
Abstract
The variant c.926C > T (p.T309I) in KCNQ1 gene was identified in 10 putatively unrelated Czech families with long QT syndrome (LQTS). Mutation carriers (24 heterozygous individuals) were more symptomatic compared to their non-affected relatives (17 individuals). The carriers showed a mild LQTS phenotype including a longer QTc interval at rest (466 ± 24 ms vs. 418 ± 20 ms) and after exercise (508 ± 32 ms vs. 417 ± 24 ms), 4 syncopes and 2 aborted cardiac arrests. The same haplotype associated with the c.926C > T variant was identified in all probands. Using the whole cell patch clamp technique and confocal microscopy, a complete loss of channel function was revealed in the homozygous setting, caused by an impaired channel trafficking. Dominant negativity with preserved reactivity to β-adrenergic stimulation was apparent in the heterozygous setting. In simulations on a human ventricular cell model, the dysfunction resulted in delayed afterdepolarizations (DADs) and premature action potentials under β-adrenergic stimulation that could be prevented by a slight inhibition of calcium current. We conclude that the KCNQ1 variant c.926C > T is the first identified LQTS-related founder mutation in Central Europe. The dominant negative channel dysfunction may lead to DADs under β-adrenergic stimulation. Inhibition of calcium current could be possible therapeutic strategy in LQTS1 patients refractory to β-blocker therapy.
Similar content being viewed by others

Introduction
Long QT syndrome (LQTS), the most often diagnosed familial electrical disease of the heart1, is characterised by a prolonged QTc interval and occurrence of polymorphic ventricular tachycardias of the torsades de pointes (TdP) type, degenerating into ventricular fibrillation in extreme cases. It results in syncopes or sudden cardiac deaths. Seventeen genes have been associated with LQTS so far2,3, while three "major" genes (KCNQ1, KCNH2, SCN5A) contribute to 75% of clinically diagnosed LQTS4,5. As confirmed recently, a substantial number of the other "minor" genes show only limited or disputed association with LQTS6.
Identification of the same mutation in apparently unrelated LQTS patients in a particular geographic region suggests derivation of the mutation from a common ancestor (a so-called “founder”). Several founder mutations in the KCNQ1 gene associated with LQTS have been reported7,8,9,10,11,12,13,14,15,16,17,18,19. Founder populations have proven to be useful for studying genotype–phenotype correlations, which are important for patients´ management as prevalence of the disease can be increased in some geographical regions20.
This study is a complex analysis of the c.926C > T variant (p.Thr309Ile or T309I), the first founder mutation in the KCNQ1 gene in Central Europe. Based on the acquired functional data, the underlying arrhythmogenic mechanism and possible alternative therapeutic way of its prevention were predicted by in silico modelling in a human ventricular cell model.
Methods
Clinical diagnostics
The study conformed to the principles outlined in the Declaration of Helsinki and was approved by the Multicenter Ethical Committee, University Hospital Brno (Brno, Czech Republic). All participants signed a written informed consent form. In the case of participants under the age of 18 years, the written informed consent was obtained from a parent and/or legal guardian.
LQTS diagnosis was established according to ESC Guidelines21. All individuals underwent clinical examination and bicycle ergometry. The initial stress was set to 0.5 W/kg, and increased by 0.5 W/kg every three minutes to achieve a heart rate higher than the submaximal value with respect to age and sex. A 12-lead ECG with Mason-Likar modification was employed. QT and RR intervals were measured manually (the end of QT interval was established using the threshold method); the Bazett’s correction formula was used. Detailed description is provided in the Supplementary Methods.
Genetic analysis
Molecular analysis of LQTS-associated genes was performed according to current practises for molecular genetics diagnostics: multiplex PCR/SSCP analysis of 3 LQTS major genes (KCNQ1, KCNH2 and SCN5A), Sanger sequencing on ABI 3100 Genetic Analyser (Applied Biosystems, Foster City, CA, USA; genes KCNQ1, KCNH2 and SCN5A), and massive parallel sequencing MPS on GS Junior (Roche, Basel, Switzerland; genes KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2) and, since 2016, on MiSeq (Illumina, San Diego, CA, USA; genes KCNQ1, KCNH2, SCN5A, AKAP9, CACNA1C, CALM1, CALM2, CAV3, KCNE1, KCNE2, KCNJ5, SCN4B, SNTA1). Genetic counselling and testing of first-degree relatives were offered to patients at risk.
The functional impact of the T309I variant was predicted with various in silico tools (SIFT, Provean22, MutationTaster, FATHMM23, and PMUT; Suppl. Tab. S1)24. Its conservation was measured with LRT and MutationAssessor. Allele frequency was determined from online databases ExAC25 and GnomAD.
For the haplotype analysis, 9 STR (short tandem repeats) markers spanning the ~ 11.9-Mb region of chromosome 11 (including the KCNQ1 gene) were chosen from the UCSC Genome Browser (Suppl. Tab. S2). Multiplex PCR with fluorescently labelled primers and fragment analysis with capillary electrophoresis were performed on SeqStudio Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The haplotype linked to the mutation was identified by studying segregation in families. In the probands, SNP (single nucleotide polymorphism) marker analysis followed; 6219 SNPs on the p-arm of chromosome 11 were analysed. Population allele frequency analysis was performed after identifying a common STR allele in the marker D11S4088 in all mutation carriers. The control group was formed by 52 unrelated patients examined at the Department of Clinical Genetics, Faculty Hospital Brno, with a signed written informed consent from each patient agreeing that their DNA samples could be used for clinical research. STR alleles (104) were analysed with capillary electrophoresis. Detailed description is provided in the Supplementary Methods.
Functional analysis
Plasmids containing wild-type (WT) human KCNQ1 in a pIRES2-eGFP vector, KCNE1 in a pKB-CMV vector, and Yotiao in a pGW1 vector were isolated from Escherichia coli using the endotoxin-free QIAprep Spin Miniprep Kit (Qiagen, Hilden, Germany). The mutation c.926C > T in the human KCNQ1 (p.T309I) was generated by site-directed mutagenesis using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, Cedar Creek, TX, USA). Detailed description is provided in the Supplementary Methods.
