
Abstract
Patients with short QT syndrome (SQTS) may present with syncope, ventricular fibrillation or sudden cardiac death. Six SQTS susceptibility genes, encoding cation channels, explain <25% of SQTS cases. Here we identify a missense mutation in the anion exchanger (AE3)-encoding SLC4A3 gene in two unrelated families with SQTS. The mutation causes reduced surface expression of AE3 and reduced membrane bicarbonate transport. Slc4a3 knockdown in zebrafish causes increased cardiac pHi, short QTc, and reduced systolic duration, which is rescued by wildtype but not mutated SLC4A3. Mechanistic analyses suggest that an increase in pHi and decrease in [Cl−]i shortened the action potential duration. However, other mechanisms may also play a role. Altered anion transport represents a mechanism for development of arrhythmia and may provide new therapeutic possibilities.
Similar content being viewed by others


Introduction
The short QT syndrome (SQTS), first recognized in 20001, is a rare, genetically determined, severe arrhythmogenic disease with a high risk of syncope, atrial and ventricular fibrillation, and sudden cardiac death (SCD)2. Cardiac events have a clear male preponderance, typically presenting during the 2nd or 3rd decade3. The treatment after aborted SCD and malignant ventricular arrhythmia is implantable cardioverter defibrillator (ICD) implantation4. SQTS has only been reported to present in an autosomal inherited dominant form. The diagnostic criteria for SQTS proposed by Gollob et al.4 were based on a combination of shortened duration of the repolarisation (QT interval), family- and clinical history. A frequency corrected QT (QTc) < 370 ms was suggested as an initial cutoff for diagnostic consideration of SQTS, whereas the HRS/EHRA/APHRS Expert Consensus Statement5 suggested QTc < 330 ms as a stand-alone criterion for the SQTS diagnosis or a possible diagnosis if QTc < 360 ms in combination with a pathogenic mutation, family- or clinical history.
Disease-causing mutations have been identified in the cardiac potassium ion channels KCNH2 6,7, KCNQ1 8 and KCNJ2 9 leading to gain of channel function and causing action potential duration (APD) shortening and elevated risk of cardiac arrhythmia. Loss-of-function mutations in genes coding for the subunits of the cardiac L-type calcium channels CACNA1C, CACNB2 and CACNA2D1, have recently been identified as separate genetic causes of SQTS10,11. So far, only these six cation channels have been implicated in SQTS, and most expanded genetic screening therefore focuses on cation channel genes. However, considering that identified cation channel mutations explain <25% of SQTS cases4, other classes of genes are likely to be associated with the development of this disease. Such genes may encode anion channels, transporters, or proteins involved in pH regulation, which could all potentially alter the QT interval. A role for changes in pH is supported by the finding that lowering intracellular pH (pHi) prolongs QT interval12; a role for Cl− is supported by the demonstration that modulation of Cl− conductance in cardiomyocytes also changes APD13.
In this study, we identify a novel genetic etiology for SQTS in a family with presumed autosomal dominant inheritance of SQTS. We find that affected family members have a mutation in the anion exchanger Solute Carrier Family 4 Member 3 (SLC4A3) gene, which encodes a Cl,HCO3-exchanger (AE3). The mutation leads to a trafficking defect, decreased Cl,HCO3-exchange over the cell membrane, increased pHi and it reduces QT-duration in zebrafish embryos. These findings represent a previously unappreciated mechanism for the development of SQTS.
Results
Cardiac manifestations in probands and relatives with SQTS
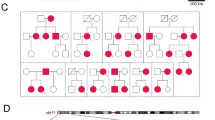
Pedigrees and demographics for the families are presented in Fig. 1a and Supplementary Table 1. The index patient (III-17) in family 1 presented 31 years old with cardiac arrest during sleep. He was successfully resuscitated and admitted to hospital where a new episode of ventricular fibrillation was converted to sinus rhythm by DC shock (Fig. 1b). In his ECG, a QTc duration of 320 ms and a J-point to T-peak interval (Jp-Tp) of 120 ms (Fig. 1c) were noted. No other cardiac abnormalities were observed. A structurally normal heart was found in all relatives, except for subject IV-13, who showed dilated cardiomyopathy with decreased systolic left ventricular ejection fraction (LVEF) of 40%. This patient was later diagnosed with Limb-Girdle muscle dystrophy type 2I14. Four relatives (II-8, II-10, IV-1, III-13) experienced syncopes in the second decade, and two relatives (III-4, III-5) died suddenly and unexplained at the age of 41 and 42 years, respectively. In family 2, the proband (II-1) died suddenly unexpectedly at rest 22 years old. His brother (II-2) was asymptomatic with a structurally normal heart and a QTc of 320 ms and a Jp-Tp < 120 ms (Fig. 1d). The parents were asymptomatic; the mother’s QTc was 355 ms. Cascade screening in family 2 revealed two more asymptomatic mutation-carrying relatives (II-2, II-3) with short QTc’s in both (Fig. 1d and Supplementary Table 1). All family members are of European descent.

Pedigrees and representative ECG recordings in two families with SQTS. a Pedigree of family 1 and 2. Proband marked with an arrow. b ECG recording showing ventricular fibrillation and DC shock conversion to sinus rhythm in the proband in family 1. c ECG-12 from the proband in family 1. d ECG-12 from patient III-2 in family 2
Whole exome sequencing of family members
Initial analysis of the family 1 index patient using a targeted Next Generation Sequencing (NGS) gene panel including the SQTS genes, known to be associated with SQTS at that time (CACNA1C, CACNB2, KCNH2, KCNQ1, and KCNJ2, Supplementary Table 2) revealed no pathogenic mutations. Exome sequencing of four SQTS-affected individuals from family 1 (III-17, IV-1, IV-3, IV-4) along with four asymptomatic family members with normal ECGs (I-6, II-6, III-16, IV-2) was performed (Fig. 1a). Three members from family 2, who were referred to the clinic at the time of the analysis, were also exome sequenced (Supplementary Table 3).
