
Abstract
Robustness is a key system-level property of living organisms to maintain their functions while tolerating perturbations. We investigate here how a regulatory network controlling multiple virulence factors impacts phenotypic robustness of a bacterial plant pathogen. We reconstruct a cell-scale model of Ralstonia solanacearum connecting a genome-scale metabolic network, a virulence macromolecule network, and a virulence regulatory network, which includes 63 regulatory components. We develop in silico methods to quantify phenotypic robustness under a broad set of conditions in high-throughput simulation analyses. This approach reveals that the virulence regulatory network exerts a control of the primary metabolism to promote robustness upon infection. The virulence regulatory network plugs into the primary metabolism mainly through the control of genes likely acquired via horizontal gene transfer, which results in a functional overlay with ancestral genes. These results support the view that robustness may be a selected trait that promotes pathogenic fitness upon infection.
Similar content being viewed by others


Introduction
Living organisms have to face changing environments and are subject to accumulation of molecular damages during their growth and development. Therefore, they need to exhibit a certain degree of robustness to sustain their resilience in response to these perturbations. Robustness is commonly defined as the capacity of a biological system to maintain its function(s) despite environmental or internal perturbations; internal perturbations referring here to deleterious mutations or stochastic gene expression1,2,3. Depending on the biological function to be maintained and the kinds of perturbations considered, robustness can arise from various types of molecular mechanisms (Fig. 1). Among them are (i) versatility, i.e., the ability of the system to collect its needs (nutrition, information) from different sources in the environment4, (ii) functional redundancy, which includes genetic redundancy5,6 and fail-safe alternatives (alternative metabolic or regulatory pathways), and (iii) the system control, i.e., the capacity of the system to sense and compensate appropriately the undergone perturbation to maintain its homeostasis. This variety of mechanisms and their tight imbrications make robustness challenging to study. Nevertheless, as these mechanisms carry a buffering capacity, studying robustness is key to understand accommodation capacity as well as evolvability of biological systems1,7. For instance, genetic redundancy is recognized as a main source of robustness with respect to genetic (internal) perturbation8. The adaptive backup capacity provided by genetic redundancy was also proposed to be a transient byproduct during the functional divergence of gene duplicates9, which eventually could promote evolvability1.

Sources of robustness in biological systems. The three main sources of phenotypic robustness in biological systems with respect to internal or environmental perturbations are: versatility, functional redundancy (genetic and pathway redundancy), and the system control. The virulence functions (virulence phenotypes) can be seen as a mechanism to modify the host environment to make it beneficial for the growth of the pathogen
Phenotypic robustness of living organisms is recognized as a network-scale property1. Robustness provided by the metabolic network has been extensively studied10,11,12,13. One challenge in studying properties of complex networks like metabolism, which harbors a high number of pathway combinations, remains in the computational capacity to explore the large solution space of the system. The development of flux balance analysis (FBA) methods14, which rely on constraint-based modeling and steady-state assumption has provided the capacity to investigate the sources of robustness at genome-scale level11,15,16,17. Modeling and experimental approaches revealed that robustness of metabolic networks challenged with genetic perturbations relies on pathway redundancy or genetic redundancy, but their extent can vary depending of the organisms10,15.
Robustness provided by regulatory networks received attention because it determines broad adjustments of cellular processes through the genetic regulation of metabolism, signal transduction, and cell differentiation processes1,18,19,20. Various types of regulation via feed-back loops within the metabolism have been discovered, most of them being aimed to maintain consistency between the biosynthesis of metabolites and their consumption upon perturbation21,22,23. There is evidence that an increase of fitness in various organisms was obtained through modifications of the regulatory network structure24,25,26,27. Indeed, inappropriate metabolic transcriptional reprogramming can lead to lower fitness with regard to the kind of environmental perturbation encountered by an organism28,29. Interactions between the regulatory and metabolic networks therefore determine the properties of the global network and the distribution of robustness or fragility points.
One highly complex biological system in which robustness is a crucial property is the host-pathogen interaction13. During interaction, the pathogen subverts the host cell machinery to collect host resources30, while the host reacts to protect its own physiological functions31. Each one will induce perturbations to the other’s system, with the aim to lessen the system’s robustness of the confronting organism13,32. Virulence functions of pathogens are controlled by sophisticated, multicomponent, interconnected regulatory networks that are responsive to environmental and internal signaling33,34. Reconstruction of metabolic models of pathogens and studies of their robustness with regard to internal perturbations were conducted with the aim to discover new drug targets35,36 but only one virulence regulatory network has been reconstructed so far37. To our knowledge, robustness analysis at the system level of a pathogen combining both metabolic and virulence regulatory networks has not yet been investigated. This information should provide new insights about how phenotypic robustness is associated to the evolution of virulence determinants.
In this study, we investigated impact of the virulence regulatory network (VRN) on the phenotypic robustness of the plant pathogen Ralstonia solanacearum. This bacterium is one of the most destructive plant pathogenic bacterium worldwide due to its unusual wide host range and its broad geographical distribution38,39,40. Several reports have already highlighted that many pathogenicity functions display a high level of robustness as evidenced by genetic or functional redundancies41,42,43,44. We recently found that key VRN components can impair the versatility of the pathogen due to a resource allocation trade-off between virulence function and bacterial growth (hereafter referred as proliferation)45. Using a reconstructed cell-scale model of R. solanacearum and modeling studies, we predicted the robustness of phenotypes under environmental as well as internal perturbations. We evaluated the functional interplay between the metabolic network and the VRN and revealed the strong impact of this VRN on various sources of robustness such as functional redundancy. Results indicate that the VRN evolved to control functionally redundant metabolic genes, which overlap some primary metabolic pathways, thus leading to increased robustness capacities under specific host environment.
Results
Hybrid cell-scale model reconstruction of R. solanacearum
We reconstructed a cell-scale model of R. solanacearum comprising three interconnected modules: a genome-scale metabolic network, a macromolecule network dedicated to virulence functions and a VRN (Fig. 2). The two first modules correspond to biochemical reaction networks previously described45, whereas the VRN is a biochemical interaction network reconstructed in this study. Thus, the modular structure of this resulting “hybrid model”46 allows computational approaches with appropriate methods for both types of networks, i.e., constraint-based modeling for the biochemical networks and multi-state logical modeling for the regulatory network, see Supplementary Note 1. Both methods do not require kinetic parameters and thus are relevant for genome-scale analysis.

