
Abstract
The volume of the lateral ventricles (LV) increases with age and their abnormal enlargement is a key feature of several neurological and psychiatric diseases. Although lateral ventricular volume is heritable, a comprehensive investigation of its genetic determinants is lacking. In this meta-analysis of genome-wide association studies of 23,533 healthy middle-aged to elderly individuals from 26 population-based cohorts, we identify 7 genetic loci associated with LV volume. These loci map to chromosomes 3q28, 7p22.3, 10p12.31, 11q23.1, 12q23.3, 16q24.2, and 22q13.1 and implicate pathways related to tau pathology, S1P signaling, and cytoskeleton organization. We also report a significant genetic overlap between the thalamus and LV volumes (ρgenetic = −0.59, p-value = 3.14 × 10−6), suggesting that these brain structures may share a common biology. These genetic associations of LV volume provide insights into brain morphology.
Similar content being viewed by others

Introduction
The volume of lateral ventricles increases in normal aging1,2,3,4. The enlargement of lateral ventricles has also been suggested in various complex neurological disorders such as Alzheimer’s disease, vascular dementia, and Parkinson’s disease5,6,7,8 as well as psychiatric disorders such as schizophrenia and bipolar disorder9,10,11. Furthermore, ventricular enlargement has been associated with poor cognitive functioning and cerebral small vessel disease pathology12,13,14. Even though it might be intuitive to interpret ventricular expansion primarily as an indicator of brain shrinkage after the onset of the disorder, recent studies have provided evidence against this notion15,16. The size of lateral ventricles is influenced by genetic factors with heritability estimated to be 54%, on average16, but changing with age, from 32–35% in childhood to about 75% in late middle and older age16. Even though the size of surrounding gray matter structures is also heritable17,18,19, ventricular volume is reported to be genetically independent of other brain regions surrounding the ventricles20. Similarly, ventricular enlargement in schizophrenia does not appear to be linked to volume reduction in the surrounding structures15.
Elucidating the genetic contribution to inter-individual variation in lateral ventricular volume can thus provide important insights and better understanding of the complex genetic architecture of brain structures and related neurological and psychiatric disorders. Candidate gene studies have identified single-nucleotide polymorphisms (SNPs) mapping to Catechol-O-Methyltransferase (COMT) and Neuregulin 1 (NRG1) genes as associated with larger lateral ventricular volume in patients with the first episode of non-affective psychosis21,22. However, a comprehensive investigation of the genetic determinants of lateral ventricular volume is lacking.
Here, we perform a genome-wide association (GWA) meta-analysis of 23,533 middle-aged to elderly individuals from population-based cohorts participating in the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium in order to identify common genetic variants that influence lateral ventricle volume. We apply a commonly used two-stage GWA design followed by a joint analysis approach that combines information across the stages and provides greater power23. We identify 7 genetic loci associated with lateral ventricular volume and report genome-wide overlap with thalamus volume.
Results
Genome-wide association results
The overview of study design is illustrated in Supplementary Fig. 1. The GWA results from 12 studies were combined in stage 1 and subsequently evaluated in an independent sample from 14 studies in stage 2. Finally, the results of stage 1 and stage 2 analyses were combined in stage 3. Detailed information on study participants, image acquisition and genotyping is provided in Supplementary Note 1 and Supplementary Data 1–3.
The results of the stage 1 meta-analysis (N = 11,396) are illustrated in Supplementary Fig. 2. The quantile-quantile plot suggests that potential population stratification and/or cryptic relatedness are well controlled after genomic correction (λ = 1.04) (Supplementary Fig. 2, Supplementary Table 1). The stage 1 meta-analysis identified 146 significant variant associations, mapping to three chromosomal regions at 3q28, 7p22.3, and 16q24.2 (Table 1). All 146 stage 1 significant associations replicated in the stage 2 meta-analysis (N = 12,137) with the same direction of effect at Bonferroni adjusted significance (p-value = 5 × 10−3, Supplementary Data 4), except one SNP (p-value = 7.6 × 10−3). Subsequently, the results from all individual studies were combined in the stage 3 GWA meta-analysis (N = 23,533). The quantile–quantile plot showed again adequate control of population stratification or relatedness (Supplementary Fig. 3). The combined stage 3 GWA meta-analysis identified 314 additional significant associations mapping to four additional chromosomal regions at 10p12.31, 11q23.1, 12q23.3, and 22q13.1 (Figs. 1, 2, Table 1). The effect size for the lead variant mapped to 10p12.31 locus was correlated with mean age of the cohort (r = 0.50, p-value = 0.03) (Supplementary Fig. 4). No correlation was found for the other lead variants (Supplementary Fig. 5–10).

Manhattan plot for stage 3 genome-wide association meta-analysis. Each dot represents a variant. The plot shows –log10 p-values for all variants. Red line represents the genome-wide significance threshold (p-value < 5 × 10−8), whereas blue line denotes suggestive threshold (p-value < 1 × 10−5)

Regional association and recombination plots in combined stage 3 GWA meta-analysis. The left axis represents –log10 p-values for association with total later ventricular volume. The right axis represents the recombination rate, and the x-axis represents chromosomal position (hg19 genomic position). The most significant SNPs of the regions are denoted with a purple diamond. Surrounding SNPs are colored according to their pairwise correlation (r2) with the top-associated SNP of the region. The gene annotations are below the figure
Even though cohorts of European (EA) and African-American (AA) ancestry were included, all significant associations were mainly driven by EA samples (Supplementary Fig. 11–12). The direction of effect size across the EA cohorts for the seven lead variants was generally concordant and showed no evidence of any single cohort driving the associations (Supplementary Fig. 11). Despite the different methods of phenotyping across the cohorts, the cohorts with different phenotyping methods showed evidence of effect suggesting that there is limited heterogeneity in effects (Supplementary Fig. 12).