TransFast Transfection Reagent (Promega, Madison, WI, USA) was used for transfection of the plasmids KCNQ1, KCNE1 and Yotiao (the molar ratio 1:2:4, total amount of KCNQ1 DNA 1 µg, ratio of DNA to transfection agent 1:1.5) into Chinese hamster ovary (CHO) cells cultured at 37 °C / 5% CO2 in the Ham’s F-12 medium supplemented with 10% foetal calf serum and 0.005% gentamycin. KCNQ1 was transfected in one of three ways: 1) WT variant alone (1 µg; WT); 2) T309I variant alone (1 µg; T309I); 3) both the WT and T309I variants cotransfected in the ratio 1:1 (0.5 µg of WT KCNQ1 and 0.5 µg of the T309I variant; WT/T309I) to mimic the heterozygous state in the mutation carriers.
Biophysical analysis was performed ~ 24 h after the transfection by the whole cell patch clamp technique at 37°C. The resistance of filled glass electrodes was < 2.5 MΩ the series resistance was compensated up to 60%. Tyrode solution of the following composition was used (in mmol/L): NaCl 132, KCl 4.8, CaCl2 2.0, MgCl2 1.2, HEPES 10, glucose 5 (pH 7.4, NaOH). The patch electrode filling solution contained (in mmol/L): K-aspartate 110, K2ATP 5, CaCl2 1, MgCl2 1, EGTA 11, HEPES 10 (pH 7.3, KOH). To simulate β-adrenergic stimulation, the pipette solution was supplemented with cyclic adenosine monophosphate (cAMP, 200 µmol/L) and okadaic acid (OA, 0.2 µmol/L). The junction potential was + 15 mV.
Considering missing proportionality between the measured current and estimated cell membrane capacitance, conversion of the magnitude of the current to the current density was avoided in this study, as recently recommended by Kula et al.26. The average cell membrane capacitance was comparable in cells expressing the wild type (WT), T309I, and WT/T309I IKs channels (13.8 ± 1.7, 13.3 ± 2.0, and 13.5 ± 1.8 pF, respectively; P > 0.05).
For the fluorescence image acquisition, a confocal laser scanning microscope Leica TCS SP8 X (Leica microsystems, Wetzlar, Germany) was used. WT and T309I human KCNQ1 tagged with GFP at the 3′-terminus, or WT-KCNQ1 without GFP in a pBK-CMV vector, as well as WT human KCNE1 in a pBK-CMV vector were transfected into CHO cells seeded on glass bottom dishes (Cellvis, Mountain View, CA, USA) coated with fibronectin ~ 48 h before the evaluation (KCNQ1 and KCNE1 in the molar ratio 1:2). In some experiments, WT with no GFP was cotransfected with the GFP-tagged T309I-KCNQ1 and KCNE1, and the cell membrane was stained (CellMask Orange Plasma Membrane Stain; Invitrogen, Carlsbad, CA, USA).
Detailed description is provided in the Supplementary Methods. The chemicals were purchased from Sigma-Aldrich (Prague, Czech Republic) unless otherwise indicated.
Mathematical modelling
A previously published model of human ventricular subepicardial myocyte27 was modified and adapted to changes accompanying β-adrenergic stimulation induced by 1 µM isoproterenol (Suppl. Fig. S1, Suppl. Tab. S3; for validation of the new model, see Suppl. Fig. S3-S5). The measured gating properties of WT and WT/T309I IKs channels were incorporated into the model (Suppl. Fig. S6). Detailed description is provided in the Supplementary Methods. The Matlab code of the control model can be downloaded at https://www.it.cas.cz/en/d3/l033/.
Statistical analysis
The data are mostly presented by the arithmetic mean (± SD from n patients, or ± SEM from n cells; Origin, version 8.5.1; OriginLab Corporation). To determine the statistical significance of the differences, paired/unpaired t-tests and one-way/repeated measures ANOVA with the Bonferroni post-test were performed using the software GraphPad Prism, version 6.05 (GraphPad Software, Inc.). The same software was used for curve fitting. If the difference between the arithmetic and geometric means was > 10% (considering a recent study by Kula et al.28), the geometric mean x/ geometric SE and the non-parametric Mann–Whitney test were used (Figs. 2B, 3B). P < 0.05 was considered statistically significant.
To compare the statistical significance of differences in relative fluorescence intensity in Fig. 4 (where several data samples did not show the normal distribution according to the Shapiro–Wilk test; the mean values are represented by the geometric mean ± 95% confidence interval), either the Friedman test or the Kruskal–Wallis test (both with the Dunn´s multiple comparison) were used, the first one in the case of paired data (comparison within individual groups), the latter in the case of unpaired data (comparison among various groups).
Following software was used to prepare the figures: Figs. 1A and 4B,D,F—GraphPad Prism, version 6.05 (GraphPad Software, Inc.); Fig. 1B,C,D—Inkscape, version 0.92.2 (Inkscape Project; https://inkscape.org/cs/about/branding/; the map in Fig. 1B, lower panel, was generated using the software QGIS, version 3.10, with data downloaded from https://www.arcdata.cz/produkty/geograficka-data/arccr-500); Figs. 2, 3 and 4A,C,E (lower panels, and 5—Origin, version 8.5.1; OriginLab Corporation); Fig. 4A,C,E (upper panels)—LAS X, version 3.5.2.18963 (Leica Microsystems CMS GmbH).

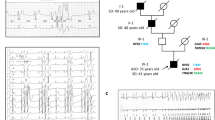
Clinical and genetic characterization of the T309I variant. (A) QTc interval length in the heterozygous T309I carriers (T309I +) and their unaffected relatives (T309I-), both at rest and in the 4th minute of exercise test recovery; ***statistical significance at P < 0.001; the graph was prepared using the software GraphPad Prism, version 6.05. (B) Scheme of T309I-Kv7.1 subunit (upper panel) and birthplaces of the oldest T309I carriers in every family (lower panel; the map was generated using the software QGIS, version 3.10, with data downloaded from https://www.arcdata.cz/produkty/geograficka-data/arccr-500). In the 2 families located in the central part of the country, some earlier ancestors from the eastern part could be identified. Nevertheless, none of them has been living at the time of the study and biological material was not available for molecular analysis. Thus, we can only speculate, whether these ancestors coming from the east really carried the variant, even if it is truly anticipated. (C) Family pedigrees; affected individuals in red, probands indicated with arrows. (D) Haplotype analysis of the KCNQ1 gene and surrounding regions (analysed STR markers shown in relative distances). The region shared by all affected individuals is framed by bold lines. Families with the same detected haplotype are divided into 3 subgroups (differentiated by colours); presumable ancestral haplotype is shown in blue. The uncoloured parts are unique in every single family; small crosses mark where crossing-overs occurred. Parts B, C, and D were prepared using the software Inkscape, version 0.92.2.