Variants were filtered to include variants shared by all six SQTS individuals but not present in any of the five healthy controls. After removal of variants observed in publicly available databases (frequency > 5%) only 11 variants remained. Nine were outside gene regions, one was intronic (chr19:52149499:c421–11A > G, SIGLEC14), not predicted to affect splicing, and one was a heterozygous missense variant (chr2:220497051:c.1109 G > A NM_201574, p.R370H) in SLC4A3 (Supplementary Table 3). The latter variant was not present in any of the publically available variant databases. The resulting amino acid change from arginine to histidine (R370H) is in the conserved E367-T-A-R-W-I-K-F-E-E376 motif of the SLC4 family. The variant was predicted as possibly damaging by PolyPhen215 and had a CADD score 28.1 also indicating a deleterious effect of the variant16.
ECG changes and manifestations in carriers and non-carriers
Cascade screening in family 1 identified a total of 23 carriers of the SLC4A3 c.1109 G > A variant. All carriers had a QTc ≤ 370 ms (mean 340 ± 18 ms) and 19 non-carriers had a QTc > 370 ms (mean 402 ± 24 ms) (p < 0.001, Students two-tailed t-test) (Fig. 1a and Table 1). Post mortem analysis based on paraffin-embedded tissue showed that subject II-2 also carried the c.1109 G > A mutation, which makes the two relatively young SCD victims (III-4 and 5) obligate carriers of the c.1109 G > A variant. All SLC4A3 c.1109 G > A carriers had Jp-Tp ≤ 140 ms (mean 121 ± 15 ms) compared to mean 173 ± 22 ms in non-variant carriers (p < 0.001, Students two-tailed t-test). In the unrelated smaller family 2, the four mutation-carrying relatives had QTc < 370 ms. Only men and no children experienced clinical symptoms. There were no differences in QTc (p = 0.78, Students two-tailed t-test) or Jp-Tp (p = 0.97, Students two-tailed t-test) between male and female carriers of the mutation (Table 1). Due to the clinical course in these two families with SCD and aborted SCD in the index patients and serious cardiac syncope in another relative (Fig. 1a and Supplementary Table 1) mutation carriers were offered implantation of a primary prophylactic ICD. After thorough information, 16 out of 25 (64%) of the asymptomatic mutation-carrying relatives, accepted ICD implantation. After a follow-up of a mean of 49 ± 9 month none of the mutation-carrying relatives including the aSCD survivor had experienced ICD therapy. In addition, the mutation-carrying relatives without an ICD were also asymptomatic.
Reduced membrane localization of the mutated AE3
We functionally characterized the mutated AE3 variant in HEK-293 cells transfected with cDNA encoding wild-type AE3 or the AE3-R370H mutant. No difference in expression of wild-type and mutated AE3 was seen in whole cell lysates (Fig. 2a and Supplementary Fig. 1), however, the surface localization of the mutated protein was significantly reduced compared to the wild-type protein (Fig. 2b). This strongly suggests that trafficking of the mutated AE3 to the membrane was compromised.

Immunoblot of AE3 and intracellular pH in HEK-293 cells. a Representative immunoblots of total and membrane localized (biotinylated) AE3 protein in AE3 transfected and AE3R370H transfected HEK-293 cells. Pan-actin was used as control/reference protein. b Average total and membrane localized AE3 protein relative to pan-actin expression (n = 4) (*p < 0.05 as determined by unpaired Student t-test). c Steady state pHi of AE3 transfected and AE3R370H transfected HEK-293 cells in CO2/HCO3 − containing media and CO2/HCO3 − free media. d Representative recording of pH in HEK-293 cells. Numbers indicate parts of the protocols used for calculations of net base transport shown in e. e Average net base uptake for AE3 transfected and AE3R370H transfected HEK-293 cells. Numbers correspond to the numbers given in c (n = 7). *p < 0.05, **p < 0.01, ***p < 0.001 as determined by unpaired Student t-test. Error bars throughout represent the s.e.m.
Reduced Cl,HCO3-exchange activity of mutated AE3
Steady state pHi was similar in HEK cells expressing wild-type AE3 and mutated AE3 in both the presence and absence of HCO3 − (Fig. 2c). We next analyzed HCO3 − transport in the two types of cells (Fig. 2d, e). When assayed in the presence of HCO3 −, the net transfer rate of acid equivalents across the cell membrane following both omission of Cl− and addition of Cl− was faster in the cells expressing wild-type AE3 compared to cells expressing the mutated variant. In the absence of HCO3 −, the transfer rates were low and no difference was seen (Fig. 2d, e). These findings indicate a loss-of-function effect of the mutation. Addition of sodium acetate (NaOAc) was associated with a reduction of pHi followed by recovery. When NaOAc was washed out, pHi increased similarly in both groups. However, the following decrease in pHi (in the presence of HCO3 −) was significantly faster in cells expressing wild-type AE3 compared to cells expressing the mutated variant (Fig. 2d, e), further supporting a loss-of-function effect.
Elevated pHi and short QTc in AE3−deficient zebrafish hearts
To determine the consequence of lacking AE3 activity in vivo, we assessed the effect of slc4a3 knockdown on systolic contraction in zebrafish. Zebrafish slc4a3 was efficiently knocked down in embryos (Supplementary Fig. 2a). Whereas heart rates were unaffected by the knockdown (Supplementary Fig. 2b and Supplementary Fig. 2c), systolic contraction duration was significantly shortened (Fig. 3a). The phenotype was rescued by mRNA encoding wild-type, but not Arg370His-mutated AE3, demonstrating that amino acid residue Arg370 of AE3 is required to maintain normal systolic duration in vivo (Fig. 3a, b). Importantly, ECG analysis (Fig. 3c) demonstrated that the observed shortening of systolic contraction corresponded to significantly shortened QTc for slc4a3 knockdown embryos (Fig. 3d) which was associated with an increased ventricular pHi (Fig. 3e).