To investigate whether seven lead variants have an effect in early life, childhood, the analyses were carried out in a children’s cohort of 1141 participants from Generation R study. The percentage of lead variants showing consistent direction of effect with stage 3 was 85.7% (6 out of 7, binomial p-value = 0.05) (Supplementary Data 4), and a variant mapped to the 12q23.3 region showed nominal association with lateral ventricular volume in the children’s cohort (Zscore = −2.56, p-value = 0.01). Additionally, three out of seven lead variants (or their proxies; r2 > 0.7) showed pleiotropic association (p-value < 5 × 10−8) with other traits according to the PhenoScanner database (Supplementary Data 5)24.
To capture gender-based differences, sex-stratified GWA analysis was performed (Nmen = 10,358; Nwomen = 12,872). None of the 15,660,719 variants that were tested for heterogeneity between men and women reached genome-wide significance threshold (Supplementary Fig. 13). However, an indel located at 4q35.2 showed suggestive evidence of association in men (4:187559262:C_CAA, p-value = 5.43 × 10−8) but not in women (p-value = 0.88).
Independent signals within loci
The conditional and joint (COJO) analysis using the Genome-wide Complex Trait Analysis (GCTA) identified no other additional variants, after conditioning on the lead variant at the locus 3q28, 7p22.3, 10p12.31, 11q23.1, 12q23.3, 16q24.2, or 22q13.1.
Functional annotation
A large proportion of genome-wide significant variants were intergenic (335/460) (Supplementary Fig. 14). Variants with the highest probability of having a regulatory function based on RegulomeDB score (Category 1 RegulomeDB score) were located at 7p22.3 and at 22q13.1 (Supplementary Data 6). Of seven lead variants, four were intergenic, four were in an active chromatin state and three showed expression quantitative trait (eQTL) effects (Supplementary Data 6). The lead SNP at 22q13.1 (rs4820299) was associated with differential expression of the largest number of genes (n = 6). In brain tissue, the alternate allele of this SNP was associated with higher expression of TRIOBP suggesting that higher expression was associated with smaller lateral ventricles (Supplementary Fig. 15).
Partitioned heritability
SNP-based heritability in the sample of European ancestry participants was estimated at 0.20 (SE = 0.02) using LD score regression, and this was higher in women 0.19 (SE = 0.04) than in men 0.15 (SE = 0.05). The seven lead variants explained 1.5% of total variance in lateral ventricular volume. Partitioning of heritability based on functional annotation using LD score regression, revealed significant enrichment of SNPs within 500 bp of highly active enhancers, where 17% of SNPs accounted for 54% of the heritability (p-value = 7.9 × 10−6, Supplementary Table 2). Significant enrichment was also found for histone marks including H3K27ac (which indicates enhancer and promoter regions), H3K9ac (which highlights promoters), H3K4me3 (which indicates promoters/transcription starts), and H3K4me1 (which highlights enhancers) (Supplementary Table 2)25,26.
Functional enrichment analysis
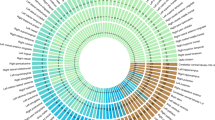
Functional enrichment analysis using regulatory regions from the ENCODE and Roadmap projects using the GWAS Analysis of Regulatory or Functional Information Enrichment with LD correction (GARFIELD) method revealed that SNPs associated with lateral ventricular volume at p-value threshold <10−5 were more often located in genomic regions harboring histone marks (H3K9ac (associated with promoters) and H3K36me3 (associated with transcribed regions))25 and DNaseI hypersensitivity sites (DHS) than a permuted background (Fig. 3, Supplementary Data 7).

Functional enrichment analysis of lateral ventricular volume loci within DNaseI hypersensitivity spots. The radial lines show fold enrichment (FE) at eight GWA p-value thresholds. The results are shown for each of 424 cell types which are sorted by tissue, represented along the outer circle of the plot. The font size is proportional to the number of cell types from the tissue. FE values are plotted with different colors with respect to different GWA thresholds. Significant enrichment for a given cell type is denoted along the outer circle of the plot from a GWA p-value threshold <10−5 (outermost) to GWA p-value threshold <10−8 (innermost). The results show ubiquitous enrichment
Integration of gene expression data
Integration of functional data from the Genotype-Tissues Expression (GTEx) project using the MetaXcan method revealed two significant associations between genetically predicted expression in brain tissue and lateral ventricular volume (Supplementary Fig. 16). Expression levels of TRIOBP at the locus 22q13.1 (p-value = 3.2 × 10−6) and MRPS16 at the locus 10q22.2 (p-value = 1.8 × 10−6) were associated with lateral ventricular volume.
Gene annotation and pathway analysis
The results of gene-based and pathway analyses are illustrated in Supplementary Table 3 and Supplementary Data 8. The pathway analysis identified “regulation of cytoskeleton organization” (GO:0051493) gene-set to be significantly enriched (p-value = 6 × 10−6). Genes of the “regulation of cytoskeleton organization” pathway have previously been implicated in various neurological or cardiovascular diseases (Supplementary Data 9). Furthermore, pathways that pointed towards sphingosine 1 phosphate (S1P) signaling showed suggestive enrichment (Supplementary Data 8).
