10% decrease following 30 h of continuous CO2 electroreduction (Fig. 4d and Supplementary Fig. 15). We attribute this superior stability to the suppressed reduction of the Cu3N support over time, such that the beneficial effect of the Cu3N support is sustained over this longer operating time. In contrast, Cu-on-Cu2O catalyst shows a loss of about 25% relative of its selectivity following 15 h, while pure Cu shows decreased C2+ production following 5 h of CO2 reduction (Fig. 4d).
Discussion
Recent studies have suggested that the CO2 reduction performance of oxide-derived Cu catalysts can be ascribed to local pH and to derived surface defects19,21,22. Compared to pure Cu, Cu-on-Cu+ catalysts display a suppression in methane selectivity, which can be attributed to increased local pH. Comparing Cu-on-Cu2O and Cu-on-Cu3N catalysts, the geometric current densities are similar (Supplementary Fig. 16), which indicates a nearly identical consumption rate of local protons during CO2 reduction. We propose therefore that differences in local pH do not account for the higher C2+ selectivity for Cu-on-Cu3N relative to Cu-on-Cu2O.
We also considered surface defects as another possible contributing mechanism. For the Cu-on-Cu2O catalyst, we believe that surface defects—grain boundaries—may influence the selectivity toward C2+ in the case of the oxide-derived process. For the Cu-on-Cu3N catalyst, surface defects can also affect the C2+ selectivity. However, compared with Cu-on-Cu2O, which was quickly derived to Cu (Fig. 3d), the Cu-on-Cu3N catalyst retained a higher C2+ selectivity under CO2 reduction. We offer that suppressed reduction of the Cu3N support thus plays a significant part in the high selectivity over the course of CO2 reduction.
Single-crystal studies have also shown that the exposed Cu facets affect selectivity45,46,47. In this work, we synthesized three catalysts using an initial electroreduction of the surface oxidation layer using a negative cyclic voltammetry (CV) scan. During this process, the Cu species possess a polycrystalline structure (Supplementary Fig. 17). These structures do not exhibit the specific facet orientation. Therefore, we would not expect that these would contribute in a quantitatively significant way to increase C2+ selectivity.
Taking these findings together with those from XPS (including ARXPS and VBS), in situ XAS, and HRTEM-EELS, we propose that the Cu+ support may play a role in selectivity toward C2+. Due to the change of surface structure with time evolution, the surface Cu layer is no longer uniform and some of Cu+ may reside in the subsurface layer during the initial reduction (Supplementary Fig. 6), favouring selectivity for C2+. Cu3N as the support stabilizes the Cu+ to a greater degree than does Cu2O during CO2 reduction (Fig. 3d), leading to heightened C2+ production.
To understand the role of Cu+ support in Cu0-on-Cu+ composite catalyst for CO2 reduction, we performed density functional theory (DFT) computations to calculate the oxidation state of the surface Cu in the models: Cu, Cu3N, Cu2O, Cu-on-Cu3N, and Cu-on-Cu2O (Supplementary Figs. 18–22). Bader charge analyses show that both Cu2O and Cu3N as Cu+ supports modulate the partial oxidation state of the surface copper layer (Supplementary Table 5), with +0.03 extra charge induced by Cu2O support and +0.25 by Cu3N support on (100) facet, respectively, different from that of pure Cu (0), pure Cu2O (+0.26), and pure Cu3N (+0.47) on (100) facet. This modulated partial oxidation state enables Cu-on-Cu3N to achieve the lowest CO dimerization barrier energy (0.884 eV) among all models (Supplementary Figs. 23–24 and Supplementary Table 5), thereby indicating promise as a candidate for C2+ production. To evaluate further the selectivity of these catalysts for C2+ products compared to the competing C1 products, we also calculated the energy barriers for CO protonation (Supplementary Methods). The results reveal that the energy barrier for the C1 pathway for Cu-on-Cu3N (0.933 eV) is higher than that of Cu-on-Cu2O (0.749 eV) and pure Cu (0.721 eV) on (100) facets (Supplementary Table 5).
Since, the stability of the sublayer Cu+ is important, we studied the diffusion free energy barrier for nitrogen and oxygen from their original positions in the Cu3N and Cu2O structures, respectively, to the surface of Cu-on-Cu3N and Cu-on-Cu2O with 4 Cu top layers. Although there is a large energy barrier (>2 eV) for both O and N to leave their original position, there is no more diffusion barrier for O from the first layer to the surface (Supplementary Figs. 25–26 and Supplementary Table 6). However, for N we observe another energy barrier (~1 eV) to diffuse to the surface (Supplementary Fig. 27 and Supplementary Table 6). This agrees with our observations throughout that N in the sublayer is more stable than O.