Loss of slc4a3 in zebrafish embryos replicates the QTc shortening observed in patients. a Systolic interval divided by heart cycle duration is significantly reduced in slc4a3 knockdown embryos 2 days post fertilization (dpf). This phenotype is rescued by overexpression of AE3 in knockdown embryos. n = 19 (Control), n = 14 (Knockdown), and n = 19 (Knockdown + AE3) compiled from three independent experiments, each including all three experimental groups. b Reduced systolic interval in slc4a3 knockdown embryos is not rescued by overexpression of R370H-mutated AE3 in knockdown embryos. n = 13 (Control), n = 16 (Knockdown), and n = 15 (Knockdown + AE3R370H) compiled from three independent experiments, each including all three experimental groups. c A raw ECG recording of a 2 dpf zebrafish embryo; P,Q,R,S,T waves, and RR and QT intervals indicated. d Corrected QT intervals (QTc) for control and slc4a3 knockdown embryos were calculated with Bazett’s formula. n = 12 (Control) and n = 11 (Knockdown) compiled from three independent experiments, each including both experimental groups. e pHi is significantly increased in hearts from slc4a3 knockdown embryos 2 days post fertilization (dpf). n = 16 (Control), n = 14 (Knockdown). * p < 0.05, **p < 0.01, ***p < 0.001 as determined by one way ANOVA with Tukey’s post-test (a, b) or unpaired student t-test (d, e). Error bars throughout represent the s.e.m.
High pHi and low [Cl−]i shorten cardiac action potential
Due to the increased pHi observed in embryonic slc4a3 knockdown zebrafish hearts, we tested whether increased pHi leads to shortened APD and QTc in the isolated heart. Addition of 10 mM NH4Cl extracellularly caused an increase of pHi in rabbit cardiomyocytes as expected (Supplementary Fig. 3). The increased pHi was associated with a reduced APD of the cardiomyocytes (Fig. 4a, b) and a shorter RT (Fig. 4c, d). Since AE3 mediates HCO3 − efflux in exchange for influx of Cl−, we further tested whether lowering of [Cl−]i shortens APD by patching rat ventricular cardiomyocytes with 40 mM or 10 mM Cl− in the pipette solution. As shown in Fig. 4e, 10 mM Cl− was associated with a shorter APD compared to 40 mM Cl−.

Intracellular alkalization and low [Cl−]i shortens the action potential duration and R-T interval. a Action potentials of rabbit hearts measured before and during application of 10 mM NH4Cl. APD: Action potential duration. b Mean APD during application of NH4Cl (n = 5). Zero indicates time of intervention. c ECGs in rabbit hearts before and during application of 10 mM NH4Cl. d Mean RT interval in rabbit hearts (n = 5). In a, c the records are aligned at the pacing stimulus. ***p < 0.001, ****p < 0.0001 as determined by one-way repeated measures ANOVA. Zero indicates time of intervention. e Bar graph showing APD90 of rat ventricular cardiomyocytes patched with 40 mM (open, n = 23) or 10 mM (black, n = 16) chloride in the pipette solution. *p < 0.05 as determined by one-way repeated measures ANOVA. Error bars throughout represent the s.e.m.
Discussion
We report that a mutation in the SLC4A3 gene, which encodes the cardiac chloride-bicarbonate exchanger AE3, is associated with SQTS. Within a large affected family, the group of carriers had experienced several serious cardiac events and SCD, whereas no events had occurred in non-carriers. All carriers had a QTc ≤ 370 ms and all non-carriers had a QTc > 370 ms. The J-point to T-peak interval was ≤ 140 ms in all carriers. Jp-Tp has been suggested as a parameter to be applied in the diagnostic score of SQTS4, and has been reported to have a high specificity for the SQTS and is clinically readily recognizable on visual ECG inspection. These ECG findings indicate a high penetrance as assessed by repolarization duration, the equivalent of the APD. In agreement with findings in other SQTS families, clinical manifestations were present in males only, and absent in children. Out of 27 living mutation carriers from the two families, six showed cardiac events considered SQTS related. The mutation leads to reduced trafficking of AE3 to the membrane, loss of Cl,HCO3-exchange and increased pHi. The role of the mutation was emphasized by the identification of the same mutation in an unrelated family with SQTS and by the finding that slc4a3 knockdown in zebrafish causes increased pHi, short QTc, and reduced systolic contraction duration, which was rescued by wildtype but not R370H-mutated AE3. Thus, our data show the involvement of an anion transporter in the development of SQTS.
Previously, other SLC4A3 variants have been associated with seizures17. Further, mice with a targeted disruption of the Cl,HCO3-exchanger AE3 display a reduced threshold for triggering an epileptic seizure18,19. However, none of the mutation-carriers in our two families has had any clinical presentations with unconsciousness suspected to be due to epileptic events. The case with aSCD had documented VF.
AE3 is a comprehensively studied Cl,HCO3-exchanger with the highest mRNA expression of the HCO3 − transporters20 in mouse hearts. AE3 is known to be important for myocardial pHi recovery from alkaline loads20,21. However, AE3 knockout mice show no overt cardiac phenotype unless crossed with Na,K,Cl−exchanger knockout mice22 or mice carrying a mutation in α-tropomyosin in a model of hypertrophic cardiomyopathy23. No electrophysiological characterization of the hearts of the AE3 knockout mice has been published though.
The identified mutation of the SLC4A3 gene causes a single-residue Arg-to-His change at amino acid position 370 of AE3, which is part of the E367-T-A-R-W-I-K-F-E-E376 motif. This motif is highly conserved in all SLC4 gene products24. Previously, a Glu-to-Arg mutation in position 91 of NBCe1/SLC4A4 (position equivalent to Glu375 of AE3)25 has been found to cause poor surface targeting upon expression in MDCK cells. By analysis of an NBCe1 homology model, Arg370 has been proposed to interact with Glu376 (equivalent to Glu92 in NBCe1) through a salt bridge26. We find by further homology modeling that in addition, Arg370 may form a cation-π interaction with Trp385 (Fig. 5). These two structural elements hint to a fixed structural environment that spatially orientates Arg370 and could lead to misfolding if disrupted with a mutation to histidine. In agreement with these data, we found that the mutated variant (Arg-370-His) showed a level of translation similar to wild-type AE3, but only poor surface targeting.