Architecture of the cell-scale model of Ralstonia solanacearum GMI1000 and the simulation pipeline for robustness analysis. The cell-scale model is composed of three modules: a genome-scale metabolic network, a macromolecule biosynthesis and secretion module45, and the virulence regulatory network (VRN). Simulation was performed using hybrid modeling, i.e., discrete logical modeling for the regulatory network and constraint-based modeling for the biochemical reaction network. Robustness quantification was performed using the BECO analysis, which evaluates impact of environmental perturbations (πe) or internal perturbations (πi) on phenotypes
R. solanacearum possesses a complex regulatory network which orchestrates the actuation of its large repertoire of virulence factors40. This VRN responds to various signals including host cell contact, environmental nutrient availability, and quorum sensing molecules to coordinate the expression of multiple virulence factors at different steps of the infectious cycle. We reconstructed the VRN of R. solanacearum strain GMI1000 by collecting bibliographical information based on genetic and genomic studies (see Methods section for details), including three transcriptomic studies47,48,49. The reconstructed R. solanacearum VRN comprises 712 genes including 29 genes coding for transcription factors and 34 proteins involved in signal transduction. This VRN perceives 86 signals and controls the expression of 606 genes. Among those genes, 444 are present in the biochemical reaction network. Overall, 86 reactions of the macromolecule network are also regulated by the VRN, and correspond to the induction of macromolecule secretion via the control of secretion systems. In total, the VRN iRP1443REG encompasses 1443 interactions generating 705 logical rules (Fig. 3a, b, Supplementary Fig. 1, Supplementary Data 1). The resulting hybrid model includes a substantial proportion of the genes annotated in the reference genome as enzymes (71.1%), transporters (65.3%), regulators (34.3%), receptors (75%), and virulence genes (90.2%), indicating a significant level of completeness (Supplementary Table 1).

Properties of the VRN and prediction performance. a Compositions and metrics of the virulence regulatory network. b View of the VRN structure with the three major regulatory hubs: PhcA, HrpG, HrpB (see detail in text). c Prediction capacity of the VRN on differential gene expression compared by an accuracy analysis to an in planta transcriptomic data set50. Environmental conditions used for simulation: MMGLU minimal medium with l-glutamate, MMGLC minimal medium with d-glucose, CASAM minimal medium with casamino acids, CM complete medium, APO tomato apoplast, XYL tomato xylem, HCD high-cell density. d Phenotype prediction of the cell-scale model (biochemical reaction network plus regulatory network) compared to experimental data for four groups of phenotypes and VRN deletion mutants in various conditions. The term “active phenotype” means that the corresponding biological function was either observed experimentally or inferred from simulation of the cell-scale model (i.e. possibility to realize a metabolic flux through the reaction associated to the phenotype). Size of the boxes is proportional to the number of phenotypes in each category (n = 43). The metrics of the model performance are reported as precision, sensitivity and accuracy. EPS Exopolysaccharide production, PCW plant cell wall
To validate the capacity of the reconstructed VRN to predict correctly gene expression in response to environmental changes, we compared the differential gene expression from an experimental transcriptomic data-set with the model predictions. These experimental data were extracted from a study using R. solanacearum grown either in complete medium in vitro or in planta during xylem colonization50. This experimental data-set was not used for the VRN reconstruction and was therefore well-suited to test the prediction capacity of the VRN module. The prediction of gene expression was performed through a discrete logical modeling method using FlexFlux51 under various environmental conditions, including in vitro and in planta conditions (see Supplementary Data 2; Supplementary Note 2). Experimental gene expression values were discretized using a cut-off threshold of 2.0 of absolute log2 expression levels to be directly compared with the model predictions (Supplementary Data 3). Among 64 conditions tested, the differential gene expression predicted in xylem tissues at high-cell density (thus similar to the experimental condition) was the closest match with experimental gene expression data, F1 score 0.60 (Supplementary Data 3), see Fig. 3c and Supplementary Note 3. To assess the predictability of the network and the sensitivity of each environmental component to trigger the specific gene transcription reprogramming, we evaluated the deviation of the prediction induced by changing each environmental component (see Methods). Simulations revealed four key environmental constraints acting on the VRN status listed in the order of importance: (i) the plant cell wall sensing, (ii) quorum sensing (i.e., high-cell density versus low-cell density), (iii) O2 limitation, and (iv) nitrate availability. We concluded from this analysis that the model reliably predicted the transcriptional responses controlled by the VRN and the major signals or constraints it perceives in its environment.
We tested the capacity of the hybrid model to predict the modulation of the phenotypic plasticity by the VRN. We performed a simulation of various phenotypes in different environmental conditions and compared them with experimental observations (list in Supplementary Data 4). In addition, we compared the predicted and experimentally observed phenotypes of 16 VRN deletion mutants (Supplementary Data 4). We found an overall accuracy of 77% of the cell-scale model prediction for phenotypic plasticity (Fig. 3d). For instance, simulations reproduced the well-known phenotypic switch induced by the quorum sensing system52,53, which controls many biological functions including production of virulence determinants or motility repression.
The capacity of the metabolic model to predict the functional redundancy of the pathogen metabolism was experimentally assessed by screening a Tn5 transposon insertion mutant library. A bank of 4046 random transposon insertions in R. solanacearum strain GMI1000 was generated and screened for gene essentiality under two growth conditions, i.e., d-glucose and l-glutamate as sole carbon and energy sources (see Methods, Supplementary Data 5, and Supplementary Note 4). Among the 176 genes present in the metabolic model and in the mutant library, 146 of them harbored a deletion phenotype accurately predicted by the model in both conditions, with an overall accuracy of 83% (see Supplementary Data 6). Among the predictions, which were confirmed experimentally, is the functional redundancy between the two copies of the malic enzymes and the phosphoenolpyruvate carboxykinase, which catalyze reactions required during growth on l-glutamate (see Supplementary Data 6).
Quantification and sources of phenotypic robustness
The challenge in apprehending robustness properties of biological networks comes from the difficulties in defining a quantitative metric of robustness1,3. We set out a metric that could be broadly used and results in the probability of the phenotype to be maintained in presence of defined perturbations. We defined the robustness (R(φ,π)) of the phenotype (φ) facing the set of perturbations (π) as the number (n(π,φ>α)) of perturbations for which the phenotype is feasible, i.e., the value of the phenotype is strictly superior to a chosen threshold (α), divided by the number (n(π)) of the faced perturbations.
Hence, this robustness value (R) ranges from 0, when each perturbation leads to a loss of function, to 1 when the phenotype is always maintained in presence of any perturbation tested. This definition is prone to each considered phenotype and perturbation. It has also the advantage to allow comparison of robustness of different phenotypes with respect to a similar set of perturbations as well as comparing the phenotypic robustness of different organisms encountering a similar set of perturbations. To perform in silico robustness quantification, we designed a global robustness analysis, called BECO for Biological Entity Clustering by their contribution to the Objective function. This algorithm performs multiple regulatory steady-state analyses51 followed by FBA of a set of phenotypes (ϕ), used as objective functions, under a number n(πe) of environmental conditions (πe) (see Methods and Supplementary Note 5 for details).
In this study, the BECO analysis that was conducted referred to the following definitions: a phenotype is considered to be feasible if a flux through its specific reaction or defined set of reactions (ϕ), i.e., an objective function, is possible (ϕ > α, with α = 0 in our study). An environment refers to different compositions and concentrations of compounds available in the medium and to physical parameters (e.g., temperature). An environmental perturbation is simulated by changing composition or concentration of any compound in an environment or any physical parameter. An internal perturbation is simulated by removing a metabolic gene, a regulator or a reaction.