Biophysical impact of T309I mutation. (A) Representative records in wild type (WT), T309I, and WT/T309I channels (KCNE1 subunit cotransfected); arrows point to the tail current. (B) Average voltage dependence of tail current activation (n = 20, 9, and 16 in WT, T309I, and WT/T309I channels, respectively); the half-maximal activation voltage V1/2 = 8.3 ± 2.2 mV in WT channels and 15.6 ± 2.1 mV in WT/T309I channels; P < 0.05). (C) Activation experimental protocol; the dashed lines—steps used for stimulation only in T309I channels. (D) Representative trace from a non-transfected CHO cell. (E) Average voltage dependence of steady-state activation. (F) Average voltage dependence of the time constant of activation (τact) and time constant of deactivation (τdeact; n = 11 and 9 in WT and WT/T309I channels, respectively); inset: deactivation experimental protocol; *, ** and ***statistical significance between WT and WT/T309I channels at P < 0.05, 0.01 and 0.001, respectively. The stimulation frequency was 0.08 Hz. All graphs were prepared using the software Origin, version 8.5.1.

Accumulation of WT and WT/T309I currents (KCNE1 subunit cotransfected). (A) Average relative rate-dependent accumulation of the currents at interpulse intervals between 75 and 800 ms (n = 10 and 8 in WT and WT/T309I channels, respectively); inset: experimental protocol applied after a 30-s pause; i.i.—interpulse interval. (B) Average absolute currents at the 1st and 100th pulse of the accumulation protocol with the interpulse interval of 100 ms. (C) Experimental protocol (the stimulation frequency 0.1 Hz), and representative traces of WT and WT/T309I currents under simulated β-adrenergic stimulation (cAMP and OA in the pipette solution); red trace—time 0, control (no cAMP/OA stimulation); green trace—time 120 s, steady-state β-adrenergic stimulation; black traces—time 10 to 110 s in 10-s intervals, development of β-adrenergic response. (D) Time course of the relative increase of the current in presence of cAMP and OA (+ cAMP + OA) in WT and WT/T309I channels (n = 14 and 8; n = 9 in WT without cAMP and OA). (E) Relative steady-state increase of the current in + cAMP + OA; *, ** and ***statistical significance at P < 0.05, 0.01 and 0.001, respectively. All graphs were prepared using the software Origin, version 8.5.1.

Subcellular localization of WT- and T309I-Kv7.1 subunits expressed in CHO cells (KCNE1 subunit cotransfected). (A) Confocal microscopic images of representative cells expressing WT-GFP (i.e. tagged with GFP at the C-terminal end), T309I-GFP, or both WT-GFP and T309I-GFP subunits (upper panels), and respective relative fluorescence intensity profiles (lower panels) under the blue lines in the upper panels; scale bar 25 µm. (B) The resulting maximal relative fluorescence intensity at the cell membrane (M1 and M2) and its mean value in the cytosol (C) in all investigated cells (n = 20, 20 and 12 in WT-GFP, T309I-GFP and WT-GFP/T309I-GFP subunits, respectively; cells from 4 to 7 transfections used in each variant); ** and ***statistical significance of the difference in fluorescence intensity among M1, M2 and C in the respective group of cells at P < 0.01 and 0.001, respectively. (C,E) Subcellular localization of the T309I-GFP subunits, either expressed alone (C) or co-expressed with non-tagged WT (no GFP; E); the cell membrane was stained with red fluorescent dye. Representative cells (upper panels) and respective relative fluorescence intensity profiles (lower panels) under the blue lines in the upper panels of the GFP signal (green lines) and of the membrane staining signal (red lines; both at the cell cross-sections indicated by the blue lines in the upper panels). (D, F) The resulting maximal relative fluorescence intensities of both GFP and membrane staining fluorescence (green and red dots, respectively) at the cell membrane (M1 and M2) and their mean values in the cytosol (C) in all investigated cells (n = 13 and 20 in D and F, respectively; cells from 3 and 7 transfections used); ***statistical significance of the difference in fluorescence intensity among M1, M2 and C at P < 0.001. Figure 4A,C, E were prepared using the software LAS X, version 3.5.2.18963 (upper panels) and Origin, version 8.5.1 (lower panels), Fig. 4B,D,F using the software GraphPad Prism, version 6.05.
Results
Clinical characteristics of T309I carriers
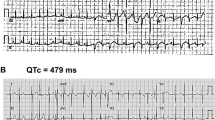
Members of 10 putatively unrelated families with occurrence of the T309I variant were investigated (Table 1). All of the 4 syncopes and 2 aborted cardiac arrests (ACA) which were reported in the investigated T309I carriers appeared during physical exertion (ice hockey, swimming, exercise at school). QTc intervals were significantly longer in T309I carriers compared to their healthy relatives, both at rest (466 ± 24 ms vs. 418 ± 20 ms, P < 0.001) and in the fourth minute of exercise test recovery (508 ± 32 ms vs. 417 ± 24 ms, P < 0.001; Fig. 1A). The QTc prolongation after exercise was significant in T309I carriers (P < 0.001). Representative ECG traces at rest and during recovery in symptomatic and asymptomatic T309I carriers are shown in Suppl. Fig. S7. In children under 16 years, the available QTc values were similar to the values in the adult population of the respective patient group (Table 1). No significant changes were apparent in PQ interval and QRS complex (Suppl. Fig. S8). Cardioverter defibrillators were implanted in the 2 ACA cases. In the other T309I carriers, β-blocker therapy (preferably with nadolol) has been administered and no syncope recurrences have been observed since.