Molecular interaction in direct contact with Arg370. Glu376 is forming a charged hydrogen bond to Arg370 (dashed black lines), while Trp385 is linked through a cation-π interaction (orange dashed line). The figure was prepared with The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC, and the modeling based on an alignment of the AE1 and AE3 sequence in the cytoplasmic region using PDB ID entry 1HYN as template
The reduced membrane trafficking is strongly supported by the reduced Cl,HCO3-exchange activity observed in transfected cells expressing the mutated variant. The data therefore strongly suggest that the mutation leads to trans-membraneous Cl− and HCO3 − transport deficits, which might increase pHi and reduce [Cl−]i.
There is some evidence that modulation of AE3 activity leads to changes in pHi and [Cl−]i. In AE3 KO mice there is a tendency to a higher steady state pHi in isolated cardiomyocytes compared to pHi in cardiomyocytes from WT mice and the recovery time from an imposed intracellular alkalosis is significantly prolonged27. This is consistent with the effect of an inhibitory AE3 antibody on pHi in rat cardiomyocytes. Thus, in the presence of endothelin-1, inhibition of AE3 increased pHi 21. Additionally, α-adrenergic receptor activation of the rat heart reduces steady state pHi most likely through activation of AE328. These findings indicate that in hearts expressing the mutated AE3, the capacity to counteract an increase in pHi is compromised, particularly under in vivo conditions where the heart is exposed to various hormones and neurotransmitters. Our finding of an increased steady state pHi in the hearts of zebrafish embryos with knockdown of AE3 is consistent with this. It has been suggested that activation of AE3 during ischemia29 and extracellular acidosis30 contribute to the increase in guinea pig and sheep ventricular muscle [Cl−]i. This indicates that the effect of AE3 on [Cl−]i may also depend on conditions. Although these findings suggest that inhibition of AE3 mediated anion transport leads to increased pHi and decreased [Cl−]i this conclusion may need further validation.
Since the AE3 mutation increased pHi and likely decreases [Cl−]i, we tested the effects of increased pHi and decreased [Cl−]i on myocyte electrophysiology. Increased pHi was associated with a shortened APD in rabbit heart. This finding is consistent with the finding of an APD prolongation by low pHi in rabbit ventricular myocytes12. It is unclear how this effect is mediated. However, high pHi activates Kv7.1 channels (KCNQ1)31,32, which may accelerate repolarization. The knowledge about the effects of high pHi on cardiac ion channels seems very limited. For KCNH2 we are not aware of studies addressing the effect of high pHi. For the L-type Ca2+ channels the effect of high pHi is likely inhibition12 which would also accelerate repolarization.
We demonstrated that also reduced [Cl−]i leads to shortened APD. A reduction of [Cl−]i will cause the equilibrium potential for Cl− to be more negative and hence increase the Cl− influx in phase 2 and the beginning of phase 3 leading to a shorter action potential. This finding is consistent with studies where different Cl− channels in cardiomyocytes are inhibited33. Together this demonstrates that low [Cl−]i due to inhibition of AE3 activity might contribute directly to the SQTS.
We suggest that both high pHi and low [Cl−]i are likely to contribute to the SQTS, but cannot exclude that other effects may also contribute.
The low event rate during follow-up indicates the difficulties in predicting the risk of ventricular arrhythmias or SCD in patients with SQTS and underscores the importance of developing risk scores. The clinical course of more families with AE3 mutations—probably to be identified in the pool of SQTS patients with hitherto negative genetic test results—will be needed to identify AE3 mutation specific clinical risk factors.
In summary, we have identified a novel mutation in AE3 in two independent families with SQTS. The mutation leads to defective targeting of AE3 to the cell membrane, reduction of Cl,HCO3-exchange and increased pHi. The impact of the mutation on QTc duration was confirmed in zebrafish and the mechanism through which a defect in Cl,HCO3-exchange leads to shortened QTc likely relates to a combined effect of a decrease in HCO3 − efflux (i.e., increased pHi) and a decrease in Cl− influx. These findings offer insight into mechanisms of arrhythmia in general and may provide new treatment possibilities in patients with SQTS. Currently, no specific factors triggering arrhythmia in patients with SQTS have been reported. Based on this study, possible triggers may include medications, physical activities, metabolic derangements or diets challenging the Cl,HCO3-exchange.
Further studies are required to determine the frequency of involvement of SLC4A3 in SQTS and to delineate details of how high pHi and low [Cl−]i may lead to the serious arrhythmia associated with SQTS.
Methods
Patients and clinical evaluation
Informed consent was obtained from all subjects. The study was approved by the Ethics Committee Central Denmark Region (no. 1-10-72-189-16) and by the Danish Data Protection Agency (record no. 1-16-02-629-16).
A 31-year old man resuscitated after cardiac arrest was diagnosed with SQTS4. Cascade screening revealed a large family with 41 relatives to the proband. Subsequently, another unrelated family with SQTS was included for verification. Clinical work-up included clinical examination, ECG, echocardiography, routine blood tests and 48-hours Holter monitoring. Bazett corrected QT interval (QTc) was used. Blood was collected from all family members for molecular genetic analysis. In two cases, post-mortem genetic testing was performed.