Then, BECO systematically tests the contribution of each biological entity (genes, regulators, and reactions), i.e., internal perturbations, on the phenotypes under several environments that correspond to environmental perturbations. This is realized by simulating series of gene and reaction knockout (KO) analyses and flux variability analyses, which leads to a classification of these entities similarly to the classification proposed by Wang and Zhang10 and Lewis et al.27 (see Methods).
Impact of the VRN on robustness upon external perturbations
As a first analysis, we assessed if R. solanacearum has a level of phenotypic robustness comparable to what is observed for other microorganisms. Hence, we determined the level of genetic redundancy in R. solanacearum and 10 other representative microorganisms. We also performed a BECO analysis on orthologs shared between R. solanacearum, E. coli, and P. aeruginosa. We found that both levels of functional and genetic redundancy ensuring robustness of the proliferation trait were similar in R. solanacearum and the other tested bacteria, see Supplementary Figs. 2 and 3, and Supplementary Note 6.
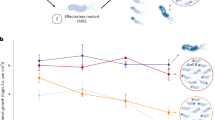
We then investigated the impact of the R. solanacearum VRN on phenotypic robustness when bacteria face an environmental perturbation. We selected a range of 14 phenotypes (listed in Fig. 4a) representative of a broad set of functions performed by bacteria, including housekeeping functions (proliferation, production of storage compounds) and known virulence and/or plant-associated functions (Type 3 effector secretion, exopolysaccharide production, phytohormone production, motility and secretion of plant cell wall degrading enzymes). The set of 14 environmental perturbations (πe) tested were representative of several environmental conditions (i.e., different carbon and nitrogen sources or presence of quorum sensing signals, etc…) encountered by the bacteria in vitro or in planta (see Supplementary Data 7). Robustness(ϕ, πe) of the phenotypes calculated by the BECO analysis, with or without considering the VRN, is presented in Fig. 4a. These simulations revealed that the different phenotypes tested followed distinct patterns: the virulence-associated functions were subjected to a strong reduction of their robustness with respect to environmental changes due to the VRN (up to 60% of reduction of their solution space) whereas housekeeping function phenotypes in similar conditions retained robustness. This difference was not due to a difference between the number of VRN-regulated genes involved in the housekeeping (14.3 ± 2.6%) and virulence phenotypes (15.1 ± 4.5%), p-value 0.628. It cannot be ruled out however that some redundant genes involved in housekeeping functions may be also controlled by regulatory networks other than the VRN, and thus could become essential under specific environments.

The VRN restricts phenotypic robustness in presence of environmental perturbations. a Impact of the VRN on the phenotypic robustness in presence of environmental perturbations. The simulations were conducted in presence or absence of the VRN in 16 different environments, including in planta (e.g., plant tissues, high and low-cell density) and in vitro (e.g., complete medium, minimal medium) conditions. The detailed list of tested environments is provided in Supplementary Data 7. b Analysis of the impact of the VRN on phenotype robustness after simulation in presence of 1000 random environments
To assess the significance of the observations made through the BECO analysis, we developed a 2D robustness analysis (ROBA), which simulates robustness of the phenotypes when considering a broader range of 1000 random environmental perturbations (see Supplementary Note 7). Here again, impact of the VRN on phenotypic robustness had a strong effect on virulence-associated functions but not on housekeeping functions, see Fig. 4b. This confirmed that in presence of the VRN the virulence-associated and housekeeping functions can be differentiated by their sensitivity to environmental perturbations (t-test, p-value 6 × 10−4). The apparent decrease of robustness for the virulence-associated phenotypes indicated that essential genes required for virulence were not induced or repressed by the VRN in some specific environments. When comparing conditions that activate virulence functions, we found that the robustness of the virulence phenotypes is higher in planta (0.741 ± 0.313) compared to in vitro conditions (0.208 ± 0.140), p-value 0.0006. On the other side, housekeeping functions were almost insensitive to the VRN control with respect to environmental perturbations, thus showing that expression of essential genes required for housekeeping functions is not repressed by the VRN in any of the tested environments.
The VRN plugs on redundant genes of the primary metabolism
The previous analysis revealed that phenotypic robustness of the housekeeping functions in presence of environmental perturbations is insensitive to the VRN control. However, it appears that among the 444 metabolic genes under control of the VRN in the reconstructed model, a large number of them code for primary metabolism enzymes, i.e., involved in the biosynthesis of cell biomass45,46. This observation suggested that the VRN-regulated genes coding for primary metabolism components can be sources of robustness through genetic redundancy or pathway redundancy. We used the BECO method to test the contribution of these VRN-regulated genes in the primary metabolism to the robustness of the proliferation phenotype. Hence, the robustness of various biological functions was assessed with respect to internal perturbations (πi), i.e., when occurs a loss of function of each gene or reaction included in the model, in a defined set of 14 different environments (see Supplementary Data 7). This simulation mimicked the occurrence of a deleterious mutation or the inhibition of an enzymatic reaction. We found that proliferation is the function the most impacted by the VRN when internal perturbations occur, thus indicating that a significant number of metabolic genes providing robustness to this phenotypic trait are under the control of the VRN (Supplementary Fig. 4, and Supplementary Note 8 for detailed analysis).
We verified that the connection of the VRN on metabolic pathways inferred from the BECO analysis was significant and therefore statistically different from a random connection. We first defined eight different classes of genes, based on how they contribute to robustness of the metabolic network (Fig. 5a), see Methods for the procedure used for gene class definition. These different classes also distinguish the contribution to robustness that are conditionally dependent (i.e., dependent upon a specific environmental condition). The output of the BECO analysis then provided for each class the proportion of genes in the network, with simulations either in presence or absence of the VRN (Fig. 5b; Supplementary Data 8). We tested if the gene distribution in the different classes was dependent on VRN control by performing χ2 tests. The statistical analysis showed that the connection of the VRN into the metabolic network is not random (p-value of 1.9 × 10−6 and 4.1 × 10−4 for the proliferation and T3SS effector production phenotypes, respectively). A tendency to an enrichment of VRN-regulated genes in conditionally dependent redundant pathways was observed (see classes OPT-C and ELE-C in Fig. 5b). Other virulence-associated traits followed the same trend, such as secretion of lectin Rsl2, extracellular proteins Tek and Pme with a p-value of 4.1 × 10−3, 6.7 × 10−4 and 3.5 × 10−3, respectively; or EPS production with a p-value of 1.9 × 10−2. Interestingly, the pattern of gene distribution in the different classes for the VNR-regulated genes differed between phenotypes, suggesting that different metabolic constraints occur depending on the phenotype. For instance, the connection of the VRN within the primary metabolism to control proliferation (housekeeping function) differs from T3SS effector production (virulence-associated function) (p-value 5.2 × 10−5), see Fig. 5.