In summary, the present work introduces a Cu-based catalyst that enables metallic Cu-on-Cu3N to promote the production of C2+ species. Cu3N was chosen as the inner support to modify the electronic structure of the surface metal, affecting thereby the adsorption and dimerization of intermediate CO properly in the CO2 reduction. Together with the suppressed reduction of Cu+ using Cu3N as the support, we were able to achieve higher selectivity for C2+ formation using Cu-on-Cu3N compared to the case of Cu-on-Cu2O and pure Cu.
Methods
Synthesis of Cu3N nanocrystals
Quantity of 0.15 g of Cu(NO3)2·3H2O and 4.3 g of 1-octadecylamine (ODA) was dissolved in 15 mL of 1-octadecene. The solution was degassed for 10 min at 150 °C. The temperature was then raised to 240 °C and kept for another 10 min. When it cooled down to room temperature, the product was collected by centrifugation, washed with hexane/acetone (1/4) three times, and finally dispersed in hexane.
Transformation of Cu3N to CuO-on-Cu3N
We used the ligand exchange method in ambient air to achieve the transformation of Cu3N to CuO-on-Cu3N. Ten milligrams of Cu3N nanocrystals with organic ligands was dissolved in 1 mL of hexane (10 mg L−1), while 10 mg of sodium azide (NaN3) was dissolved in 1 mL of NMF (10 mg L−1). The two solutions were then mixed and stirred overnight. The nanocrystals gradually transferred to NMF. The bottom phase was extracted and washed with hexane three times. The N3−-capped Cu3N nanocrystals were then precipitated out using chloroform as the anti-solvent. The precipitate was dried in vacuum for 15 min and then stored. In the ligand exchange process, we deliberately exposed the materials to ambient air to introduce an oxide layer at the surface of Cu3N nanocrystals.
Transformation of CuO-on-Cu3N to Cu-on-Cu3N
We conducted the initial electroreduction for the CuO-on-Cu3N sample by sweeping the cyclic voltammetry (CV) curve from 0 to −1.75 V vs RHE at a rate of 50 mV s−1, yielding the Cu-on-Cu3N catalyst.
Synthesis of control Cu-on-Cu2O and pure Cu catalysts
Cu2O and Cu nanocrystals were synthesized using 0.5 g of Cu(NO3)2·3H2O and 0.05 g of Cu(NO3)2·3H2O instead, respectively, while keeping other experimental conditions the same as in the synthesis of Cu3N nanocrystals. The ligand exchange and initial electroreduction processes were the same as in the case of the Cu-on-Cu3N catalyst.
Working electrode preparation
Ten milligrams of the catalyst was dispersed in 1 mL of methanol, including with 20 μL of Nafion solution (anhydrous, 5 wt %) by sonicating for 30 min. Twenty microliter of the homogeneous solution was then loaded on a glassy carbon electrode. The geometric surface area was 0.19 cm2. The electrode was dried in ambient air for the subsequent CO2 electroreduction test.
Electrochemical measurement
Electrochemical tests were performed in a two-compartment H-cell. A proton exchange membrane (Nafion 117) was used. The electrolyte was 30 mL of 0.1 M KHCO3 solution saturated with CO2 gas in the cathode part for at least 30 min prior to the CO2 reduction test. Platinum was used as the counter electrode and Ag/AgCl as the reference electrode (saturated with 3.0 M KCl, BASi). The glassy carbon electrode loaded with the catalyst served as the working electrode. Liner sweep voltammetry (LSV) with a scan rate of 50 mV/s was conducted first. The gas products were detected using a gas chromatograph (GC, PerkinElmer Clarus 600) equipped with a thermal conductivity detector (TCD) for hydrogen (H2) quantification and a flame ionization detector (FID) for methane (CH4) and ethylene (C2H4). Liquid products were quantified using 1H nuclear magnetic resonance (NMR, Agilent DD2 500). The NMR samples were prepared by mixing 0.5 mL of electrolyte with 0.1 mL of deuterated water (D2O), and 0.02 μL of dimethyl sulfoxide (DMSO) was added as an internal standard. Potential E was converted to the RHE reference electrode using:
Electrochemical active surface area (ECSA) measurement
We used the double layer capacitance method to measure the surface roughness factors (Rf) for the samples relative to polycrystalline Cu (Rf = 1) foil. Rf was calculated from the ratio of the double-layer capacitance C of the catalyst electrode and Cu foil electrode (CCu foil = 29 μF), that is, Rf = C/CCu foil. C was determined by measuring the geometric current density at a potential at which no Faradaic process was occurring when we varied the scan rate of the CV. CV was performed in the same electrochemical cell with 0.1 M KHCO3 electrolyte separated with a Nafion proton exchange membrane. The linear slope provides C. ECSA = Rf × S, where S stands for the geometric area of the glassy carbon electrode (S = 0.19 cm2 in this work).