Genetic characteristics of families with T309I variant
The same substitution c.926C > T (NM_000218.2) in the KCNQ1 gene (Suppl. Fig. S9) resulting in T309I amino acid change in the P-loop of Kv7.1 protein (Fig. 1B, upper panel) was identified in 10 unrelated probands, and in 30 carriers in total (Fig. 1C; heterozygous carriers in all cases). The birthplaces of the oldest known T309I carrier in every family are shown in Fig. 1B, lower panel.
The in silico predictive tools which were used suggested a damaging or disease-causing effect of T309I variant and evolutionary analysis showed high conservation of threonine at this position across species (Suppl. Fig. S9). The T309I variant was found neither in the ExAC nor in the GnomAD online databases.
The same haplotype was identified by STR analysis in the region nearest to the mutation spot in all T309I carriers (Fig. 1D). The same allele was detected in 100 SNPs surrounding the variant in all probands with SNP analysis, which also delimited the maximal size of the area shared by all affected individuals to 658,407 bp. This area contained the whole KCNQ1 gene.
STR and SNP analysis of markers lying more distant from the KCNQ1 gene identified 3 subgroups of families sharing the same haplotype spanning longer chromosome regions (Fig. 1D). SNP analysis also identified possible crossing-over spots (crosses in Fig. 1D). The maximum size of the area shared by two families (8 and 10) was delimited with 1847 SNPs and measured 12,633,501 bp. Genealogical analysis also revealed that the oldest carriers of T309I variant in these two families came from two neighbouring villages.
In the marker D11S4088 (the closest one to the KCNQ1 gene), the same fragment with a length of 211 bp segregating with T309I variant was detected in all families. This marker was highly polymorphic in a control population; the fragment lengths varied between 203 and 253 bp. The 211-bp fragment was detected in 13 out of 104 chromosomes, resulting in allele frequency of 0.125. The probability that 10 unrelated people would have this allele is negligible (9.5 × 10–7).
Impact of T309I variant on IKs channel function
All functional data were measured in the presence of KCNE1 subunits. If not otherwise stated, the stimulation frequency was 0.08 Hz. Considering the study by Kula et al.26, conversion of the magnitude of the measured current to the current density was avoided (for details, see Methods).
The homozygous T309I channels resulted in a complete absence of IKs (Fig. 2A,B), even if stimulated up to + 140 mV (for the experimental protocol, see Fig. 2C). The membrane current responses in non-transfected CHO cells did not differ from those in homozygous T309I channels (Fig. 2D).
In the heterozygous WT/T309I channels, IKs amplitude was significantly decreased compared to WT channels (Fig. 2A,B). The dysfunction showed characteristics of dominant negativity—WT/T309I current was supressed by 82 to 55% at voltages between -30 and + 20 mV (i.e. at voltages relevant for the cardiac action potential). The voltage dependence of steady-state activation was significantly shifted to the right in WT/T309I channels (Fig. 2E; the half-maximal activation voltage V1/2 = 8.3 ± 2.2 mV in WT channels and 15.6 ± 2.1 mV in WT/T309I channels, P < 0.05); the slope factors k remained unaltered (15.7 ± 0.7 and 17.3 ± 0.8). The time course of activation was significantly decelerated in WT/T309I channels at voltages between 0 and + 50 mV (Fig. 2F; the time constant of activation τact at + 20 mV: 1740 ± 110 ms in WT channels and 2254 ± 240 ms in WT/T309I channels, P < 0.05). No significant changes of the time constant of deactivation (τdeact) were observed (Fig. 2F; 220.5 ± 26.9 ms in WT channels and 253.6 ± 33.8 ms in WT/T309I channels at − 80 mV; the experimental protocol shown in the inset of Fig. 2F).
Subsequently, sets of hundred 200-ms pulses from -80 to + 20 mV were applied after a 30-s pause (inset in Fig. 3A). IKs amplitude was not significantly different during the 1st and 100th pulse during stimulation with the interpulse interval of 800 ms (Fig. 3A). At shorter interpulse intervals, IKs accumulated in both WT and WT/T309I channels, up to a similar extent in the relative scale (Fig. 3A; for data in absolute values at the interpulse interval of 100 ms, see Fig. 3B). Accumulation of IKs during simulated β-adrenergic stimulation (cAMP and OA in the pipette solution) was preserved in WT/T309I channels, being similar in WT and WT/T309I channels in the relative scale (reaching relative steady-state increase of the tail current by 36.2% in WT/T309I channels on average, vs. 35.6% in WT channels; Fig. 3C–E). As demonstrated in Fig. 3B,C, the preserved reactivity of WT/T309I channels to both the high-rate and β-adrenergic stimulations was not sufficient to reach adequate IKs magnitude (not reaching even the level of the WT current before start of the stimulations).
Subcellular localization of IKschannels with T309I variant
Since IKs was absent in the homozygous T309I channels (Fig. 2A,B), the subcellular localization of WT and T309I subunits (in the presence of KCNE1) was studied. WT-GFP channels showed clear fluorescence preferentially localized at the cell membrane (Fig. 4A,B, left panels; P < 0.001). In contrast, T309I-GFP channels seemed to be retained inside of the cell (Fig. 4A,B, middle panels). To prove this hypothesis, experiments with cells expressing T309I-GFP channels were repeated in a new set of cells, with the cell membrane marked by a red fluorescent dye. GFP fluorescence tagged with T309I subunits (green line/dots) was detected only inside of the cells, i.e. not in the cell membrane (red line/dots; Fig. 4C,D).
When WT-GFP and T309I-GFP subunits were co-transfected, the GFP fluorescence signal was present both on the cell membrane and inside the cells (Fig. 4A,B, right panels). It was significantly more intense on the cell membrane (P < 0.001). The relative intensity of the fluorescence signal at cytosol was significantly different in all tested transfection variants (Fig. 4B; P < 0.001 in WT vs. T309I, P < 0.01 in WT/T309I vs. T309I, and P < 0.05 in WT vs. WT/T309I). This suggests that trafficking is impaired in the WT/T309I channels; however, contribution of T309I subunits to formation of the cell membrane channels is likely. To prove this hypothesis, channels composed of WT subunits without GFP and T309I subunits tagged with GFP (WT/T309I-GFP) were prepared, and the cell membrane was marked by red fluorescent dye. The fluorescence intensity profile peaks of the cell membrane staining (red line/dots) and of the GFP fluorescence tagged with T309I subunits (green line/dots) overlapped (Fig. 4E,F). This implies that T309I subunits contributed to the formation of IKs channels localized in the cell membrane when co-expressed with the WT subunits.