Genetic correlation
Additionally, we examined the genetic overlap between lateral ventricular volume and other traits (Table 2). We found that genetically determined components of thalamus and lateral ventricular volumes appear to be negatively correlated (ρgenetic = −0.59, p-value = 3.14 × 10−6). This finding was also confirmed at the phenotype level (Supplementary Table 4). Weaker genetic overlap was observed with infant head circumference (ρgenetic = 0.28, p-value = 8.7 × 10−3), intracranial volume (ρgenetic = 0.35, p-value = 9 × 10−3), height (ρgenetic = −0.14, p-value = 5.7 × 10−3), and mean pallidum (ρgenetic = −0.29, p-value = 2.5 × 10−2), whereas no significant genetic overlap was found with neurological diseases, psychiatric diseases, or personality traits.
Genetic risk score
We next examined the association of genetic risk scores (GRS) for Alzheimer’s disease, Parkinson’s disease, schizophrenia, bipolar disorder, cerebral small vessel disease, and tau-related pathology, including tau and phosphorylated tau levels in cerebrospinal fluid, amyotrophic lateral sclerosis (ALS), and progressive supranuclear palsy (PSP), using the lead SNPs from the largest published GWA study and lateral ventricular volume (Supplementary Data 10). We found a suggestive association of GRS for tau levels in cerebrospinal fluid (p-value = 9.59 × 10−3) and lateral ventricular volume (Supplementary Table 5). The association was driven by one SNP (Supplementary Table 6). No association was observed with other examined phenotypes (Supplementary Table 5).
Discussion
We have performed the first genome-wide association study of lateral ventricular volume including up to 23,533 individuals. We identified statistically significant association between lateral ventricular volume and variants at 7 loci. Additionally, we found that genetically determined components of thalamus and lateral ventricular volume are correlated.
The strongest association was observed at the intergenic 3q28 locus between non-coding RNA SNAR-I and OSTN. This region has previously been associated with cerebrospinal fluid tau/ptau levels and Alzheimer’s disease risk, tangle pathology and cognitive decline27. Similarly, the genome-wide significant locus at 12q23.3 encompasses NUAK1, which has also been associated with tau pathology. Nuak1 modulates tau levels in human cells and animal models and associates with tau accumulation in different tauopathies28. NUAK1 is most prominently expressed in the brain where it has a role in mediating axon growth and branching in cortical neurons29. The lead SNP of the 12q23.3 locus mapped to an intron of NUAK1. This SNP is among the top 1% of most deleterious variants in the human genome based on its Combined Annotation Dependent Depletion (CADD) score of 21.5 and is located in an enhancer region (Supplementary Data 6). Interestingly, this variant also showed an effect in early life.
In our data, the significant variants of 7p22.3 region had the highest probability of being regulatory based on the RegulomeDB score (1b). The lead variant at 7p22.3 was in an active chromatin state and was associated with differential expression of GNA12 (Supplementary Data 6). The GNA12 gene is involved in various transmembrane signaling systems30,31,32,33. Interestingly, this gene was part of S1P signaling pathways identified to be enriched among genes associated with lateral ventricular volume. S1P, a bioactive sphingolipid metabolite, regulates nervous system development34 such as neuronal survival, neurite outgrowth, and axon guidance35,36, and plays a role in neurotransmitter release37. It also plays a role in regulating the development of germinal matrix (GM) vasculature38. Disruption of S1P regulation results in defective angiogenesis in GM, hemorrhage, and enlarged ventricles38.
The other identified locus, 16q24.2, has previously been connected with small vessel disease and white-matter lesions formation39. Further, the alternate allele of the lead SNP at 22q13.1 in TRIOBP is associated with higher expression of the same gene in basal ganglia and brain cortex, and the same allele is associated with smaller lateral ventricular volume. Interestingly, predicted expression of this gene in cerebral cortex was significantly associated with lateral ventricular volume, suggesting a causal functional role of the gene. The same analysis revealed significant association of the expression of MRPS16 in frontal cortex with lateral ventricular volume. This gene was previously related to agenesis/hypoplasia of corpus callosum and enlarged ventricles40.
Finally, the lead intergenic SNP at 11q23.1 maps between C11orf53 and ARHGAP20, whereas the 10p12.31 region encompasses MLLT10 which has been linked to various leukemias, ovarian cancer, and meningioma41,42. The effect size of this variant on lateral ventricular volume was correlated with mean cohort age, with the effect being near zero at younger age and larger at older ages.
The gene-enrichment analysis highlighted “regulation of cytoskeleton organization” (GO:0051493) pathway. Genes that are part of this pathway have previously been implicated in various neurological diseases such as Parkinson’s disease (PARK2), frontotemporal dementia (MAPT), neurofibromatosis 2 (NF2), tuberous sclerosis (TSC1) (Supplementary Data 9). The cytoskeleton is essentially involved in all cellular processes, and therefore crucial for processes in the brain such as cell proliferation, differentiation, migration, and signaling. Dysfunction of cytoskeleton has been associated with neurodevelopmental, psychiatric and neurodegenerative diseases43,44,45.
Previous studies showed significant sex-specific differences in lateral ventricular volume46,47. In our study we did not observe sex-specific differences; as for the lead seven variants, both males and females were contributing to the association signal. However, we observed only one suggestive association at 4q35.2 that showed association in men only. The lead variant (indel) is mapped to FAT1 which encodes atypical cadherins. Mutation in this gene causes a defect in cranial neural tube closure in a mouse model and an increase in radial precursor proliferation in the cortex48. However, the SNP-based heritability estimates were slightly higher in females. This may be explained by the differences in sample size in male and female-specific analyses implying that there is lower precision.