Characterization
XRD were measured on a Philips X’Pert Pro Super X-ray diffractometer equipped with graphite-monochromatized Cu Ka radiation. X-ray photoelectron spectroscopy (XPS) was carried out on an ESCA Lab MKII X-ray photoelectron spectrometer. The source for excitation is Mg Ka radiation. For angle-resolved XPS (ARXPS), the samples were fixed on a rotatable holder, which enables measurement for take-off angles θ of 20° measured relative to the surface normal. Low-resolution transmission electron microscopy (TEM) studies were performed on JEOL-2010F with an acceleration voltage of 200 kV. High-angle annular dark field scanning transmission electron microscopy (HAADF- STEM) and high-resolution transmission electron microscope electron energy loss spectroscopy (HRTEM-EELS) were carried out using a cold-field emission Cs-corrected JEOL ARM-200F Atomic Resolution Analytical Microscope operating at an accelerating voltage of 200 kV. In situ X-ray absorption of the Cu K-edges was performed at the Soft X-ray Microcharacterization Beamline (SXRMB) at Canadian Light Source (CLS). A homemade in situ electrochemical cell was used, with platinum as the counter electrode and Ag/AgCl as the reference electrode. The electrolyte is CO2-purged 0.1 M KHCO3. The acquisition of each spectrum took 15 min.
Data availability
The data that support the findings of this study are available within the article and its Supplementary Information files. All other relevant source data are available from the corresponding author upon reasonable request.
References
Qiao, J., Liu, Y., Hong, F. & Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 43, 631–675 (2014).
Wang, Z.-L., Li, C. & Yamauchi, Y. Nanostructured nonprecious metal catalysts for electrochemical reduction of carbon dioxide. Nano Today 11, 373–391 (2016).
Li, F., MacFarlane, D. R. & Zhang, J. Recent advances in the nanoengineering of electrocatalysts for CO2 reduction. Nanoscale 10, 6235–6260 (2018).
Hori, Y. I. in Modern aspects of electrochemistry 89–189 (Springer, 2008).
Kuhl, K. P., Cave, E. R., Abram, D. N. & Jaramillo, T. F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 5, 7050–7059 (2012).
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315 (2010).
Britovsek, G. J. et al. Iron and cobalt ethylene polymerization catalysts bearing 2, 6-bis (imino) pyridyl ligands: synthesis, structures, and polymerization studies. J. Am. Chem. Soc. 121, 8728–8740 (1999).
Keskin, A. & Gürü, M. The effects of ethanol and propanol additions into unleaded gasoline on exhaust and noise emissions of a spark ignition engine. Energy Sourc. A, Recovery Util. Environ. Eff. 33, 2194–2205 (2011).
Papa, A. J. in Ullmann’s Encyclopedia of Industrial Chemistry (Wiley-VCH Verlag GmbH & Co. KGaA, South Charleston, 2000).
Atsumi, S., Hanai, T. & Liao, J. C. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451, 86–89 (2008).
Yanowitz, J., Christensen, E. & McCormick, R. L. Utilization of renewable oxygenates as gasoline blending components. (National Renewable Energy Laboratory (NREL), Golden, CO, USA, 2011).
Masum, B., Kalam, M., Masjuki, H., Palash, S. & Fattah, I. R. Performance and emission analysis of a multi cylinder gasoline engine operating at different alcohol–gasoline blends. RSC Adv. 4, 27898–27904 (2014).
Singh, M. R., Kwon, Y., Lum, Y., Ager, J. W. III & Bell, A. T. Hydrolysis of electrolyte cations enhances the electrochemical reduction of CO2 over Ag and Cu. J. Am. Chem. Soc. 138, 13006–13012 (2016).
Mistry, H. et al. Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 7, 12123 (2016).
Ren, D., Wong, N. T., Handoko, A. D., Huang, Y. & Yeo, B. S. Mechanistic insights into the enhanced activity and stability of agglomerated Cu nanocrystals for the electrochemical reduction of carbon dioxide to n-propanol. J. Phys. Chem. Lett. 7, 20–24 (2015).