Functional impact of T309I variant in human cardiac cell model
At slow steady-state stimulation (cycle length, CL, 1000 ms), the action potential duration at 90%-repolarization (APD90) was only slightly prolonged in the model with WT/T309I channels in control conditions (by 3.8% vs. the WT model; Fig. 5A, left panel). This agrees with the well-known limited impact of IKs on action potential configuration at low stimulation rates29,30,31. Under β-adrenergic stimulation (Fig. 5A, right panel), APD90 decreased by 6.3% in the WT model (as usual32) but increased by 1.6% in the WT/T309I model. This resulted in substantially longer APD90 in the WT/T309I vs. WT model at β-adrenergic stimulation (by 12.5%, or, during the first action potential after start of β-adrenergic stimulation, even by 13.9%).

Impact of WT/T309I dysfunctional channels on action potential (AP) configuration in a human ventricular cell model. (A) APs (upper panels) and IKs (lower panels) at steady-state stimulation at the cycle length (CL) 1000 ms in control conditions and during β-adrenergic stimulation; APD90—AP duration at 90%-repolarization. (B) APs (upper panels) and IKs (lower panels) at extrastimulation within the vulnerable period of AP in the WT/T309I model during β-adrenergic stimulation—CL suddenly shortened from 1000 to 268 ms. (C) Intracellular Ca2+ concentration at the end of stimulation cycle ([Ca2+]i,end; left upper panel), APs (right upper and middle panels) and intracellular Ca2+ transients ([Ca2+]i; lower panel) after change of CL from 1000 to 500 ms and contemporary start of β-adrenergic stimulation (for sequence of changes, note the time axes in all graphs); arrows—stimuli at CL 500 ms, *delayed afterdepolarization (DAD) or AP activated by DAD. All graphs were prepared using the software Origin, version 8.5.1.
If extrastimulation was applied within the vulnerable period of the first WT/T309I action potential after the start of β-adrenergic stimulation, the first action potential was suppressed and the following one was extremely prolonged in the WT/T309I model (it was longer by 31% in comparison with WT model; Fig. 5B). Irregular suppression of action potentials and action potential alternans followed (not illustrated). To provoke this, CL had to be decreased below 269 ms in the WT/T309I model, and below 238 ms in the WT model.
If CL was shortened to 500 ms and β-adrenergic stimulation was started at the same time (i.e. conditions comparable to the start of physical activity), the prolonged action potential in the WT/T309I model under β-adrenergic stimulation resulted in Ca2+ overload and development of delayed afterdepolarizations (DADs), often eliciting a premature action potential, within 540 s (Fig. 5C).
Discussion
In this study, we performed a detailed description of the c.926C > T-KCNQ1 (p.T309I-Kv7.1) variant, the first identified founder LQTS mutation in Central Europe. The dominant negative dysfunction of T309I IKs channels was observed in the heterozygous setting. Mathematical modelling demonstrated formation of DADs and action potential alternans at short CLs during β-adrenergic stimulation.
Founder effect of T309I variant
The T309I variant was identified almost simultaneously in a Taiwanese family33 and in our earlier report34. No functional data has been available so far.
As the T309I variant was detected in 10 probands in our cohort of 44 LQTS1 families, its prevalence is unusually high in our region25. Majority of these 10 carrier families is geographically clustered (Fig. 1B, lower panel). Such characteristics are typical for mutations derived from the founder effect. Using detailed haplotype analysis of both STR and SNP markers, we confirmed that all affected individuals shared the same haplotype spanning ~ 658 kbp including the complete coding region of the KCNQ1 gene (Fig. 1D). Allelic variability at a greater distance from the mutation spot can be explained by crossing overs in T309I ancestors. We identified three subgroups of haplotypes among the 10 investigated families, which are shared also in distant markers. The maximum length of the shared haplotype between two families (8 and 10) was ~ 12.63 Mb, and was represented by the same allele in the 1847 SNPs analysed (Fig. 1D). Hence, these two families are more closely related, which also correlates with their geographical origin. A close common ancestor could be also predicted in two other subgroups of families with shared haplotypes.
Functional defect in T309I IKs channels
Considering the position of the T309I-Kv7.1 variant in a highly conserved pore helix35 and the replacement of the small polar residue by the bigger non-polar one, serious functional defect may be expected.
IKs was completely missing in the homozygous T309I channels (Fig. 2A,B) due to impaired channel trafficking (Fig. 4A,B), similarly as in the near variant A302V36 and in some other pore-region LQTS1 variants37,38. However, other IKs pore region variants showed unaffected trafficking39,40. This implies that only specific residues within the IKs pore play a role in the process.
In the heterozygous WT/T309I channels, dominant negativity was observed (Fig. 2A,B), as is often found in the pore-region LQTS1 variants36,37,38,39,40,41,42. A rightward shift of the voltage dependence of channel activation (Fig. 2E), which was also detected in some other pore-region LQTS1 variants37,38,40,41,42, suggests that even pore residues can modify the voltage sensitivity of the IKs channel. It has to be noted that variable expression ratio of KCNQ1 and KCNE1 in WT and WT/T309I channel complexes might contribute to the rightward activation shift. In WT/T309I channels, we also observed significantly decelerated activation kinetics (Fig. 2F). No other study analysing functional defects in the pore-region LQTS1 variants that we have found reported data on the activation kinetics; thus, we cannot assume that these variants generally exert an analogical effect. Response to β-adrenergic stimulation was preserved in WT/T309I channels (Fig. 3C–E), comparable to that reported by Spätjens et al.31 under similar conditions. Since the P-loop mutations studied so far showed unaltered β-adrenergic IKs accumulation43, P-loop is not likely involved in this essential regulation of IKs channel function.