We estimated that 20% of genetic variance in lateral ventricular volume could be explained by common genetic variants, suggesting that common variants represent a substantial fraction of overall genetic component of variance. Moreover, the most statistically significant effect occurred in the regions of highly active enhancers and histone marks, suggesting their involvement in gene expression. Using the LD score regression method, we found a significant negative genetic correlation between lateral ventricular volume and thalamus volume. However, these may not be independent events, but inverse reflections of the same biology. Even though not strictly significant, we also observed trends for genetic correlations with other brain volumetric measures. Furthermore, no genome-wide overlap was found between lateral ventricular volume and various neurological or psychiatric diseases. Given that enlargement of lateral ventricles has been suggested in Alzheimer’s disease, we examined the association of APOE alleles and found no association between the APOE ɛ4 (p-value = 0.86) or APOE ɛ2 (p-value = 0.81) and lateral ventricular volume in our study population.
As we identified loci underlying lateral ventricular volume at the genome-wide level, but also genes and common pathways, our results provide various insights into the genetic contribution to lateral ventricular volume variability and a better understanding of the complex genetic architecture of brain structures. The genes with variants that we found to be associated with lateral ventricular volume are relevant to neurological aging given the characteristics of the study population which is relatively free from the disease as participants with stroke, traumatic brain injury and dementia at the time of magnetic resonance imaging (MRI) were excluded. This is in line with the previously published work of Pfefferbaum et al. who showed that the stability of lateral ventricles is genetically determined, whereas other factors such as normal aging or trauma and disease play a role in its change1,16.
However, while studying genetic overlap of lateral ventricular volume and various neurological or psychiatric disorders at multiple levels (LD score regression/polygenic, GRS/oligogenic, GWA hits/monogenic), we found evidence that some single genetic variants have pleiotropic effect on lateral ventricular volume and biochemical markers for a neurological disease (AD) or meningioma (Supplementary Data 5), while no evidence was found for genetic overlap with other neurological or psychiatric disorders (Table 2, Supplementary Table 5). The pattern of association between lateral ventricular volume and psychiatric disorder, i.e., schizophrenia on multiple scales is similar to the findings of Franke et al. who evaluated association of various subcortical brain volumes and schizophrenia and reported no evidence of genetic overlap49. Even though our study does not provide a definite statement regarding the relationship between lateral ventricular volume and neurological or psychiatric disorders, it lays the foundation for future studies which should disentangle whether lateral ventricular volume is genetically related or unrelated to various neurological and psychiatric disorders (e.g., result from reverse causation). Novel insights may be revealed by improving the power of the studies, studying homogeneous samples with harmonized phenotype assessment methods along with evaluation of common and rare variants.
The strengths of our study are the large sample, population-based design and the use of quantitative MRI. Our study also has several limitations. Despite the effort to harmonize phenotype assessment, the methods used to quantify lateral ventricular volume differ across cohorts. Because of this phenotypic heterogeneity, association results of participating cohorts were combined using a sample-size weighted meta-analysis, thus limiting discussion on effect sizes. Secondly, phenotypic heterogeneity may have caused the loss of statistical power. However, despite heterogeneity in the phenotype assessment, the association signals were coming from several studies irrespective of the method of phenotype assessment, which suggests robustness of our findings. Furthermore, although we made an effort to include cohorts of EA and AA ancestry, the study comprised predominately of individuals of European origin (22,045 individuals of EA and 1488 of AA ancestry). Given the disparity in sample size, it is difficult to distinguish whether any inconsistency in results between the two groups stems from true genetic differences or from differential power to detect genetic effects. Indeed, this is also exemplified by the plots of the Z-scores (Supplementary Fig. 11) showing that direction of effect size in AA cohorts is often inconsistent with the direction of effect size in EA cohorts. However, the same inconsistency can be observed with European cohorts of equally small sample size. This inconsistency may be due to small sample size rather than ethnic background but we cannot rule out that racial-ethnic specific effects may exit. This limitation underscores the need for expanding research studies in non-European populations. Finally, as some loci only reached the genome-wide significance in the combined meta-analysis, they should be considered as highly probable findings and would still require independent replication.
To conclude, we identified genetic associations of lateral ventricular volume with variants mapping to 7 loci and implicating several pathways, including pathway related to tau pathology, cytoskeleton organization, and S1P signaling. These data provide new insights into understanding brain morphology.
Methods
Study design
The overview of study design is illustrated in Supplementary Fig. 1. We performed a GWA meta-analysis of 11,396 participants of mainly European ancestry from 12 studies (stage 1) that contributed summary statistic data before a certain deadline. The deadline was set prior to data inspection and was not influenced by the results of the GWA meta-analysis. Variants that surpassed the genome-wide significance threshold (p-value < 5 × 10−8) were subsequently evaluated in an independent sample of 12,137 participants of mainly European ancestry from 14 studies (stage 2). Finally, we performed a meta-analysis of all stage 1 and stage 2 studies (stage 3).
Study population
All participating studies are part of the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium50. A detailed description of participating studies can be found in Supplementary Note 1. General characteristics of study participants are provided in Supplementary Data 1. Written informed consent was obtained from all participants. Each study was approved by local ethical committees or the institutional review boards (see Supplementary Note 1 for details).