Exome sequencing and data analysis
Genomic DNA was extracted from peripheral leucocytes or paraffin tissue blocks on the QiaSymphony using standard protocols (Qiagen). DNA from the index patient was initially analyzed with the MOMA NGS Heart panel v1 (Custom EZ in solution DNA Capture, Nimblegen), see Supplementary Table 2 for gene list using MiSeq sequencing (Illumina). For whole exome sequencing 1 µg DNA was used for library preparation using the TruSeq DNA LT Sample Prep Kit v2 according to the manufacturer’s instructions (Illumina) followed by hybridization using Nimblegen SeqCap EZ Exome v3 (Roche) and Paired-end Sequencing (2 × 100 bp) on the Illumina HiSeq 2000 with TruSeq v3 chemistry (Illumina). CLC BioMedical Genomics Workbench 2.1.1 (Qiagen) was used for data analysis and data was trimmed for adaptor sequence and ambiguous bases before alignment to GRCh37, duplicate removal and SNV and indel calling with the fixed ploidy variant caller. Variants were uploaded to Ingenuity Variant Analysis tool (Qiagen) and filtered to exclude variants with an allele frequency of at least 5% in either 1000 Genomes Project34, ExAC35 or NHLBI ESP exomes (http://evs.gs.washington.edu/EVS). Finally, the variants shared by all individuals with SQTS but not observed in any of the control samples were subjected to further analysis. PCR amplification and subsequent Sanger sequencing of exon 8 containing the variant was used in all included family members for cascade screening for the SLC4A3 variant.
Construction of AE3 R370H and gene expression
Human SLC4A3 (NM_201574) cDNA clone (Origen; Clone SC308062, US) was used as template for introducing the mutation. The point mutation, was introduced using a phusion site-directed mutagenesis kit (Thermo Scientific) and amplified with modified DNA oligonucleotides (Europhins MWG) holding the mutation. Plasmid DNA carrying either AE3 or AE3R370H (controlled for amplication errors) was transfected into HEK-293 cells (ATCC, catalogue number CRL-1573) using TransIT-LT1 transfection reagent (Mirus). Cells were shifted to selection media containing 600 µg ml−1 G418 (Sigma Aldrich) 48 h posttransfection. Cells for pHi experiment were grown in poly-Lysine coated dishes.
AE3 surface processing and western blot analysis
Surface proteins were isolated from HEK-293 cells using the biotinylation based Pierce cell surface protein isolation kit (Thermo Scientific). Total protein cell lysate was prepared by resuspending and homogenizing cells in lysis buffer (10 mM Tris–HCl, 250 mM sucrose, 1 mM EDTA, 1 mM EGTA, 2% Triton X-100) containing Complete Mini, EDTA-free protease inhibitor (Roche Applied Science, Germany, 1 tablet per 10 ml solution). Protein concentration was assayed (Pierce BCA Protein Assay kit; Thermo Scientific) and 10 µg of protein were solubilized in SDS-buffer and separated by SDS-page. Proteins were transferred to PVDF membranes (Amersham;GE Healthcare). Membranes were incubated with primary antibodies (Rabbit anti-AE3 (1:2000), kindly provided by Professor S. Alper, (Renal Division and Division of Molecular and Vascular Medicine, Beth Israel Deaconess Medical Center, Boston, USA), Rabbit-anti-pan-actin (1:2000), Cell Signaling Technology Inc., USA [Catalog no. 4968]) overnight. Secondary antibody (goat-anti rabbit; Millipore, US (1:2000)) linked to horseradish peroxidase were used to detect binding with the ECL chemiluminescence system (Amersham; GE Healthcare).
pHi measurements in HEK-293 cells
Cells were loaded with 2 µM of the pH sensitive dye BCECF-AM for 15 min in a physiological salt solution (PSS) at 37 °C. The cells were placed on an Olympus IX70 microscope with an Olympus LUCPlanFL N 20× objective (N.A. 0.45) and an EasyRatioPro fluorescence imaging system (Photon Technology International). The cells were excited alternately at 485 and 440 nm and emission collected at 550 nm. The BCECF ratio between emission light collected during excitation at 485 and 440 nm, respectively, was calculated after subtraction of background fluorescence and calibrated to pH units using the high-K+ nigericin method, as previously described36. PSS contained in mM: 139 Na+, 5.9 K+, 1.6 Ca2+, 1.2 Mg2+, 122 Cl−, 25 HCO3 −, 1.2 SO4 2−, 1.2 H2PO4 −, 10 HEPES, 5.5 glucose and 0.026 EDTA. In HCO3 − free solutions HCO3 − was replaced by Cl−. In Cl− free solution Cl− was substituted with aspartate. HCO3 − containing solutions were gassed with 5% CO2 in air; HCO3 − free solutions were gassed with air. All solutions were titrated to pH 7.4. After washout of BCECF-AM steady state pHi was recorded and the solutions modified, as shown in Fig. 2d. Intracellular buffering capacity was calculated from the change in pHi following washing and washout of NaAcetate (NAOAc). The transport rate of acid equivalents across the membrane was calculated from the change in pHi per unit time multiplied by the buffering capacity.
Animal ethics
All animal experimental procedures were conformed to guidelines from the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes and were approved by and conducted with permission from the Animal Experiments Inspectorate of the Danish Ministry of Environment and Food, and the State Food and Veterinary Service of the Republic of Lithuania and Ethics Committee of the Lithuanian University of Health Sciences.
Zebrafish knockdown of slc4a3 and phenotyping
Adult zebrafish of the AB wildtype strain were fed four times daily and maintained on recirculating housing systems on a 14 h light per 10 h darkness cycle at 28 °C. Embryos were obtained by natural crosses and maintained in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM MgSO4, 10–5% (w/w)methylene blue, 0.33 mM CaCl2, 2 mM Hepes pH 7.4) at 28 °C. Knockdown was performed by microinjection into zygotes of slc4a3-targeted morpholino e7i7 (TCAGAAGGCTCAGCTCTCACCTCAT) or standard control morpholino (CCTCTTACCTCAGTTACAATTTATA, Gene Tools, LLC). e7i7 efficiency was assessed by reverse transcription PCR from cDNA synthesized from total RNA extracted from groups of ten microinjected embryos two days post fertilization (2 dpf) using primers 5′-CTTCGAGGACAATCCTGGGG-3′/5′-CTACGGAAGGACAGTGAGGC-3′ (slc4a3) and 5′-CACGAGACCACCTTCAACT-3′/5′-ATCCAGACGGAGTATTTGC-3′ (β-actin). Human SLC4A3 mRNA was in vitro synthesized using the mMessage mMachine T7 ULTRA kit (Ambion) with linearized wild type (NM_201574) or R370H-mutated AE3-encoding plasmid as template. Similar stability of microinjected wild type and mutant mRNA was verified by RT-PCR on 2 dpf zebrafish embryos using the human AE3-specific primer set 5′-ATCATCACCCAACTCCCAGC-3′/5′- GGCACTTAGGAGGTCTTGCC-3′.