Considering the data presented in Fig. 4E,F, a reverse way of cotransfection of WT and T309I subunits (WT tagged with GFP and T309I without GFP) might help to elucidate if a trafficking defect of WT subunits contributes to the observed IKs channel dysfunction described in Fig. 2. Comparing the intracellular GFP signals in Fig. 4B (right panel) and 4F (being not significantly different, P > 0.05), it seems that most of the subunits retained inside the cell are T309I subunits if both WT and T309I subunits are coexpressed. However, IKs channel is a heterotetrameric channel that is indeed formed by various combinations of WT and T309I subunits if both are transfected (and if even the mutated T309I subunit is present on the cell membrane, Fig. 4E,F). Taking all this into account, we speculate that a slight intracellular retention of WT subunits might happen if WT and T309I subunits are coexpressed.
Genotype—phenotype correlation in the T309I variant
A mild LQTS phenotype was observed in the heterozygous T309I carriers, similarly to many other founder variants14,44 and also to some other pore region LQTS1 variants36,40,42. Surprisingly, a dominant negative dysfunction was revealed in the heterozygous T309I channels (Figs. 2 and 3). A similar contradiction between the channel dysfunction and clinical phenotype was described in the near A302V variant36. The mild T309I phenotype likely results from the preserved reactivity of the heterozygous mutated channels to high-rate and β-adrenergic stimulations (Fig. 3). This may partially compensate the dominant negative dysfunction at situations when IKs is essential for proper cardiac cell repolarization, namely during exercise or stress.
As predicted by in silico modelling on a human ventricular cell model, stimulation with shortened CL during β-adrenergic stimulation may trigger proarrhythmic activity in patients with WT/T309I IKs channels (Fig. 5B,C). DADs were observed, which even promoted premature action potentials. This observation may seem unexpected considering previously published studies which typically demonstrated occurrence of EADs in LQTS1 under β-adrenergic stimulation, including studies performed on patient-specific derived cardiomyocytes45,46,47 and on animal models48,49,50. According to other studies, EADs as an arrhythmogenic mechanism seem to be preferentially connected to LQTS type 2 which is based on mutations in the KCNH2 gene (the rapid delayed rectifier current IKr is affected) and in which the TdP arrhythmia is usually pause-dependent51,52. In contrast, pause-independent TdP has been observed in LQTS152. The fast heart rate preceding TdP (absence of pause) in LQTS1 is compatible with the mechanism of genesis of DADs53. Genesis of DADs at fast rates during β-adrenergic stimulation accompanied by IKs dysfunction (both pharmacologically-induced) was demonstrated in canine cardiac cells and tissue54,55, similarly as we have seen it in our human ventricular cell model with WT/T309I channels (Fig. 5C). The above listed contradictory data on LQTS1 arrhythmogenic mechanism suggest necessity of a further detailed study within this field.
In our model, the development of DADs was caused by an increase of sodium-calcium exchange current and calcium-activated non-specific current, due to a premature release of Ca2+ from the sarcoplasmic reticulum at intracellular Ca2+ overload, which in turn was caused by prolonged action potentials at the fast heart rate (for details, see Suppl. Fig. S10). This proarrhythmic events predicted by in silico modelling may explain the occurrence of arrhythmias during physical exertion in T309I carriers (see “Clinical characteristics of T309 I carriers” ).
The essential proarrhythmogenic role of Ca2+ overload suggested by in silico modelling should be considered in clinical practise in the future. It implies that, beside the standard treatment with β-blocking agents, additional or modified treatment may be beneficial in some LQTS1 patients, namely those who insufficiently respond to the standard β-blocking therapy. As demonstrated in Suppl. Fig. S11, a slight (5%) inhibition of the cardiac calcium current ICa is able to prevent development of DADs in our WT/T309I model during the tested period of 600 ms with a negligible effect on the magnitude of the intracellular Ca2+ transient (thus, likely not significantly impairing cardiac contractility). It implies that possible new treatment strategy of low doses of verapamil might be relevant in some LQTS1 patients, similarly as in patients with refractory catecholaminergic polymorphic ventricular tachycardia56. In patients with compromised cardiac contractility (in whom even a mild ICa inhibition would be contraindicated), a decrease of the late sodium current (INa,late) might be beneficial57,58.
Limitations of the study
Use of the human ionic channels heterogeneously expressed in a cell line limits possibilities of direct experimental investigation of impact of a particular mutation on complex cardiac cell electrophysiology. Therefore, it might be beneficial to prove the data in another model, for example in isolated cardiomyocytes of the affected patient/s (which is however very problematic, if not impossible, from ethical reasons), or in patient-specific cardiomyocytes derived from the induced pluripotent stem cells. Likewise, such more complex in vitro models might be helpful to better validate the human ventricular cell in silico model. In addition, introduction of the experimental data presented here into a mathematical model of electromechanical activity of the left ventricle or even of the whole heart might reveal the effects of IKs suppression on propensity of the heart to development of reentry-based arrhythmias and on ECG under control conditions and at adrenergic stimulation. In coexpression of WT and T309I subunits, a slight trafficking defect of WT subunits might contribute to the observed IKs channel dysfunction. Unfortunately, a direct proof of this suspicion is missing in the study.
Conclusions
The c.926C > T-KCNQ1 pore variant (p.T309I-Kv7.1) is the first LQTS-related founder mutation in Central Europe. Its dominant negative effect on IKs channel function is in contrast with the mild phenotype. As predicted by in silico modelling, the dysfunction may result in genesis of proarrhythmic DADs and action potential alternans at fast rates during β-adrenergic stimulation. Elimination of DADs by ICa modulation might be a possible new treatment strategy in some LQTS1 patients.
Data availability
The relevant data are available from the authors upon reasonable request.
Change history
29 April 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41598-021-89234-z
References
Schwartz, P. J. et al. Prevalence of the congenital long QT syndrome. Circulation 120, 1761–1767 (2009).
Schwartz, P. J., Crotti, L. & Insolia, R. Long-QT syndrome: from genetics to management. Circ. Arrhythm. Electrophysiol. 5, 868–877 (2012).
Crotti, L. et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 127, 1009–1017 (2013).
Tester, D. J., Will, M. L., Haglund, C. M. & Ackerman, M. J. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2, 507–517 (2005).
Kapplinger, J. D. et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION® long QT syndrome genetic test. Heart Rhythm. 6, 1297–1303 (2009).