Imaging
Each study performed MRI and estimated the volume of the lateral ventricles and intracranial volume (ICV). The field strength of scanners ranged from 0.35 to 3 T. Information on scanner manufacturers and measurement methods is provided in Supplementary Data 2. While most of the studies quantified lateral ventricular volume using validated automated segmentation methods, some studies used validated visual grading scales. The visual and volumetric scales were compared previously and showed high agreement for lateral ventricular volume2. The assessment of consistency of lateral ventricular volume on volumetric scale across time and different versions of software (freesurfer v4.5, v5.1, and v6.0), revealed high intraclass correlation (ICC > 0.98) in a subset of participants from the Rotterdam Study. Participants with dementia at the time of MRI, traumatic brain injury, prior or current stroke or intracranial tumors were excluded.
Genotyping and imputation
Information on genotyping platforms, quality control procedures and imputations methods for each participating study are provided in Supplementary Data 3. All studies used commercially available genotyping arrays, including Illumina or Affymetrix arrays. Similar quality control procedures were applied for each study (Supplementary Data 3). Using the validated software (Minimac51, IMPUTE52, BEAGLE53), each study performed genotype imputations using mostly the 1000 Genome phase 1 v3 reference panel.
Genome-wide association (GWA) analysis
Each participating study performed the GWA analysis of total lateral ventricular volume under an additive model using variant allele dosage as predictors and natural logarithm of the total lateral ventricular volume as the dependent variable. Transformation of the lateral ventricular volume was applied to obtain approximately normal distribution (Supplementary Fig. 17). The association analyses were adjusted for age, sex, total intracranial volume, age2 if significant, population stratification, familial relationship (family-based studies) or study site (multi-site studies). Population stratification was controlled for by including principal components derived from genome-wide genotype data. Study-specific details on covariates and software used are provided in Supplementary Data 3. Quality control (QC) was conducted for all participating studies using a standardized protocol provided by Winkler et al.54. Variants with low imputation quality r2 < 0.3 or minor allele count (MAC) ≤ 6 were filtered out. The association results of participating studies were combined using a fixed-effect sample-size weighted Z-score meta-analysis in METAL because of the difference in measurement methods of lateral ventricular volume55. Genomic control was applied to account for small amounts of population stratification or unaccounted relatedness. After the meta-analysis, variants with information in less than half the total sample size were excluded. Meta-analyses were performed separately for each of the stages. In the stage 1 meta-analysis, a p-value < 5 × 10−8 was considered significant. Variants that surpassed the threshold were evaluated in the stage 2 meta-analysis. In order to model linkage disequilibrium (LD) between those variants, we first calculated the number of independent tests using the eigenvalues of a correlation matrix using the Matrix Spectral Decomposition (matSpDlite) software56. Subsequently, a Bonferroni correction was applied for the effective number of independent tests (0.05/10 independent SNPs = 5 × 10−3). Additionally, all analyses were stratified by sex. Following the same QC steps as for overall analyses, the sex-stratified association results of participating studies were combined using a fixed-effect sample-size weighted Z-score meta-analysis in METAL while applying genomic control55. The variants were assessed only if test statistics (Z-score) were heterogeneous between males and females (p-value < 0.1) and if the association in a sex-combined analysis did not reach genome-wide significance threshold57.
Conditional analysis
In order to identify variants that were independently associated with lateral ventricular volume, we performed conditional and joint (COJO) GWA analysis using Genome-wide Complex Trait Analysis (GCTA), version 1.26.058. LD pattern was calculated based on 1000 Genome phase 1v3 imputed data of 6291 individuals from the Rotterdam Study I.
Functional annotation
To annotate genome-wide significant variants with regulatory information, we used HaploReg v4.159, RegulomeDB v1.160, and Combined Annotation Dependent Depletion (CADD) tools61. To determine whether they have an effect on gene expression, we used GTEx data62. For the lead variants, we explored 5 chromatin marks assayed in 127 epigenomes (H3K4me3, H3K4me1, H3K36me3, H3K27me3, H3K9me3) of RoadMap data63. To search for pleiotropic associations between our lead variants and their proxies (r2 > 0.7) with other traits, we used the PhenoScanner database designed to facilitate the cross-referencing of genetic variants with many phenotypes9. The association results with genome-wide significance at 5 × 10−8 were extracted.
Variance explained
The proportion of variance in lateral ventricular volume explained by each lead variant was calculated using Pearson’s phi coefficient squared as explained in Draisma et al.64. The total proportion of variance in lateral ventricular volume was calculated by adding up the proportions of variance in lateral ventricular volume explained by each lead association signal.
Partitioned heritability
SNP-based heritability and partitioned heritability analyses were performed using LD score regression following the previously described method65. Partitioned heritability analysis determines enrichment of heritability in SNPs partitioned into 24 functional classes as reported in Finucane et al.65. To avoid bias, an additional 500 bp window was included around the variants included in the functional classes. Only the HapMap3 variants were included as these seem to be well-imputed across cohorts.
Functional enrichment analysis
We performed functional enrichment analysis using regulatory regions from the ENCODE and Roadmap projects using GWAS Analysis of Regulatory or Functional Information Enrichment with LD correction (GARFIELD) method66. The method provides fold enrichment (FE) statistics at various GWA p-value thresholds after taking into account LD, minor allele frequency, and local gene density66. The FE statistics were calculated at eight GWA p-value thresholds (0.1 to 1 × 10−8). The associations were tested for various regulatory elements including DNase-I hypersensitivity sites, histone modifications, chromatin states and transcription factor binding sites in over 1000 cell and tissue-specific annotations66. The significance threshold calculated based on the number of annotations used was set at 4.97 × 10−5.