For pHi measurements in isolated zebrafish hearts, 2 dpf zebrafish embryos were briefly anesthetized by rapid cooling on wet ice prior to the dissection of the heart from the embryo. The isolated heart was immediately transferred to fish buffer and the embryo was euthanized. The hearts were loaded with 5 µM BCECF-AM for 5–15 min in fish buffer at room temperature. After washout of BCECF-AM the recording and calibration of the fluorescence signal from the ventricles of the hearts were made as described for pHi measurements in HEK cells. The fish buffer contained in mM: 131 Na+, 4.0 K+, 1.6 Ca2+, 127 Cl−, 10 HCO3 −, 1.0 H2PO4 −, and 5.0 glucose. The solution was gassed with 1% CO2 in air and pH was 7.65.
For assessment of cardiac function, 2 dpf embryos were anaesthetized using buffered 150 ng mL−1 Ethyl 3-aminobenzoate methanesulfonate (MS-222, Sigma-Aldrich) and staged in 4% methyl cellulose (average M n ~ 86,000, Sigma-Aldrich) in E3. To obtain systolic and diastolic durations, high-speed video recordings of eight seconds duration at 100 frames per second were made. Video analysis was performed, as previously described37,38 using an algorithm written in MATLAB (The MathWorks Inc.) allowing for evaluation of ventricular contraction and relaxation by quantification of changing pixel intensity. Recordings were excluded from analysis in cases where (1) atrial contraction/relaxation caused movement of the ventricle interfering with quantification of ventricular contraction/relaxation and (2) the quality of recording, e.g., signal-to-noise ratio, did not allow quantitative analysis. All embryos were euthanized by MS-222 overdose immediately after phenotyping.
For ECG recordings two dpf zebrafish embryos were placed in E3 medium with MS-222 (150 ng mL−1) for ECG at room temperature (22–24 °C). The recordings were made using a potassium acetate (3 M) filled borosilicate glass micropipette (PG15OT-7.5; Harvard Apparatus) with tip resistances 5–10 MΩ at 10 kHz using an Axopatch 200B amplifier (Axon Instruments, Inc.) in a current-clamp configuration. The recording electrode was positioned on embryo skin surface near the heart (no penetration). Data acquisition was done with the software package Clampex 7 for Windows (Axon Instruments Inc.). Data were analyzed in LabChart 7 (ADInstruments) after low-pass digital filtering with cut-off frequency 50 Hz. The ECG data were only excluded if the quality of recording, i.e., signal-to-noise ratio, did not allow quantitative analyzes of QT intervals. QT and RR intervals were calculated for each embryo and converted to QTc.
Measurement of action potentials and ECG in rabbit heart
Experiments were performed on six months old male New Zealand white rabbits (weight: 2.95 ± 0.18 kg, n = 5). Animals received 10 mg kg−1 xylazine (s.c.) following i.v. injection of ketamine (10 mg kg−1) with heparin (1000 U kg−1). The heart was excised and coronary arteries perfused in the Langendorff-system with oxygenated Tyrode solution (in mM): 135 NaCl, 5.4 KCl, 1.8 CaCl2, 0.9 MgCl2, 0.33 NaH2PO4, 10 glucose, 10 HEPES, pH 7.4, 37 °C. In total 20 μM (−)-blebbistatin and 5 mM 2,3-butanedione monoxime were added to stop contraction. The heart was stimulated with bipolar silver electrodes in the left ventricle (LV) at 300 ms interval and 2 ms square pulses of about twice diastolic threshold. ECG was recorded with two ECG electrodes placed on LV (near the apex and near the base) using FE136 amplifier (ADInstruments) and digitized at 20 kHz.
The heart was stained with 50 μM di-4-ANBDQBS (JPW-6033) by slow 10 ml bolus injection. Optical recordings of action potentials (OAP), as described previously39 were acquired with a cooled (−100 °C) 14-bit-EMCCD camera (iXonEM + DU-860, Andor Technology) with a 50-mm focal length objective (Navitar) and using the imaging software (Andor SOLIS). Collimated LED of 660 nm wavelength (M660L3, Thorlabs) was used for excitation of di-4-ANBDQBS. Fluorescence emission was collected using long-pass filter of 720 nm (HQ720lp, Chroma) with a sampling rate of 500 Hz. Background fluorescence was subtracted and 16 consequent recordings were averaged and normalized to the control level. No exclusion criteria were applied to the data from the rabbit hearts. Di-4-ANBDQBS was obtained from Dr L. Loew (University of Connecticut, USA).
LabChart8 Pro software (ADInstruments) was used to analyze the R-T interval between R wave and 50% of T wave amplitude decrease. The optical movies were preprocessed with the use of ImageJ software and OAPs were analyzed using MS Excel Activation time of OAP was calculated as time between stimulus and 50% of OAP upstroke amplitude; duration of OAP (OAPD)—time between 50% of OAP upstroke amplitude and 50% of OAP repolarization time.