Adler, A. et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 141, 412–428 (2020).
Russell, M. W., Dick, M., Collins, F. S. & Brody, L. C. KVLQT1 mutations in three families with familial or sporadic long QT syndrome. Hum. Mol. Genet. 5, 1319–1324 (1996).
Piippo, K. et al. A founder mutation of the potassium channel KCNQ1 in long QT syndrome: Implications for estimation of disease prevalence and molecular diagnostics. J. Am. Coll. Cardiol. 37, 562–568 (2001).
Brink, P. A. et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation 112, 2602–2610 (2005).
Fodstad, H. et al. Molecular characterization of two founder mutations causing long QT syndrome and identification of compound heterozygous patients. Ann. Med. 38, 294–304 (2006).
Arbour, L. et al. A KCNQ1 V205M missense mutation causes a high rate of long QT syndrome in a First Nations community of northern British Columbia: a community-based approach to understanding the impact. Genet. Med. 10, 545–550 (2008).
Bhuiyan, Z. A. et al. Clinical and genetic analysis of long QT syndrome in children from six families in Saudi Arabia: Are they different?. Pediatr. Cardiol. 30, 490–501 (2009).
Marjamaa, A. et al. High prevalence of four long QT syndrome founder mutations in the Finnish population. Ann. Med. 41, 234–240 (2009).
Winbo, A., Diamant, U.-B., Stattin, E.-L., Jensen, S. M. & Rydberg, A. Low incidence of sudden cardiac death in a Swedish Y111C type 1 long-QT syndrome population. Circ. Cardiovasc. Genet. 2, 558–564 (2009).
Eldstrom, J. et al. Mechanistic basis for LQT1 caused by S3 mutations in the KCNQ1 subunit of IKs. J. Gen. Physiol. 135, 433–448 (2010).
Gray, C. et al. Expression of a common LQT1 mutation in five apparently unrelated families in a regional inherited arrhythmia clinic. J. Cardiovasc. Electrophysiol. 21, 296–300 (2010).
Hofman, N. et al. Recurrent and founder mutations in the Netherlands: the long-QT syndrome. Neth. Heart J. 19, 10–16 (2011).
Stattin, E.-L. et al. Founder mutations characterise the mutation panorama in 200 Swedish index cases referred for Long QT syndrome genetic testing. BMC Cardiovasc. Disord. 12, 95. https://doi.org/10.1186/1471-2261-12-95 (2012).
Zafari, Z. et al. Identification and characterization of a novel recessive KCNQ1 mutation associated with Romano-Ward Long-QT syndrome in two Iranian families. J. Electrocardiol. 50, 912–918 (2017).
Offerhaus, J. A., Bezzina, C. R. & Wilde, A. A. M. Epidemiology of inherited arrhythmias. Nat. Rev. Cardiol. 17, 205–215 (2020).
Priori, S. G. et al. ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 36, 2793–2867 (2015).
Choi, Y. & Chan, A. P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 (2015).
Shihab, H. A. et al. Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum. Genomics. 8, 11. https://doi.org/10.1186/1479-7364-8-11 (2014).
López-Ferrando, V., Gazzo, A., de la Cruz, X., Orozco, M. & Gelpí, J. L. PMut: a web-based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Res. 45, W222–W228 (2017).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Kula, R., Bébarová, M., Matejovič, P., Šimurda, J. & Pásek, M. Current density as routine parameter for description of ionic membrane current: is it always the best option? Prog. Biophys. Mol. Biol. 157, 24–32 (2020).
Hrabcová, D., Pásek, M., Šimurda, J. & Christé, G. Effect of ion concentration changes in the limited extracellular spaces on sarcolemmal ion transport and Ca2+ turnover in a model of human ventricular cardiomyocyte. Int. J. Mol. Sci. 14, 24271–24292 (2013).
Kula, R., Bébarová, M., Matejovič, P., Šimurda, J. & Pásek, M. Distribution of data in cellular electrophysiology: Is it always normal? Prog. Biophys. Mol. Biol. 157, 11–17 (2020).
Jost, N. et al. Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation 112, 1392–1399 (2005).
Roden, D. M. & Yang, T. Protecting the Heart Against Arrhythmias: Potassium Current Physiology and Repolarization Reserve. Circulation 112, 1376–1378 (2005).
Spätjens, R. L. H. M. G. et al. Long-QT mutation p.K557E-Kv7.1: dominant-negative suppression of IKs, but preserved cAMP-dependent up-regulation. Cardiovasc. Res. 104, 216–225 (2014).
Szentandrássy, N. et al. Role of action potential configuration and the contribution of Ca 2+ and K + currents to isoprenaline-induced changes in canine ventricular cells: Isoprenaline in canine heart. Br. J. Pharmacol. 167, 599–611 (2012).
Ko, Y. L. et al. Linkage and mutation analysis in two Taiwanese families with long QT syndrome. J. Formos. Med. Assoc. Taiwan Yi. Zhi. 100, 767–771 (2001).
Andrsova, I. et al. Clinical characteristics of 30 Czech families with long QT syndrome and KCNQ1 and KCNH2 gene mutations: importance of exercise testing. J. Electrocardiol. 45, 746–751 (2012).
Bohnen, M. S. et al. Molecular pathophysiology of congenital long QT syndrome. Physiol. Rev. 97, 89–134 (2017).
Steffensen, A. B. et al. IKs gain- and loss-of-function in early-onset lone atrial fibrillation. J. Cardiovasc. Electrophysiol. 26, 715–723 (2015).
Mousavi, N. A., Gharaie, S. & Jeong, K. H. Cellular mechanisms of mutations in Kv7.1: auditory functions in Jervell and Lange-Nielsen syndrome vs Romano-Ward syndrome. Front. Cell. Neurosci. 9, 32. https://doi.org/10.3389/fncel.2015.00032 (2015).
Burgess, D. E. et al. High-risk long QT syndrome mutations in the Kv71 (KCNQ1) pore disrupt the molecular basis for rapid K+ Permeation. Biochemistry (Mosc). 51, 9076–9085 (2012).