Integration of gene expression
To integrate functional data in the context of our meta-analysis results, we used the MetaXcan method, which evaluated the association between lateral ventricular volume and brain-specific gene-expression levels predicted by genetic variants using the data from GTEx project62,67. This method is an extension of PrediXcan method modified to use summary statistic data from meta-analysis67. Based on a total number of genes tested, the Bonferroni-corrected significance threshold was set to 0.05/12,379 = 4 × 10−6.
Gene annotation and pathway-based analysis
The gene-based test statistics were computed using VEGAS2 software which tests for enrichment of multiple single variants within the genes while accounting for LD structure68. LD structure was computed based on the 1000 Genomes phase 3 population. Variants within 10 kb of the 5′ and 3′ untranslated regions were included in this analysis in order to maintain regulatory variants68. Subsequently, the gene-based scores were used to perform gene-set enrichment analysis using VEGAS2pathway69. VEGAS2Pathway approach accounts for LD between variants within a gene, and between neighboring genes, gene size, and pathway size69. It uses computationally predicted Gene Ontology pathways and curated gene-sets from the MSigDB, PANTHER, and pathway commons databases69. The pathway-based significance threshold was set to the p-value = 1 × 10−5 while taking into account the multiple testing of correlated pathways (0.05/5000 independent tests)69.
Genetic correlation
We used the LD score regression method to estimate genetic correlations between lateral ventricular volume and various traits including anthropometric traits, brain volumes, neurological and psychiatric diseases and personality traits. The analyses were performed using a centralized database of summary-level GWA study results and a web interface for LD score regression, the LD-hub70. Summary-level GWA study results for white matter hyperintensities were obtained from the CHARGE consortium71 and the analyses were performed using the ldsc tool (https://github.com/bulik/ldsc).
Genetic risk scores
We generated genetic risk scores (GRS) for Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), Parkinson’s disease, bipolar disorder, schizophrenia, white matter lesions and tau-related phenotypes. The tau-related phenotypes, including tau and phosphorylated tau levels in cerebrospinal fluid, and progressive supranuclear palsy (PSP), were studied in relatively small sample and are therefore not appropriate for LD score regression. We extracted the lead genome-wide significantly associated SNPs and their effect estimates from the largest published GWA studies (Supplementary Data 10). For white matter lesions burden, effect estimate and standard errors were estimated from Z-statistics using the previously published formula72. The allele associated with an increased risk in corresponding traits was considered to be the effect allele. The weighted GRS was constructed as the sum of products of effect sizes as weights and respective allele dosages from 1000 Genome imputed data of Rotterdam Study using R software version 3.2.5 (https://www.R-project.org). Variants with low imputation quality (r2 < 0.3) were excluded. Subsequently, the GRS was tested for association with lateral ventricular volume in three cohorts of Rotterdam Study while adjusting for age, sex, total intracranial volume, age2 and population stratification. The significance threshold for genetic risk score association was set to p-value = 5 × 10−3 (0.05/10) based on the number of genetic risk scores tested.
Data availability
The summary statistics will be made available upon publication on the CHARGE dbGaP site under the accession number phs000930.v7.p1.
References
Pfefferbaum, A., Sullivan, E. V. & Carmelli, D. Morphological changes in aging brain structures are differentially affected by time-linked environmental influences despite strong genetic stability. Neurobiol. Aging 25, 175–183 (2004).
Carmichael, O. T. et al. Ventricular volume and dementia progression in the Cardiovascular Health Study. Neurobiol. Aging 28, 389–397 (2007).
Apostolova, L. G. et al. Hippocampal atrophy and ventricular enlargement in normal aging, mild cognitive impairment (MCI), and Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 26, 17–27 (2012).
Long, X. et al. Healthy aging: an automatic analysis of global and regional morphological alterations of human brain. Acad. Radiol. 19, 785–793 (2012).
Nestor, S. M. et al. Ventricular enlargement as a possible measure of Alzheimer’s disease progression validated using the Alzheimer’s disease neuroimaging initiative database. Brain 131, 2443–2454 (2008).
Kuller, L. H. et al. Determinants of vascular dementia in the Cardiovascular Health Cognition Study. Neurology 64, 1548–1552 (2005).
Mak, E. et al. Longitudinal whole-brain atrophy and ventricular enlargement in nondemented Parkinson’s disease. Neurobiol. Aging 55, 78–90 (2017).
Kuller, L. H., Lopez, O. L., Becker, J. T., Chang, Y. & Newman, A. B. Risk of dementia and death in the long-term follow-up of the Pittsburgh Cardiovascular Health Study-Cognition Study. Alzheimers Dement. 12, 170–183 (2016).
Vita, A., De Peri, L., Silenzi, C. & Dieci, M. Brain morphology in first-episode schizophrenia: a meta-analysis of quantitative magnetic resonance imaging studies. Schizophr. Res. 82, 75–88 (2006).
Kempton, M. J., Geddes, J. R., Ettinger, U., Williams, S. C. & Grasby, P. M. Meta-analysis, database, and meta-regression of 98 structural imaging studies in bipolar disorder. Arch. Gen. Psychiatry 65, 1017–1032 (2008).
Olabi, B. et al. Are there progressive brain changes in schizophrenia? A meta-analysis of structural magnetic resonance imaging studies. Biol. Psychiatry 70, 88–96 (2011).