Recording of action potentials and ECG in rat cardiomyocytes
Ventricular cardiomyocytes were isolated from male Sprague-Dawley rats (weight: 325 ± 8.3 g, n = 13) (Taconic, Ll. Skensved, Denmark). Rats were anaesthetized using isoflurane (5% in 100% O2), hearts were quickly excised and the aorta cannulated. Hearts were retrogradely perfused by a solution containing in mM: 136 NaCl, 2.68 KCl, 1.47 KH2PO4, 8.06 Na2HPO4, 0.9 CaCl2, 0.9 MgCl2, 6 glucose, pH 7.4, equilibrated with 100% O2 for 5 minutes, followed by 2 minutes of perfusion with the same solution without calcium. Subsequently, perfusion was changed to a solution containing (in mM) 20 NaCl, 120 K-gluconate, 1 MgCl2, 10 HEPES, 10 glucose, pH 7.4 equilibrated with 100% O2 for 2 min and perfusion with the same buffer added 96 U ml−1 collagenase (Type IV, Worthington), 25 µM CaCl2 and 0.1% BSA until the tissue was digested. After the perfusion process, the heart was transferred to a petri dish, the atria were discarded and the ventricles were cut into pieces. A solution containing (in mM) 100 K-glutamate, 10 K-aspartate, 25 KCl, 10 KH2PO4, 2 MgSO4, 20 Taurine, 5 Creatine, 20 glucose, 0.5 EGTA, 5 HEPES, 0.1% BSA, pH 7.4 was used to cleanse the remaining pieces from collagenase and to stop the digestive process. The pieces of ventricle were gently bubbled with 100% O2 to dissolve the tissue and release the cardiomyocytes. Hereafter the solution was filtered using a metal grid (50 mesh) and washed. Ca2+ was gradually reintroduced to a final concentration of 0.72 mM and the cells were stored at 5 °C, until use on the day of isolation. Action potentials were recorded in the whole-cell zero current patch clamp configuration using a SEC-05LX amplifier (npi electronics, Tamm, Germany). Signals were low-pass filtered (current: 2 kHz; potential: 1 kHz) and sampled at 10 kHz using CellWorks (npi electronics, Tamm, Germany).
The cardiomyocytes were superfused at room temperature with solution containing in mM: 136 NaCl, 2.68 KCl, 1.47 KH2PO4, 8.06 Na2HPO4, 0.9 CaCl2, 0.9 MgCl2, 6 glucose; pH adjusted to 7.4. Borosilicate patch pipettes (3–5 MΩ) were filled with either 40 or 10 mM Chloride containing solution. The 40 mM Chloride solution contained (in mM): 105 K-gluconate, 40 KCl, 9 Na-gluconate, 5 Na-pyruvate, 1 Na2ATP, 1.92 MgSO4, 0.353 Ca-gluconate2, 1 EGTA, 5 HEPES, pH adjusted to 7.2. For 10 mM Chloride, 30 mM KCl was exchanged with K-gluconate. Action potentials were elicited at 1 Hz by current injection through the pipette for ten minutes. The reported APD90 value is the average time to 90% repolarization of the last 20 stimuli. Analyses were performed using a custom written script in MatLab (MatLab R2013a, MathWorks, Inc.). Data from the patch clamp analysis were excluded if the resting potential was unstable, in such cases depolarization most often led to hypercontracture and loss of seal stability. These are standard criteria for patch clamp experiments in our laboratory.
Statistics
Clinical data are presented as mean values ± SD. Statistical significant differences between patient groups were ascertained by Students two-tailed t-test for un-paired observations. Bonferroni’s correction was applied to correct for multiple comparisons. Experimental data are presented as mean values ± s.e.m. Differences between means in the HEK cell and rat cardiomyocyte experiments were tested with unpaired Student t-test. Differences between means in the zebrafish embryos were tested by unpaired Student t-test or one-way ANOVA with Tukey’s post-test. Differences between means in the rabbit heart intracellular alkalization experiments were tested with one-way repeated measures ANOVA. p < 0.05 was considered significant. All clinical and zebrafish embryo data were tested and found to be normally distributed. The pH measurements and the action potential measurements are known from other experiments to have a normal distribution.
Sample sizes were estimated based on previously published data for the experiments on zebrafish embryos, rabbit hearts, and rat cardiomyocytes. No statistical method was used to determine sample size. No randomization or blinding was applied to experiments involving zebrafish embryos, rabbit hearts, and rat cardiomyocytes.
Data availability
Exome sequencing data have been deposited in The European Genome-phenome Archive under accession code EGAS00001002632. Access to the sequence data needs to be approved by the local Data Access Committee.
References
Gussak, I. et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 94, 99–102 (2000).
Giustetto, C. et al. Long-term follow-up of patients with short QT syndrome. J. Am. Coll. Cardiol. 58, 587–595 (2011).
Mazzanti, A. et al. Novel insight into the natural history of short QT syndrome. J. Am. Coll. Cardiol. 63, 1300–1308 (2014).
Gollob, M. H., Redpath, C. J. & Roberts, J. D. The short QT syndrome: proposed diagnostic criteria. J. Am. Coll. Cardiol. 57, 802–812 (2011).
Priori, S. G. et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 10, 1932–1963 (2013).
Brugada, R. et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 109, 30–35 (2004).
Hong, K., Bjerregaard, P., Gussak, I. & Brugada, R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J. Cardiovasc. Electrophysiol. 16, 394–396 (2005).
Bellocq, C. et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 109, 2394–2397 (2004).
Priori, S. G. et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 96, 800–807 (2005).
Antzelevitch, C. et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115, 442–449 (2007).
Templin, C. et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur. Heart J. 32, 1077–1088 (2011).
Saegusa, N., Moorhouse, E., Vaughan-Jones, R. D. & Spitzer, K. W. Influence of pH on Ca(2)(+) current and its control of electrical and Ca(2)(+) signaling in ventricular myocytes. J. Gen. Physiol. 138, 537–559 (2011).
Adkins, G. B. & Curtis, M. J. Potential role of cardiac chloride channels and transporters as novel therapeutic targets. Pharmacol. Ther. 145, 67–75 (2015).
Sveen, M. L., Schwartz, M. & Vissing, J. High prevalence and phenotype-genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann. Neurol. 59, 808–815 (2006).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Vilas, G. L., Johnson, D. E., Freund, P. & Casey, J. R. Characterization of an epilepsy-associated variant of the human Cl−/HCO3(−) exchanger AE3. Am. J. Physiol. Cell. Physiol. 297, C526–C536 (2009).
Hentschke, M. et al. Mice with a targeted disruption of the Cl−/HCO3− exchanger AE3 display a reduced seizure threshold. Mol. Cell Biol. 26, 182–191 (2006).