Ikrar, T. et al. A double-point mutation in the selectivity filter site of the KCNQ1 potassium channel results in a severe phenotype, LQT1, of long QT syndrome. J. Cardiovasc. Electrophysiol. 19, 541–549 (2008).
Bianchi, L. et al. Mechanisms of I(Ks) suppression in LQT1 mutants. Am. J. Physiol. Heart Circ. Physiol. 279, H3003–H3011 (2000).
Li, W. et al. The G314S KCNQ1 mutation exerts a dominant-negative effect on expression of KCNQ1 channels in oocytes. Biochem. Biophys. Res. Commun. 383, 206–209 (2009).
Aidery, P. et al. Impaired ion channel function related to a common KCNQ1 mutation: implications for risk stratification in long QT syndrome 1. Gene 511, 26–33 (2012).
Policarová, M., Novotný, T. & Bébarová, M. Impaired Adrenergic/Protein Kinase A Response of Slow Delayed Rectifier Potassium Channels as a Long QT Syndrome Motif: Importance and Unknowns. Can. J. Cardiol. 35, 511–522 (2019).
Winbo, A. et al. Phenotype, origin and estimated prevalence of a common long QT syndrome mutation: a clinical, genealogical and molecular genetics study including Swedish R518X/KCNQ1 families. BMC Cardiovasc. Disord. 14, 22. https://doi.org/10.1186/1471-2261-14-22 (2014).
Moretti, A. et al. Patient-specific induced pluripotent stem cell models for long-QT syndrome. N. Engl. J. Med. 363, 1397–1409 (2010).
Sogo, T. et al. Electrophysiological properties of iPS cell-derived cardiomyocytes from a patient with long QT syndrome type 1 harboring the novel mutation M437V of KCNQ1. Regen. Ther. 4, 9–17 (2016).
Wang, Z. et al. Pathogenic mechanism and gene correction for LQTS-causing double mutations in KCNQ1 using a pluripotent cell model. Stem Cell Res. 38, 101483. https://doi.org/10.1016/j.scr.2019.101483 (2019).
Liu, G. X. et al. Differential conditions for early after-depolarizations and triggered aktivity in cardiomyocytes derived from transgenic LQT1 and LQT2 rabbits. J. Physiol. 590, 1171–1180 (2012).
Kim, T. Y. et al. Complex excitation dynamics underlie polymorphic ventricular tachycardia in a transgenic rabbit model of long QT syndrome type 1. Heart Rhythm. 12, 220–228 (2015).
ter Bekke, R. M. A. et al. Proarrhythmic proclivity of left-stellate ganglion stimulation in a canine model of drug-induced long-QT syndrome type 1. Int. J. Cardiol. 286, 66–72 (2019).
Liu, J. & Laurita, K. R. The mechanism of pause-induced torsades de pointes in long QT syndrome. J. Cardiovasc. Electrophysiol. 16, 981–987 (2005).
Tan, H. L. et al. Genotype-specific onset of arrhythmias in congenital long-QT syndrome: possible therapy implications. Circulation 114, 2096–2103 (2006).
Marban, E., Robinson, S. W. & Wier, W. G. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J. Clin. Invest. 78, 1185–1192 (1986).
Burashnikov, A. & Antzelevitch, C. Block of I(Ks) does not induce early afterdepolarization activity but promotes beta-adrenergic agonist-induced delayed afterdepolarization activity. J. Cardiovasc. Electrophysiol. 11, 458–465 (2000).
Johnson, D. M. et al. Diastolic spontaneous calcium release from the sarcoplasmic reticulum increases beat-to-beat variability of repolarization in canine ventricular myocytes after β-adrenergic stimulation. Circ. Res. 112, 246–256 (2013).
Swan, H., Laitinen, P., Kontula, K. & Toivonen, L. Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia patients with RyR2 mutations. J. Cardiovasc. Electrophysiol. 16, 162–166 (2005).
Shryock, J. C., Song, Y., Rajamani, S., Antzelevitch, C. & Belardinelli, L. The arrhythmogenic consequences of increasing late INa in the cardiomyocytes. Cardiovasc. Res. 99, 600–611 (2013).
Belardinelli, L. et al. A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J. Pharmacol. Exp. Ther. 344, 23–32 (2013).
Acknowledgements
This work was supported by the Ministry of Health of the Czech Republic, Grant No. 16-30571A. The authors thank prof. Paul G.A. Volders (Maastricht University, Netherlands) and prof. Robert S. Kass (Columbia University, USA) for kindly providing the plasmids, to Mrs. B. Vyoralová for technical assistance, and to Joseph Lennon, BA, PhD for English proofreading.
Author information
Authors and Affiliations
Contributions
I.S.—data acquisition and evaluation (genetic analysis including the haplotype analysis), preparation of Fig. 1, Suppl. Fig. S9, and of Suppl. Tabs. S1 and S2, writing of the manuscript; M.B.—study design, data acquisition and evaluation (whole cell patch clamp), preparation of Figs. 1, 2, 3, 4 and 5, Suppl. Fig. S8, and of Table 1, writing of the manuscript; I.A.—data acquisition and evaluation (clinical analysis in adults), preparation of Table 1; L.C.—data acquisition and evaluation (confocal microscopy), writing of the manuscript; O.Š.—data acquisition and evaluation (whole cell patch clamp, confocal microscopy), preparation of Fig. 4; J.H.—data acquisition and evaluation (site direction mutagenesis and amplification of plasmids), writing of the manuscript; M.P.—data acquisition and evaluation (mathematical modelling), preparation of Fig. 5, Suppl. Figs. S1-S6 and S10-S11, and of Suppl. Tab. S3, writing of the manuscript; P.V.—data acquisition and evaluation (clinical analysis in children); I.V.—data acquisition (genetic analysis); R.G.—data acquisition (genetic analysis); R.N.—data acquisition (haplotype analysis); T.N.—study design, data acquisition and evaluation (clinical analysis in adults), preparation of Suppl. Fig. S7, writing of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Synková, I., Bébarová, M., Andršová, I. et al. Long-QT founder variant T309I-Kv7.1 with dominant negative pattern may predispose delayed afterdepolarizations under β-adrenergic stimulation. Sci Rep 11, 3573 (2021). https://doi.org/10.1038/s41598-021-81670-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-81670-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