Mosley, T. H. Jr. et al. Cerebral MRI findings and cognitive functioning: the Atherosclerosis Risk in Communities study. Neurology 64, 2056–2062 (2005).
Appelman, A. P. et al. White matter lesions and lacunar infarcts are independently and differently associated with brain atrophy: the SMART-MR study. Cerebrovasc. Dis. 29, 28–35 (2010).
Geerlings, M. I. et al. Brain volumes and cerebrovascular lesions on MRI in patients with atherosclerotic disease. The SMART-MR study. Atherosclerosis 210, 130–136 (2010).
Horga, G. et al. Correlations between ventricular enlargement and gray and white matter volumes of cortex, thalamus, striatum, and internal capsule in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 261, 467–476 (2011).
Kremen, W. S. et al. Heritability of brain ventricle volume: converging evidence from inconsistent results. Neurobiol. Aging 33, 1–8 (2012).
Peper, J. S., Brouwer, R. M., Boomsma, D. I., Kahn, R. S. & Hulshoff Pol, H. E. Genetic influences on human brain structure: a review of brain imaging studies in twins. Hum. Brain Mapp. 28, 464–473 (2007).
Schmitt, J. E. et al. Review of twin and family studies on neuroanatomic phenotypes and typical neurodevelopment. Twin. Res. Hum. Genet. 10, 683–694 (2007).
Kremen, W. S. et al. Genetic and environmental influences on the size of specific brain regions in midlife: the VETSA MRI study. Neuroimage 49, 1213–1223 (2010).
Eyler, L. T. et al. Genetic patterns of correlation among subcortical volumes in humans: results from a magnetic resonance imaging twin study. Hum. Brain. Mapp. 32, 641–653 (2011).
Mata, I. et al. A neuregulin 1 variant is associated with increased lateral ventricle volume in patients with first-episode schizophrenia. Biol. Psychiatry 65, 535–540 (2009).
Crespo-Facorro, B. et al. Low-activity allele of Catechol-O-Methyltransferase (COMTL) is associated with increased lateral ventricles in patients with first episode non-affective psychosis. Prog. Neuro-Psychoph 31, 1514–1518 (2007).
Skol, A. D., Scott, L. J., Abecasis, G. R. & Boehnke, M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 38, 209–213 (2006).
Staley, J. R. et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics 32, 3207–3209 (2016).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Cruchaga, C. et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 78, 256–268 (2013).
Lasagna-Reeves, C. A. et al. Reduction of Nuak1 decreases tau and reverses phenotypes in a tauopathy mouse model. Neuron 92, 407–418 (2016).
Courchet, J. et al. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell 153, 1510–1525 (2013).
Yanamadala, V., Negoro, H., Gunaratnam, L., Kong, T. & Denker, B. M. Galpha12 stimulates apoptosis in epithelial cells through JNK1-mediated Bcl-2 degradation and up-regulation of IkappaBalpha. J. Biol. Chem. 282, 24352–24363 (2007).
Kelly, P. et al. The G12 family of heterotrimeric G proteins promotes breast cancer invasion and metastasis. Proc. Natl Acad. Sci. USA 103, 8173–8178 (2006).
Krakstad, B. F., Ardawatia, V. V. & Aragay, A. M. A role for Galpha12/Galpha13 in p120ctn regulation. Proc. Natl Acad. Sci. USA 101, 10314–10319 (2004).
Zhu, D., Kosik, K. S., Meigs, T. E., Yanamadala, V. & Denker, B. M. Galpha12 directly interacts with PP2A: evidence FOR Galpha12-stimulated PP2A phosphatase activity and dephosphorylation of microtubule-associated protein, tau. J. Biol. Chem. 279, 54983–54986 (2004).
Blaho, V. A. & Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 55, 1596–1608 (2014).
Strochlic, L., Dwivedy, A., van Horck, F. P., Falk, J. & Holt, C. E. A role for S1P signalling in axon guidance in the Xenopus visual system. Development 135, 333–342 (2008).
Herr, D. R. et al. Sphingosine 1-phosphate (S1P) signaling is required for maintenance of hair cells mainly via activation of S1P2. J. Neurosci. 27, 1474–1478 (2007).
Shen, H. et al. Coupling between endocytosis and sphingosine kinase 1 recruitment. Nat. Cell Biol. 16, 652–662 (2014).
Ma, S., Santhosh, D., Kumar, T. P. & Huang, Z. A brain-region-specific neural pathway regulating germinal matrix angiogenesis. Dev. Cell 41, 366–381 (2017).
Traylor, M. et al. Genetic variation at 16q24.2 is associated with small vessel stroke. Ann. Neurol. 81, 383–394 (2017).
Miller, C. et al. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann. Neurol. 56, 734–738 (2004).
Pharoah, P. D. P. et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat. Genet. 45, 362–370 (2013).
Egan, K. M. et al. Brain tumor risk according to germ-line variation in the MLLT10 locus. Eur. J. Hum. Genet. 23, 132–134 (2015).
Paus, T., Pesaresi, M. & French, L. White matter as a transport system. Neuroscience 276, 117–125 (2014).
McMurray, C. T. Neurodegeneration: diseases of the cytoskeleton? Cell Death Differ. 7, 861–865 (2000).
Cairns, N. J., Lee, V. M. & Trojanowski, J. Q. The cytoskeleton in neurodegenerative diseases. J. Pathol. 204, 438–449 (2004).
Hasan, K. M., Moeller, F. G. & Narayana, P. A. DTI-based segmentation and quantification of human brain lateral ventricular CSF volumetry and mean diffusivity: validation, age, gender effects and biophysical implications. Magn. Reson. Imaging 32, 405–412 (2014).