Sources of phenotypic robustness mobilized by the VRN with respect to internal and external perturbation. a Sources of phenotype robustness in a metabolic network. b Classification and distribution of genes contributing to robustness of the proliferation phenotype (n = 78 for VRN-regulated genes and 839 for VRN-independent genes) and the T3SS-dependent secretion of the effector RipG7 (n = 78 for VRN-regulated genes and 521 for VRN-independent genes) obtained with the BECO analysis. Dependency of the two distributions, VRN-regulated vs VRN-independent, is shown and significance of the differences was assessed by performing a χ2-test. In total 919 metabolic genes were classified by the BECO analysis
Next, we ran a BECO analysis to identify under which conditions were mobilized the VRN-regulated genes that provide πi-robustness for the two phenotypes (T3SS and proliferation). We determined the Robustness under three conditions: an environment not activating the VRN used as control, and two environments with activating signals encountered by the bacteria during infection: the presence of plant cell wall components54 and the quorum sensing signal53. The simulations predicted that the robustness of both phenotypes increased when the VRN was activated, either by the plant cell wall or the quorum sensing signal (Supplementary Fig. 5). The decrease of the number of essential genes indicated that other network components were activated: the activation of redundant genes or pathways by the VRN could complement some reactions which are otherwise essential when the VRN is not activated. This analysis also suggested that phenotypic robustness is higher in planta compared to in vitro conditions. We conducted simulations to test the robustness of the proliferation function in different environments: this indeed revealed that robustness was significantly higher in planta (0.783 ± 0.004, for six environments tested) compared to in vitro (0.751 ± 0.016, for eight environments tested), t-test p-value 0.0005.
VRN-regulated genes acquired via horizontal gene transfer
Our analysis revealed that several VRN-regulated genes within the primary metabolism provide robustness to proliferation as well as virulence functions. To get evolutionary insights on how phenotypic robustness was shaped by the VRN, we performed phylogenetic analyses using homology groups containing the genes from 28 representatives of the beta-Proteobacteria (including R. solanacearum) and some gamma and alpha-class species as outgroups (Supplementary Table 2). We focused our analysis on the amino-acid biosynthesis pathways, which are primary metabolism pathways broadly shared between organisms. Many of these pathways possess VRN-regulated genes and contribute to phenotypic robustness for both the proliferation and virulence functions. We compared the presence of homologs of the R. solanacearum genes providing robustness in those organisms, as well as the presence of homologs of key regulators of the VRN, like hrpG and hrpB40. We found that the VRN plugs into six different R. solanacearum amino-acid biosynthetic pathways and mainly controls genes likely acquired via horizontal gene transfer (Supplementary Fig. 6). This is illustrated with the tryptophan biosynthesis pathway: two reactions of the pathway are catalyzed by redundant enzymes, some of them being regulated by the VRN (Fig. 6a). The distribution of the genes encoding these enzymes was performed among the 28 Proteobacteria and this analysis revealed that only the R. solanacearum species possesses the VRN-regulated genes among beta-Proteobacteria. The VRN-independent gene trpD1 (locus tag RSc2884) belongs to the highly conserved tryptophan biosynthesis operon found in most bacterial species and is likely the most ancestral gene (Fig. 6b). The VRN-regulated gene trpD2 (locus tag RSp0681) is suspected to have been acquired through horizontal gene transfer as it is not found in others beta-Proteobacteria outside of the R. solanacearum species and a blast analysis first identified an homologous gene in alpha-Proteobacteria (62% identity at the protein level with trpD2) instead of R. solanacearum trpD1 (45% identity). This observation was supported by a phylogenetic analysis of trpD sequences, which revealed incongruence on taxonomy for trpD2 (Fig. 6c). For another reaction of the pathway, the VRN-regulated gene trpC2 (RSp0680) also reveals incongruence of the phylogeny with the taxonomy (Fig. 6b; Supplementary Fig. 7) and best blast search pinpoints homologs in alpha-proteobacteria (Methylocystis; 55% identity) and gamma-Proteobacterium (Pseudomonas syringae; 54% identity) rather than the R. solanacearum ancestral gene trpC1 (RSc2885, 48.4% identity). These data were again suggestive of a horizontal acquisition of trpC2. A similar situation was also observed for other VRN-regulated genes in the l-proline (RSp0418), l-Lysine (RSp0424), l-cysteine (RSp0417), l-aspartate (RSp0943), and l-methionine (RSp0781) biosynthesis pathways, see Supplementary Fig. 6. All these VRN-regulated genes provide genetic redundancy, except RSp0781 in the l-methionine biosynthesis pathway, which illustrates a case of pathway redundancy.

Orthology analysis of VRN-regulated and VRN-independent genes contributing to robustness within the tryptophan biosynthesis pathways. a The R. solanacearum tryptophan biosynthesis pathway and the corresponding in planta gene expression profiles. The values correspond to the absolute fold change expression levels (from Jacobs et al. 50). b Presence of homologs of the R. solanacearum genes in the various taxa is displayed by boxes. Number in the boxes corresponds to the number of paralogs present in the organism. Closest homologs to a VRN-regulated gene are colored in red, whereas the closest homologs of a VRN-independent one are in gray. In the case of two genes belonging to the same homology group, the closest homolog to a R. solanacearum gene was defined as the gene showing the highest blast score to a R. solanacearum gene. c Phylogeny analysis of the trpD1 and trpD2 homolog groups. Tree scale corresponds to 0.1 mutation
Phylogenetic relationships analyses of the VRN-regulated genes in amino-acids biosynthesis pathways revealed an absence of orthologs in other beta-Proteobacteria (Supplementary Fig. 6). The hypothesis of a horizontal gene transfer origin for the acquisition of the VRN-regulated genes is also supported by a clear enrichment of VRN-regulated genes present only in R. solanacearum strains among beta-Proteobacteria: homology searches revealed that 30% of the VRN-regulated genes are specific to the R. solanacearum species whereas the proportion is only 3% for the VRN-independent genes (Supplementary Fig. 8). Moreover, a bias in GC content was detected in the total number of VRN-regulated genes compared to the VRN-independent genes, p-value 3 × 10−4 (Supplementary Fig. 9; Supplementary Data 9), again suggestive of horizontal acquisition. Several other VRN-regulated genes involved in primary metabolism pathways, such as the TCA cycle, were also identified to provide robustness patterns, and also presumably acquired through horizontal transfer, see Supplementary Data 10.
Discussion
Phenotypic robustness is a remarkable property of biological systems, allowing them to maintain their functions with respect to perturbations1,3,55. Here we developed a system modeling-based approach to study how phenotypic robustness upon internal and environmental perturbations is reprogrammed by the VRN in the plant pathogen R. solanacearum. The proliferation in the literature of the term “robustness” applied to biological systems has led to inappropriate or confusing usage of this term3,56. As stated before, the notion of robustness requires to specify which phenotypic trait is robust, to which perturbation, and to provide quantification. We provided a metric useful for a quantitative estimation of the robustness of phenotypes with respect to perturbations. Such a metric was lacking to perform deep comparisons of phenotypic robustness within biological systems1. Our metric has the advantage to be generic in the sense that it can be applied to any phenotype of any biological system facing any perturbation. The use of this robustness metric was associated with the development of a bioinformatics pipeline to calculate the impact of the internal and/or the environmental perturbations on several biological functions at genome-scale level.