Ren, D., Ang, B. S.-H. & Yeo, B. S. Tuning the selectivity of carbon dioxide electroreduction toward ethanol on oxide-derived CuxZn catalysts. ACS Catal. 6, 8239–8247 (2016).
De Luna, P. et al. Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction. Nat. Catal. 1, 103–110 (2018).
Li, C. W. & Kanan, M. W. CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 134, 7231–7234 (2012).
Verdaguer-Casadevall, A. et al. Probing the active surface sites for CO reduction on oxide-derived copper electrocatalysts. J. Am. Chem. Soc. 137, 9808–9811 (2015).
Li, C. W., Ciston, J. & Kanan, M. W. Electroreduction of carbon monoxide to liquid fuel on oxide-derived nanocrystalline copper. Nature 508, 504–507 (2014).
Roberts, F. S., Kuhl, K. P. & Nilsson, A. Electroreduction of carbon monoxide over a copper nanocube catalyst: surface structure and pH dependence on selectivity. ChemCatChem 8, 1119–1124 (2016).
Gupta, N., Gattrell, M. & MacDougall, B. Calculation for the cathode surface concentrations in the electrochemical reduction of CO2 in KHCO3 solutions. J. Appl. Electrochem. 36, 161–172 (2006).
Gao, D. et al. Plasma-activated copper nanocube catalysts for efficient carbon dioxide electroreduction to hydrocarbons and alcohols. ACS Nano 11, 4825–4831 (2017).
Lee, S., Kim, D. & Lee, J. Electrocatalytic production of C3‐C4 compounds by conversion of CO2 on a chloride‐induced bi‐phasic Cu2O‐Cu catalyst. Angew. Chem. 127, 14914–14918 (2015).
Favaro, M. et al. Subsurface oxide plays a critical role in CO2 activation by Cu (111) surfaces to form chemisorbed CO2, the first step in reduction of CO2. Proc. Natl Acad. Sci. USA 114, 6706–6711 (2017).
Xiao, H., Goddard, W. A., Cheng, T. & Liu, Y. Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc. Natl Acad. Sci. USA 114, 6685–6688 (2017).
Lum, Y. & Ager, J. W. Stability of residual oxides in oxide‐derived copper catalysts for electrochemical CO2 reduction investigated with 18O labeling. Angew. Chem. Int. Ed. 57, 551–554 (2018).
Zhuang, T.-T. et al. Steering post-C-C coupling selectivity enables high efficiency electroreduction of carbon dioxide to multi-carbon alcohols. Nat. Catal. 1, 421–428 (2018).
Wang, D. et al. Structurally ordered intermetallic platinum-cobalt core-shell nanoparticles with enhanced activity and stability as oxygen reduction electrocatalysts. Nat. Mater. 12, 81–87 (2013).
Sasaki, K. et al. Core‐protected platinum monolayer shell high‐stability electrocatalysts for fuel‐cell cathodes. Angew. Chem. Int. Ed. 49, 8602–8607 (2010).
Strasser, P. et al. Lattice-strain control of the activity in dealloyed core-shell fuel cell catalysts. Nat. Chem. 2, 454–460 (2010).
Lv, H. et al. A new core/shell NiAu/Au nanoparticle catalyst with Pt-like activity for hydrogen evolution reaction. J. Am. Chem. Soc. 137, 5859–5862 (2015).
Chen, Z. et al. Core-shell MoO3-MoS2 nanowires for hydrogen evolution: a functional design for electrocatalytic materials. Nano Lett. 11, 4168–4175 (2011).
Wang, D. et al. Pt-decorated PdCo@ Pd/C core-shell nanoparticles with enhanced stability and electrocatalytic activity for the oxygen reduction reaction. J. Am. Chem. Soc. 132, 17664–17666 (2010).
Wen, Z. et al. Nitrogen‐enriched core‐shell structured Fe/Fe3C‐C nanorods as advanced electrocatalysts for oxygen reduction reaction. Adv. Mater. 24, 1399–1404 (2012).
Giordano, C. & Antonietti, M. Synthesis of crystalline metal nitride and metal carbide nanostructures by sol-gel chemistry. Nano Today 6, 366–380 (2011).
Wang, D. & Li, Y. Controllable synthesis of Cu-based nanocrystals in ODA solvent. Chem. Commun. 47, 3604–3606 (2011).
Liu, P. & Hensen, E. J. Highly efficient and robust Au/MgCuCr2O4 catalyst for gas-phase oxidation of ethanol to acetaldehyde. J. Am. Chem. Soc. 135, 14032–14035 (2013).