Pfefferbaum, A. et al. Variation in longitudinal trajectories of regional brain volumes of healthy men and women (ages 10 to 85 years) measured with atlas-based parcellation of MRI. Neuroimage 65, 176–193 (2013).
Badouel, C. et al. Fat1 interacts with Fat4 to regulate neural tube closure, neural progenitor proliferation and apical constriction during mouse brain development. Development 142, 2781–2791 (2015).
Franke, B. et al. Genetic influences on schizophrenia and subcortical brain volumes: large-scale proof of concept. Nat. Neurosci. 19, 420–431 (2016).
Psaty, B. M. et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ. Cardiovasc. Genet. 2, 73–80 (2009).
Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G. R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 (2012).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Browning, S. R. & Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007).
Winkler, T. W. et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc. 9, 1192–1212 (2014).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Li, J. & Ji, L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity 95, 221–227 (2005).
Zeggini, E. & Ioannidis, J. P. Meta-analysis in genome-wide association studies. Pharmacogenomics 10, 191–201 (2009).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934 (2012).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 (2012).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Ernst, J. & Kellis, M. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012).
Draisma, H. H. M. et al. Genome-wide association study identifies novel genetic variants contributing to variation in blood metabolite levels. Nat. Commun. 6, 7208 (2015).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228 (2015).
Iotchkova, V. et al. GARFIELD - GWAS Analysis of Regulatory or Functional Information Enrichment with LD correction. Preprint at https://www.biorxiv.org/content/early/2016/11/07/085738 (2016).
Barbeira, A., et al. MetaXcan: summary statistics based gene-level association method infers accurate PrediXcan results. Preprint at https://www.biorxiv.org/content/early/2016/03/23/045260 (2016).
Mishra, A. & Macgregor, S. VEGAS2: software for more flexible gene-based testing. Twin. Res. Hum. Genet. 18, 86–91 (2015).
Mishra, A. & MacGregor, S. A novel approach for pathway analysis of GWAS data highlights role of BMP signaling and muscle cell differentiation in colorectal cancer susceptibility. Twin. Res. Hum. Genet. 20, 1–9 (2017).
Zheng, J. et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017).
Verhaaren, B. F. et al. Multiethnic genome-wide association study of cerebral white matter hyperintensities on MRI. Circ. Cardiovasc Genet 8, 398–409 (2015).
Chauhan, G. et al. Association of Alzheimer’s disease GWAS loci with MRI markers of brain aging. Neurobiol. Aging 36, 1765.e7–1765.e16 (2015).
Acknowledgements
See Supplementary Note 2 for information on funding sources.
Author information
Authors and Affiliations
Contributions
Conceived the study and drafted the manuscript: D.V., H.H.A., M.A.I., S.S., M.F. Performed statistical analyses: D.V., H.H.A., X.J., Q.Y., A.V.S., J.C.B., A.T., M.S., N.J.A., E.H., Y.S., M.L., M.B., S.T., J.Y., N.A.G., M.S.P., S.J.L., A.N., L.R.Y., and S.L. Acquired data: O.A.A., D.A., N.A., K.A., M.B., D.M.B., A.B., F.B., H.B., R.N.B., R.B., A.M.D., P.L.J., I.J.D., C.D., D.A.F., R.F.G., J.G., V.G., T.B.H., G.H., D.S.K., J.B.K., C.E.L., M.L., W.T.L., O.L.L., P.M., P.A.N., H.M., K.A.M., T.H.M., R.M., M.N., M.S.P., Z.P., B.M.P., K.R., C.L.S., R.S., P.S.S., P.J.S., S.S.S., D.J.S., A.T., A.G.U., M.C.V.H., M.W.V., W.W., T.W., A.V.W., K.W., M.J.W., H.T., W.K., D.A.B., J.W. J., T.P., J.A.W., H.S., P.S.S., A.V., H.J.G., J.I.R., C.M.D., L.J.L., S.S., M.A.I., M.F. All authors critically reviewed the manuscript for important intellectual content.
Corresponding authors
Ethics declarations
Competing interests
A.M.D. is a Founder of and holds equity in CorTechs Labs, Inc, and serves on its Scientific Advisory Board. He is a member of the Scientific Advisory Board of Human Longevity, Inc. and receives funding through research agreements with General Electric Healthcare and Medtronic, Inc. The terms of these arrangements have been reviewed and approved by UCSD in accordance with its conflict of interest policies. The remaining authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vojinovic, D., Adams, H.H., Jian, X. et al. Genome-wide association study of 23,500 individuals identifies 7 loci associated with brain ventricular volume. Nat Commun 9, 3945 (2018). https://doi.org/10.1038/s41467-018-06234-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-06234-w
This article is cited by
-
Longitudinal assessment of ventricular volume trajectories in early-stage schizophrenia: evidence of both enlargement and shrinkage
BMC Psychiatry (2024)
-
The effects of genetic and modifiable risk factors on brain regions vulnerable to ageing and disease
Nature Communications (2024)
-
The AMPK-related kinase NUAK1 controls cortical axons branching by locally modulating mitochondrial metabolic functions
Nature Communications (2024)
-
Genetic architectures of cerebral ventricles and their overlap with neuropsychiatric traits
Nature Human Behaviour (2023)
-
Genome-wide meta-analysis for Alzheimer’s disease cerebrospinal fluid biomarkers
Acta Neuropathologica (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