A second achievement of this study was the reconstruction of a R. solanacearum genome-scale cell model integrating a metabolic network, a macromolecule secretion module and a regulatory network. An originality of this hybrid cell model is the implementation of a VRN comprising 712 genes, which is responsive to 86 environmental signals. Thus, this is one of the most complex VRNs reconstructed so far in bacterial pathogens40, comparable in complexity to the P. aeruginosa VRN33,37. The quality and performance of this hybrid cell model was validated through different assessment steps, which include gene essentiality analyses using a transposon insertion mutant library, comparison with -omics data sets and mutant phenotyping.
The large repertoire of genes regulated by the R. solanacearum VRN (up to 444 into the metabolic and macromolecule modules) opened an unprecedented possibility to evaluate how metabolic network components from a pathogen are specifically controlled by the VRN. Our results reveal that the R. solanacearum VRN controls phenotypic robustness through two different means: (i) as expected, by activating essential reactions required to produce virulence phenotypes in specific environment, (ii) more surprisingly, by activating functionally redundant reactions within primary metabolism, presumably with the aim to specifically sustain virulence (see Fig. 7). Obviously in the first case, the VRN optimizes the network state to avoid the expression of costly virulence functions (such as the EPS biosynthetic pathway) in inappropriate conditions. Such a resource allocation trade-off between virulence and proliferation exists in R. solanacearum, due to a probable limitation of nutritional resources in the host environment45. In addition, the biosynthesis of biomass and virulence-associated traits involves many common metabolic precursors. The burden for the primary metabolism in supporting simultaneously multiple objectives therefore imposes an optimization constraint on the activation of virulence pathways, a process tightly regulated by the VRN26,45,50. This explains why control exerted by the VRN could restrict in a large extent the phenotypic robustness of virulence functions with respect to environmental perturbations. Because the R. solanacearum global regulatory network (including metabolic regulation, stress response, etc…) is not yet reconstructed, the impact of this network could not be evaluated. However, this study revealed the direct effect of the VRN on phenotypic robustness as the analyses were conducted in a straight comparison between activated/inactivated VRN states independently of the global regulatory network; future investigations are required to undercover potential cross talks between the R. solanacearum VRN and the global regulatory network.

Functional connections of the VRN with the metabolic network and their impact on the control of phenotypic robustness. Rectangles, diamonds, and circles symbolize genes, enzymatic reactions, and metabolite products, respectively. The action of the VRN is either on the control of robust nodes of the primary metabolic network (blue arrows) or on essential required for expression of virulence phenotypes (red arrow). Examples of the first class (genes colored in blue) are the tryptophan biosynthetic pathway (genetic redundancy) and the methionine biosynthetic pathway (pathway redundancy), see details in text
The resource allocation trade-off between virulence and proliferation traits may also generate perturbations on the regulation of some primary metabolic pathways as the corresponding genes are subjected to the basal metabolic regulation dedicated to the homeostasis of biomass precursors, but also to regulation by the VRN. Interestingly, the VRN plugs on different genes that apparently overlap genes or operons involved in amino-acids biosynthesis. This control therefore provides phenotypic robustness to internal perturbations during infection and could also be a potent way to increase some metabolic fluxes required for the efficient synthesis of virulence factors. There is indeed increasing evidence for the contribution of primary metabolism genes to support both virulence and growth traits in various bacterial pathogens57,58,59,60, and our report suggests that some primary metabolic pathways are specifically relevant for virulence purposes in R. solanacearum.
Phylogenetic analyses support the view that many of these primary metabolism VRN-regulated genes were probably acquired via horizontal gene transfer to supply a functional overlay to ancestral genes. This appears to be the case for several amino-acid biosynthetic pathways (tryptophan, proline, lysine, cysteine, and aspartate). Such apparent redundancy allows uncoupling the VRN control from the basal homeostatic control on these amino-acid biosynthetic pathways. This probably provides an advantage for the pathogen and may compensate the cost of possessing two functionally redundant genes. This process of metabolic gene acquisition and recruitment in the virulence regulon thus appears to be a key mechanism contributing to the phenotypic robustness of R. solanacearum. Another situation is observed in the case of the methionine biosynthetic pathway: here the metE gene encoding the cobalamin-independent methionine synthase was recruited by the VRN, whereas the functionally redundant metH gene encoding the cobalamin-dependent enzyme was insensitive to VRN regulation (Supplementary Fig. 6). MetE provides robustness to proliferation because it was previously shown to be required for full pathogenesis, being critical for infection as cobalamin is not produced by the plant host61. Significantly, our analysis shows that the VRN-regulated genes in amino-acid biosynthetic pathways, which provide robustness were most probably acquired through horizontal acquisition rather than duplication of ancestral genes. This highlights horizontal gene transfer as a major mechanism promoting evolution of virulence and suggests that robustness is shaped through evolutionary selection.
The robustness of biological systems implies that a limited number of internal perturbations can substantially disrupt the system13 and the identification of sources of robustness of pathogens will be a major issue for medical or agricultural applications. The R. solanacearum cell model is a valuable hypothesis-testing tool for further functional approaches in a biological system where phenotypic robustness potentially hampers the identification of virulence/plant adaptation determinants32.
Methods
Construction of a Tn5 transposon insertion mutant library
A R. solanacearum mutant database was generated through random insertion of the transposon EZ-Tn5<KAN-2> (EPICENTER® Biotechnologies) in strain GMI100039. The EZ-Tn5<KAN-2> was electroporated in strain GMI1000 competent cells following the protocol of the supplier and mutants were isolated on BG medium supplemented with Kanamycin. Localization of each individual insertion in the genome was determined by sequencing of its flanking region (which may differ from the mutated gene) using an adaptation of the TAIL (Thermal asymmetric interlaced)-PCR protocol from Qian et al.62 and automated BlastN comparison with the genome sequence. A total of 4046 transposon insertions in the GMI1000 genome were accurately mapped (Supplementary Data 5) and their list and position can be visualized on a dedicated webpage: https://iant.toulouse.inra.fr//bacteria/annotation/site/prj/ralso/tools/mutants_db/cgi/EZLucene.cgi?lucenedb=mutants&simpleform=1.
Virulence regulatory network reconstruction
The VRN was reconstructed following the recommendations for Transcriptional regulatory network reconstruction63 but extended to consider the entire set of components involved in a regulatory process, i.e., from signal molecule to transduction cascade and transcription activation. The reconstruction was performed using bibliographic information based on genetic and genomic studies, including three transcriptomic studies available in the literature47,48,49. This model includes the list of interactions (activation or repression) involving one regulator and one target such as the binding of transcription factor to target gene promoters, the activation of a receptor (protein) by metabolites, protein-protein interactions, etc…. These various kinds of interactions were annotated following Systems Biology Ontology recommendations64. The interactions also contain information of the state of the regulator leading to the change of the target status. Indeed, when a quantitative value is available, like affinity of a receptor for a metabolite or quantitative response of the target to a transcription factor, the value is converted into discrete thresholds (discrete logical relationship). The interactions were then converted in logical rules describing for each target the rule of activation, i.e., state of the regulators and their combination leading to the change of the target state. Then, they were assembled to generate a file in sbml-qual format (Supplementary Software 1) suitable for model exchange and computation65.