Wu, H. & Chen, W. Copper nitride nanocubes: size-controlled synthesis and application as cathode catalyst in alkaline fuel cells. J. Am. Chem. Soc. 133, 15236–15239 (2011).
Avanzino, S. C. & Jolly, W. L. A simple method for obtaining X-ray photoelectron spectra of species in liquid solution. J. Am. Chem. Soc. 100, 2228–2230 (1978).
Lin, Q. et al. Phase-transfer ligand exchange of lead chalcogenide quantum dots for direct deposition of thick, highly conductive films. J. Am. Chem. Soc. 139, 6644–6653 (2017).
Manthiram, K., Beberwyck, B. J. & Alivisatos, A. P. Enhanced electrochemical methanation of carbon dioxide with a dispersible nanoscale copper catalyst. J. Am. Chem. Soc. 136, 13319–13325 (2014).
Chiang, W.-H. et al. C/BCN core/shell nanotube films with improved thermoelectric properties. Carbon 109, 49–56 (2016).
Cumpson, P. J. Angle-resolved XPS and AES: depth-resolution limits and a general comparison of properties of depth-profile reconstruction methods. J. Electron. Spectrosc. Relat. Phenom. 73, 25–52 (1995).
Schouten, K. J. P., Qin, Z., Pérez Gallent, E. & Koper, M. T. Two pathways for the formation of ethylene in CO reduction on single-crystal copper electrodes. J. Am. Chem. Soc. 134, 9864–9867 (2012).
Hori, Y., Takahashi, I., Koga, O. & Hoshi, N. Selective formation of C2 compounds from electrochemical reduction of CO2 at a series of copper single crystal electrodes. J. Phys. Chem. B 106, 15–17 (2002).
Durand, W. J., Peterson, A. A., Studt, F., Abild-Pedersen, F. & Nørskov, J. K. Structure effects on the energetics of the electrochemical reduction of CO2 by copper surfaces. Surf. Sci. 605, 1354–1359 (2011).
Acknowledgements
This work was supported financially by the Ontario Research Fund Research-Excellence Program, the Natural Sciences and Engineering Research Council (NSERC) of Canada, and the CIFAR Bio-Inspired Solar Energy program. Computations were performed on the SOSCIP Consortium’s Blue Gene/Q computing platform. SOSCIP is funded by the Federal Economic Development Agency of Southern Ontario, the Province of Ontario, IBM Canada Ltd., Ontario Centres of Excellence, Mitacs and 15 Ontario academic member institutions. X-ray absorption spectra were performed on SXRMB beamlines at the Canadian Light Source (CLS), which is supported by the Canada Foundation for Innovation, Natural Sciences and Engineering Research Council of Canada, the University of Saskatchewan, the Government of Saskatchewan, Western Economic Diversification Canada, the National Research Council Canada, and the Canadian Institutes of Health Research. Z.L. acknowledges a scholarship from the China Scholarship Council (CSC) (201607090041) and Basic and Innovation Program, Beijing Jiaotong University (KSJB17016536). A.S. acknowledges Fonds de Recherche du Quebec-Nature et Technologies (FRQNT) for the postdoctoral fellowship award. P.D.L. acknowledges support from NSERC in the form of the Canada Graduate Scholarship. H.T. acknowledges the Netherlands Organisation for Scientific Research (NWO) for a Rubicon grant (680-50-1511) to support his postdoctoral research at University of Toronto. The authors thank C. Q. Zou, M. X. Liu, F. F. Fan, J. Xing and L. Gao from University of Toronto for fruitful discussions.
Author information
Authors and Affiliations
Contributions
E.H.S. supervised the project. Z.L. and T.Z. conceived the ideas, conducted the experiments, analyzed the results, and wrote the manuscript. A.S. carried out simulations. C.H. and P.H. conducted the TEM measurements. J.L., Y.H. and Q.X. assisted in measuring XAS and analyzing the results. S.L., L.C. and C.T. assisted in analyzing the TEM results. R.Q.-B. and Y.L. performed XPS measurements. Y.Z. assisted in analyzing the XPS results. Y.W. and F.L. assisted in discussing mechanisms. P.D.L., C.T., H.T. and Y.P. assisted in revising the manuscript. P.C., Z.X., S.Z. and D.S. assisted in reviewing the manuscript. All authors read and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liang, ZQ., Zhuang, TT., Seifitokaldani, A. et al. Copper-on-nitride enhances the stable electrosynthesis of multi-carbon products from CO2. Nat Commun 9, 3828 (2018). https://doi.org/10.1038/s41467-018-06311-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-06311-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