Salameh, A. I., Hubner, C. A. & Boron, W. F. Role of Cl–HCO3− exchanger AE3 in intracellular pH homeostasis in cultured murine hippocampal neurons, and in crosstalk to adjacent astrocytes. J. Physiol. 595, 93–124 (2017).
Wang, H. S., Chen, Y., Vairamani, K. & Shull, G. E. Critical role of bicarbonate and bicarbonate transporters in cardiac function. World J. Biol. Chem. 5, 334–345 (2014).
Chiappe de Cingolani, G. E. et al. Involvement of AE3 isoform of Na(+)-independent Cl(−)/HCO(3)(−) exchanger in myocardial pH(i) recovery from intracellular alkalization. Life. Sci. 78, 3018–3026 (2006).
Prasad, V. et al. Impaired cardiac contractility in mice lacking both the AE3 Cl−/HCO3− exchanger and the NKCC1 Na+−K+−2Cl− cotransporter: effects on Ca2+ handling and protein phosphatases. J. Biol. Chem. 283, 31303–31314 (2008).
Al Moamen, N. J. et al. Loss of the AE3 anion exchanger in a hypertrophic cardiomyopathy model causes rapid decompensation and heart failure. J. Mol. Cell. Cardiol. 50, 137–146 (2011).
Parker, M. D. & Boron, W. F. The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiol. Rev. 93, 803–959 (2013).
Li, H. C. et al. Missense mutations in Na+:HCO3− cotransporter NBC1 show abnormal trafficking in polarized kidney cells: a basis of proximal renal tubular acidosis. Am. J. Physiol. Renal. Physiol. 289, F61–F71 (2005).
Chang, M. H., DiPiero, J., Sonnichsen, F. D. & Romero, M. F. Entry to “formula tunnel” revealed by SLC4A4 human mutation and structural model. J. Biol. Chem. 283, 18402–18410 (2008).
Sowah, D., Brown, B. F., Quon, A., Alvarez, B. V. & Casey, J. R. Resistance to cardiomyocyte hypertrophy in ae3−/− mice, deficient in the AE3 Cl−/HCO3− exchanger. BMC Cardiovasc. Disord. 14, 89 (2014).
Desilets, M., Puceat, M. & Vassort, G. Chloride dependence of pH modulation by beta-adrenergic agonist in rat cardiomyocytes. Circ. Res. 75, 862–869 (1994).
Lai, Z. F. & Nishi, K. Intracellular chloride activity increases in guinea pig ventricular muscle during simulated ischemia. Am. J. Physiol. 275, H1613–H1619 (1998).
Vaughan-Jones, R. D. An investigation of chloride-bicarbonate exchange in the sheep cardiac Purkinje fibre. J. Physiol. 379, 377–406 (1986).
Kerst, G. et al. Properties and function of KCNQ1 K+ channels isolated from the rectal gland of Squalus acanthias. Pflugers Arch. 443, 146–154 (2001).
Unsold, B. et al. KCNE1 reverses the response of the human K+ channel KCNQ1 to cytosolic pH changes and alters its pharmacology and sensitivity to temperature. Pflugers Arch. 441, 368–378 (2000).
Duan, D. Phenomics of cardiac chloride channels: the systematic study of chloride channel function in the heart. J. Physiol. 587, 2163–2177 (2009).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Bouzinova, E. V. et al. Na+− dependent HCO3− uptake into the rat choroid plexus epithelium is partially DIDS sensitive. Am. J. Physiol. Cell Physiol. 289, C1448–C1456 (2005).
Sondergaard, M. T. et al. Calmodulin mutations causing catecholaminergic polymorphic ventricular tachycardia confer opposing functional and biophysical molecular changes. FEBS J. 282, 803–816 (2015).
Fink, M. et al. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. Biotechniques 46, 101–113 (2009).
Martisiene, I. et al. Voltage-sensitive fluorescence of indocyanine green in the heart. Biophys. J. 110, 723–732 (2016).
Acknowledgements
AE3-antibody was kindly provided by Professor S. Alper, (Renal Division and Division of Molecular and Vascular Medicine, Beth Israel Deaconess Medical Center, Boston, USA). This work was supported by the Danish Council for Independent Research (12–126232 to C.A.).
Author information
Authors and Affiliations
Contributions
H.K.J., K.T., H.B. and C.A. conceived, designed and directed the study. L.N.P. and K.T. performed genetic analyses. V.S.D. and C.A. performed the cell culture experiments. K.K-S. and C.O. contributed with zebrafish experiments. V.V.M. performed zebrafish ECG recordings. V.A.S., J.J. and R.T. performed the rabbit heart experiments. J.P.M. contributed with the molecular interaction data. I.M.B.S.P and M.S.N. contributed with rat cardiomyocyte experiments. H.K.J. and H.B. contributed clinical evaluations. All authors approved the final manuscript and contributed critical revisions to its intellectual content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thorsen, K., Dam, V.S., Kjaer-Sorensen, K. et al. Loss-of-activity-mutation in the cardiac chloride-bicarbonate exchanger AE3 causes short QT syndrome. Nat Commun 8, 1696 (2017). https://doi.org/10.1038/s41467-017-01630-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-01630-0
This article is cited by
-
Kardiogenetik in Deutschland – ein (Rück‑)Blick
Herzschrittmachertherapie + Elektrophysiologie (2024)
-
Dosage of the pseudoautosomal gene SLC25A6 is implicated in QTc interval duration
Scientific Reports (2023)
-
New drug discovery of cardiac anti-arrhythmic drugs: insights in animal models
Scientific Reports (2023)
-
Computational analysis of arrhythmogenesis in KCNH2 T618I mutation-associated short QT syndrome and the pharmacological effects of quinidine and sotalol
npj Systems Biology and Applications (2022)
-
Genetic alteration of human MYH6 is mimicked by SARS-CoV-2 polyprotein: mapping viral variants of cardiac interest
Cell Death Discovery (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