Simulation pipeline of the hybrid-cell scale model
The workflow to combine the R. solanacearum GMI1000 VRN and the biochemical network45 in one simulation pipeline first requires computing the regulatory network status under the considered environmental constraints. The gene status set by the regulatory network is then added as a constraint on the biochemical network in addition to environmental constraints. Then the flux balance analyses are performed (see Fig. 2). Each type of simulations was done as described below and done using the software FlexFlux51. The method used and described below is similar to the rFBA method66 as both constrain flux bounds by regulatory network outputs. However, RSA does not require time-dependent simulations. Instead, the outputs of the regulatory network are computed by looking for an attractor in the network states. These network states can be considered as a steady state of the regulatory network (see ref. 51).
The simulation of the regulatory network state was performed using the regulatory steady-state analysis (RSA) which is based on multi-state logical modeling51. Briefly, the network component status was assessed by computing synchronous update of the component status following their logical rules until an attractor was reached. The attractor, called steady-state, could be a point attractor or a cyclic attractor. The initial state of network components used to initiate the simulation were either (i) determined from the environment, (ii) set to 0 in case of gene KO mutants or internal network component disruption analysis, (iii) set using the default network component state (see Supplementary Note 1). If an attractor is found, the arithmetic mean value of each component through the attractor was computed and used as network status value determining the constraints for subsequent biochemical network computation.
Simulation of the biochemical reaction network state containing the genome-scale metabolic network module plus the macromolecule network module was performed using constraint- based modeling. Flux distributions were simulated by FBA and lower and upper values of the fluxes were determined using Flux Variability Analysis. Various constraints were applied on simulations: i) environmental conditions were defined by constraining lower and/or upper bound of exchange fluxes depending on the availability of the metabolite/macromolecule, or experimentally measured fluxes, ii) gene KO mutants or internal network component disruption were simulated by setting flux (or the gene involved in a GPR) value to 0, iii) the regulatory network constraints were taken into account by setting a gene involved in a GPR or reaction flux (lower bound and upper bound) to appropriate value depending on the regulatory network states solved previously. A broad diversity of phenotypes were simulated depending on the biological function (reaction flux) optimized in FBA.
Biological entity clustering by contribution to the objective function
The Biological Entity Clustering (BECO) method has been designed to classify the biological entities (reactions, genes, regulators) depending on their contribution to the optimization of different cellular objectives in different conditions. For each pair condition/objective, a classification of the metabolic genes depending on their contribution to the objective function (see supplementary Note 5 for details) was performed. The classification used in BECO is inspired by the gene classification proposed by Lewis et al.27 The gene categories depending on their contribution to the optimality of the objective function value are (see Fig. 5):
ess: essential gene whose inactivation makes null the objective function value.
red: redundant gene associated to an essential reaction which is associated to other genes.
opt: optimal gene which is not essential but required for the optimality of the objective function value.
ele: less efficient enzymatically gene whose the activation implies a number of activated enzymes (reactions) greater than the absolute minimum number of enzymes required for objective function optimality.
The same categories (with the suffix _c) are extended to condition-dependent categories. For instance, a gene in the category ess-c is essential for the objective function only in specific conditions.
Model performance in predicting phenotypes
Qualitative evaluations of the cell scale model performance in predicting the bacterial phenotypes were performed by comparing the model predictions with experimental measurements. Phenotype predictions in various environments were performed by in silico simulation as described in the previous section by optimizing the biochemical reaction corresponding to the investigated phenotype. If the reaction flux > α (α = 0) the phenotype is considered as performed by the bacteria and thus the prediction is classified as active phenotype (1), or if the reaction flux ≤ α the prediction is classified as not active phenotype (0). Then a binary matrix (i×j) containing the contingency of the experimental phenotype (i) and network prediction phenotypes (j) with the two classes (0, 1) are built and the network performance metrics are calculated as following. The true positive (TP) phenotypes correspond to identical value of 1 for i and j. The false positive (FP) phenotypes correspond to a value of 0 for i and 1 for j. The true negative (TN) phenotypes correspond to identical value of 0 for i and j, and the false negative (FN) phenotypes correspond to a value of 1 for i and 0 for j. The sensitivity (Sn) of the model prediction capacity, which corresponds to the true positive rate is calculated as following, where (P) is the number of experimentally active results (i = 1):
The precision (Pr) which is the positive predictive value is calculated as following:
The faux positive rate (FPR) is calculated as following:
The accuracy (Acc) of the model prediction capacity is calculated as following:
Model performance in predicting differential gene expression
Evaluation of the VRN performance in predicting differential gene expression was done by comparing the model prediction with the experimental transcriptomic data-set produced by Jacobs et al.50 For the in silico analysis, state of the regulatory network was predicted following the RSA method described above in different environments (see Supplementary Data 2 for the list and composition). Differences in gene states predicted between one tested environment and the complete medium used as reference in Jacobs et al.50 were calculated. Then, genes were classified in three discrete classes (k), differentially repressed (−1), not changed (0), activated (1). For the experimental data analysis, transcriptomics data were discretized in three similar classes (l) (−1, 0, 1) considering differential gene expression by using a threshold of 2, i.e., twice repressed or twice more expressed in planta vs complete medium. Only genes harboring a differential expression with a p-value <0.01 were considered. Then, metrics of the model prediction were computed for each class. Subsequently the global mean prediction (M) was calculated by averaging the metrics of the three classes, see Supplementary Note 2 for details calculation. Briefly, performance of the model prediction was assessed by calculating F1 score (mean) (F1(M)).
Pr(M) is the mean precision calculated as the average precision of the prediction for the three classes, where n is the number of classes:
Sn(M) is the mean sensitivity, or true positive rate, calculated as the average sensitivity of the network prediction for the three classes:
The sensitivity of network prediction to environmental changes, i.e., the deviation of the network prediction due to false assignment of environmental conditions or environmental perturbation was assessed as following. We computed deviation of the network performance due to environment parameters by randomly modifying one of them and comparing the obtained network performance to the one obtained in the original environment.
Detail of the algorithms for in silico analyses can be found in the Supplementary Note 2; scripts are freely available and can be downloaded at the following location (http://lipm-bioinfo.toulouse.inra.fr/systemsbiology/models/rsolanacearum).
Phylogeny analyses
Homology groups of proteins sequences in 29 bacterial genomes downloaded on NCBI database (see Supplementary Table 2 for the list) were assigned using OrthoMCL67 via Family-Companion (http://family-companion.toulouse.inra.fr). Sequence clustering was performed using a p-value cut-off of 1 × 10−5, and a percent match cut-off of 80. Sequences were aligned with Mafft v7.27168. Columns with more than 50% of gaps were removed. Phylogenetic trees were computed with IQ-tree (automatic mode)69 and drawn with Itol (https://itol.embl.de/).
Code availability
The BECO method is available in the FlexFlux library (http://lipm-bioinfo.toulouse.inra.fr/flexflux/documentation.html#beco). Codes for VRN validation are available from the corresponding author upon request.
Data availability
The data that support the findings of this study are available in the website dedicated to the cell model of Ralstonia solanacearum (http://lipm-bioinfo.toulouse.inra.fr/systemsbiology/models/rsolanacearum/download.html). The metabolic network is available in BioModels70 with the identifier MODEL1612020000. The VRN model was deposited also in BioModels and assigned the identifier MODEL1710170000.
References
Kitano, H. Biological robustness. Nat. Rev. Genet. 5, 826–837 (2004).
Koonin, E. V. & Wolf, Y. I. Constraints and plasticity in genome and molecular-phenome evolution. Nat. Rev. Genet. 11, 487–498 (2010).
Félix, M.-A. & Barkoulas, M. Pervasive robustness in biological systems. Nat. Rev. Genet. 16, 483–496 (2015).
Guieysse, B. & Wuertz, S. Metabolically versatile large-genome prokaryotes. Curr. Opin. Biotechnol. 23, 467–473 (2012).
Li, J., Yuan, Z. & Zhang, Z. The cellular robustness by genetic redundancy in budding yeast. PLoS Genet. 6, e1001187 (2010).
Gu, Z. et al. Role of duplicate genes in genetic robustness against null mutations. Nature 421, 63–66 (2003).
Ho, W.-C. & Zhang, J. Adaptive genetic robustness of Escherichia coli metabolic fluxes. Mol. Biol. Evol. 33, 1164–1176 (2016).
Keane, O. M., Toft, C., Carretero-Paulet, L., Jones, G. W. & Fares, M. A. Preservation of genetic and regulatory robustness in ancient gene duplicates of Saccharomyces cerevisiae. Genome Res. 24, 1830–1841 (2014).
Fares, M. A., Keane, O. M., Toft, C., Carretero-Paulet, L. & Jones, G. W. The roles of whole-genome and small-scale duplications in the functional specialization of Saccharomyces cerevisiae genes. PLoS Genet. 9, e1003176 (2013).
Wang, Z. & Zhang, J. Abundant indispensable redundancies in cellular metabolic networks. Genome Biol. Evol. 1, 23–33 (2009).
Blank, L. M., Kuepfer, L. & Sauer, U. Large-scale 13C-flux analysis reveals mechanistic principles of metabolic network robustness to null mutations in yeast. Genome Biol. 6, R49 (2005).
Lee, Y., Lafontaine Rivera, J. G. & Liao, J. C. Ensemble modeling for robustness analysis in engineering non-native metabolic pathways. Metab. Eng. 25, 63–71 (2014).
Kitano, H. & Oda, K. Robustness trade-offs and host-microbial symbiosis in the immune system. Mol. Syst. Biol. 2, 2006.0022 (2006).
Orth, J. D., Thiele, I. & Palsson, B. Ø. What is flux balance analysis? Nat. Biotechnol. 28, 245–248 (2010).
Güell, O., Sagués, F. & Serrano, M. Á. Essential plasticity and redundancy of metabolism unveiled by synthetic lethality analysis. PLoS Comput. Biol. 10, e1003637 (2014).
Suthers, P. F., Zomorrodi, A. & Maranas, C. D. Genome-scale gene/reaction essentiality and synthetic lethality analysis. Mol. Syst. Biol. 5, 301 (2009).
Nakahigashi, K. et al. Systematic phenome analysis of Escherichia coli multiple-knockout mutants reveals hidden reactions in central carbon metabolism. Mol. Syst. Biol. 5, 306 (2009).
Masel, J. & Siegal, M. L. Robustness: mechanisms and consequences. Trends Genet. 25, 395–403 (2009).
Sato, P. M., Yoganathan, K., Jung, J. H. & Peisajovich, S. G. The robustness of a signaling complex to domain rearrangements facilitates network evolution. PLoS Biol. 12, e1002012 (2014).
Garfield, D. A. et al. The impact of gene expression variation on the robustness and evolvability of a developmental gene regulatory network. PLoS Biol. 11, e1001696 (2013).
Serganov, A. & Nudler, E. A decade of riboswitches. Cell 152, 17–24 (2013).
Grundy, F. J. & Henkin, T. M. tRNA as a positive regulator of transcription antitermination in B. subtilis. Cell 74, 475–482 (1993).
Traxler, M. F. et al. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol. Microbiol. 68, 1128–1148 (2008).
Chou, H.-H., Marx, C. J. & Sauer, U. Transhydrogenase promotes the robustness and evolvability of E. coli deficient in NADPH production. PLoS Genet. 11, e1005007 (2015).
Philippe, N., Crozat, E., Lenski, R. E. & Schneider, D. Evolution of global regulatory networks during a long-term experiment with Escherichia coli. BioEssays 29, 846–860 (2007).
Perrier, A. et al. Enhanced in planta fitness through adaptive mutations in EfpR, a dual regulator of virulence and metabolic functions in the plant pathogen Ralstonia solanacearum. PLoS Pathog. 12, e1006044 (2016).
Lewis, N. E. et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 6, 390 (2010).
Silo-Suh, L., Suh, S.-J., Phibbs, P. V. & Ohman, D. E. Adaptations of Pseudomonas aeruginosa to the cystic fibrosis lung environment can include deregulation of zwf, encoding glucose-6-phosphate dehydrogenase. J. Bacteriol. 187, 7561–7568 (2005).
New, A. M. et al. Different levels of catabolite repression optimize growth in stable and variable environments. PLoS Biol. 12, e1001764 (2014).
Abu Kwaik, Y. & Bumann, D. Microbial quest for food in vivo: ‘nutritional virulence’ as an emerging paradigm. Cell Microbiol. 15, 882–890 (2013).
Brown, S. A., Palmer, K. L. & Whiteley, M. Revisiting the host as a growth medium. Nat. Rev. Microbiol. 6, 657–666 (2008).
Peyraud, R. et al. Advances on plant-pathogen interactions from molecular toward systems biology perspectives. Plant J. 90, 720–737 (2017).
Balasubramanian, D., Schneper, L., Kumari, H. & Mathee, K. A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Res. 41, 1–20 (2013).
Yuan, X. et al. Cross-talk between a regulatory small RNA, cyclic-di-GMP signalling and flagellar regulator FlhDC for virulence and bacterial behaviours. Environ. Microbiol. 17, 4745–4763 (2015).
Chavali, A. K., D’Auria, K. M., Hewlett, E. L., Pearson, R. D. & Papin, J. A. A metabolic network approach for the identification and prioritization of antimicrobial drug targets. Trends Microbiol. 20, 113–123 (2012).
Rienksma, R. A., Suarez-Diez, M., Spina, L., Schaap, P. J. & Martins dos Santos, V. A. Systems-level modeling of mycobacterial metabolism for the identification of new (multi-)drug targets. Semin. Immunol. 26, 610–622 (2014).
Galán-Vásquez, E., Luna, B. & Martínez-Antonio, A. The regulatory network of Pseudomonas aeruginosa. Microb. Inform. Exp. 1, 3 (2011).
Mansfield, J. et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 13, 614–629 (2012).
Salanoubat, M. et al. Genome sequence of the plant pathogen Ralstonia solanacearum. Nature 415, 497–502 (2002).
Genin, S. & Denny, T. P. Pathogenomics of the Ralstonia solanacearum species complex. Annu. Rev. Phytopathol. 50, 67–89 (2012).
Cunnac, S., Occhialini, A., Barberis, P., Boucher, C. & Genin, S. Inventory and functional analysis of the large Hrp regulon in Ralstonia solanacearum: identification of novel effector proteins translocated to plant host cells through the type III secretion system. Mol. Microbiol. 53, 115–128 (2004).
Deslandes, L. & Genin, S. Opening the Ralstonia solanacearum type III effector tool box: insights into host cell subversion mechanisms. Curr. Opin. Plant Biol. 20, 110–117 (2014).
Remigi, P., Anisimova, M., Guidot, A., Genin, S. & Peeters, N. Functional diversification of the GALA type III effector family contributes to Ralstonia solanacearum adaptation on different plant hosts. New Phytol. 192, 976–987 (2011).
Solé, M. et al. The awr gene family encodes a novel class of Ralstonia solanacearum type III effectors displaying virulence and avirulence activities. Mol. Plant-Microbe Interact. 25, 941–953 (2012).
Peyraud, R., Cottret, L., Marmiesse, L., Gouzy, J. & Genin, S. A resource allocation trade-off between virulence and proliferation drives metabolic versatility in the plant pathogen Ralstonia solanacearum. PLoS Pathog. 12, e1005939 (2016).
Le Novère, N. Quantitative and logic modelling of molecular and gene networks. Nat. Rev. Genet. 16, 146–158 (2015).
Occhialini, A., Cunnac, S., Reymond, N., Genin, S. & Boucher, C. Genome-wide analysis of gene expression in Ralstonia solanacearum reveals that the hrpB gene acts as a regulatory switch controlling multiple virulence pathways. Mol. Plant-Microbe Interact. 18, 938–949 (2005).
Plener, L., Manfredi, P., Valls, M. & Genin, S. PrhG, a transcriptional regulator responding to growth conditions, is involved in the control of the type III secretion system regulon in Ralstonia solanacearum. J. Bacteriol. 192, 1011–1019 (2010).
Valls, M., Genin, S. & Boucher, C. Integrated regulation of the type III secretion system and other virulence determinants in Ralstonia solanacearum. PLoS Pathog. 2, e82 (2006).
Jacobs, J. M., Babujee, L., Meng, F., Milling, A. & Allen, C. The in planta transcriptome of Ralstonia solanacearum: conserved physiological and virulence strategies during bacterial wilt of tomato. mBio 3 e00114-12 (2012).
Marmiesse, L., Peyraud, R. & Cottret, L. FlexFlux: combining metabolic flux and regulatory network analyses. BMC Syst. Biol. 9, 93 (2015).
Brumbley, S. M., Carney, B. F. & Denny, T. P. Phenotype conversion in Pseudomonas solanacearum due to spontaneous inactivation of PhcA, a putative LysR transcriptional regulator. J. Bacteriol. 175, 5477–5487 (1993).
Flavier, A. B., Clough, S. J., Schell, M. A. & Denny, T. P. Identification of 3-hydroxypalmitic acid methyl ester as a novel autoregulator controlling virulence in Ralstonia solanacearum. Mol. Microbiol. 26, 251–259 (1997).
Aldon, D., Brito, B., Boucher, C. & Genin, S. A bacterial sensor of plant cell contact controls the transcriptional induction of Ralstonia solanacearum pathogenicity genes. EMBO J. 19, 2304–2314 (2000).
Mestek Boukhibar, L. & Barkoulas, M. The developmental genetics of biological robustness. Ann. Bot. 117, 699–707 (2015).
Larhlimi, A., Blachon, S., Selbig, J. & Nikoloski, Z. Robustness of metabolic networks: a review of existing definitions. Biosystems 106, 1–8 (2011).
Rieck, B. et al. PknG senses amino acid availability to control metabolism and virulence of Mycobacterium tuberculosis. PLoS Pathog. 13, e1006399 (2017).
Ziveri, J., Barel, M. & Charbit, A. Importance of metabolic adaptations in Francisella pathogenesis. Front. Cell Infect. Microbiol 7, 96 (2017).
Chakravarthy, S. et al. Virulence of Pseudomonas syringae pv. tomato DC3000 is influenced by the catabolite repression control protein Crc. Mol. Plant-Microbe Interact. 30, 283–294 (2017).
Bartell, J. A. et al. Reconstruction of the metabolic network of Pseudomonas aeruginosa to interrogate virulence factor synthesis. Nat. Commun. 8, 14631 (2017).
Plener, L. et al. Metabolic adaptation of Ralstonia solanacearum during plant infection: a methionine biosynthesis case study. PLoS ONE 7, e36877 (2012).
Qian, W. et al. Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris. Genome Res. 15, 757–767 (2005).
Feist, A. M., Herrgård, M. J., Thiele, I., Reed, J. L. & Palsson, B. Ø. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 7, 129–143 (2009).
Courtot, M. et al. Controlled vocabularies and semantics in systems biology. Mol. Syst. Biol. 7, 543 (2011).
Chaouiya, C. et al. SBML qualitative models: a model representation format and infrastructure to foster interactions between qualitative modelling formalisms and tools. BMC Syst. Biol. 7, 135 (2013).
Covert, M. W., Xiao, N., Chen, T. J. & Karr, J. R. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 24, 2044–2050 (2008).
Chen, F., Mackey, A. J., Stoeckert, C. J. & Roos, D. S. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34, D363–D368 (2006).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Chelliah, V. et al. BioModels: ten-year anniversary. Nucleic Acids Res. 43, D542–D548 (2015).
Acknowledgements
R.P. was supported by EMBO (Long-Term Fellowship ALTF 1627–2011) and Marie Curie Actions (EMBOCOFUND2010, GA-2010-267146) and European Research Council (ERC-StG336808 project VariWhim). We thank Christian Boucher and Patrick Barberis for their contribution to the construction and screen of the Tn5 insertion mutant library. We acknowledge funding from the Institut National de la Recherche Agronomique (Plant Health Division grant AAP SPE 2012) and the French Laboratory of Excellence project TULIP (ANR-10-LABX-41; ANR-11-IDEX-0002-02).
Author information
Authors and Affiliations
Contributions
R.P., L.C. and S.G. conceived and designed the study. R.P. reconstructed the VRN model and performed the experimental validation of model predictions. R.P. and L.C. conducted simulations and computational analyses. R.P., L.C. and L.M. conceived algorithms. L.C. and L.M. developed bioinformatic tools and the website. R.P., L.C. and S.G. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peyraud, R., Cottret, L., Marmiesse, L. et al. Control of primary metabolism by a virulence regulatory network promotes robustness in a plant pathogen. Nat Commun 9, 418 (2018). https://doi.org/10.1038/s41467-017-02660-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-02660-4
This article is cited by
-
Genome-wide association of rice response to blast fungus identifies loci for robust resistance under high nitrogen
BMC Plant Biology (2021)
-
The relative importance of soil moisture in predicting bacterial wilt disease occurrence
Soil Ecology Letters (2021)
-
High abundance of Ralstonia solanacearum changed tomato rhizosphere microbiome and metabolome
BMC Plant Biology (2020)
-
Phage combination therapies for bacterial wilt disease in tomato
Nature Biotechnology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
