
Abstract
Here we describe three novel TCOF1 mutations found in unrelated patients with Treacher Collins syndrome. These mutations include one deletion, NM_001135243.2:c.2604_2605delAG (p.Gly869Glufs*3), and two substitutions, NM_001135243.2:c.2575C>T (p.Gln859*) and NM_001135243.2:c.4111G>T (p.Glu1371*). These mutations cause shortening of a protein called Treacle in patients with features typical of TCS. Continuous identification of new mutations is important to expand the mutation base, which is helpful in the diagnosis of both patients and their families
Similar content being viewed by others

Treacher Collins syndrome (TCS, OMIM 154500), also called mandibulofacial dysostosis (MFD), was described in 1900 and extensively examined by Franceschetti and Klein in 19491. It is a rare developmental disorder with an incidence rate of ~1 in 50,000 live births. Forty percent of cases are related to family history, whereas the remaining incidences occur due to de novo mutations. The literature describes four clinical TCS subtypes: TCS1 (OMIM 154500) is a result of mutations in the TCOF1 gene (OMIM 606847), TCS2 (OMIM 613717) is caused by mutations in the POLR1D gene (OMIM 613715), TCS3 (OMIM 248390) is caused by POLR1C gene mutations (OMIM 610060), and TCS4 (OMIM 618939) is caused by mutations in the POLR1B gene (OMIM 602000). TCOF1 and POLR1B sequence variants have autosomal dominant inheritance, variants in POLR1C are autosomal recessive, and variants in POLR1D can be either autosomal dominant or autosomal recessive. The TCOF1 gene has been cloned by the Treacher Collins Syndrome Collaborative Group2. It maps to 5q32-q33.1. So et al.3 discovered two additional exons: 6A (231 nt) and 16A (108 nt). Some of the reported transcripts lack exon 19 (114 nt). Dauwerse et al.4 detected mutations in the POLR1C gene, which maps to 6p21.1, whereas POLR1D maps to 13q12.2 in TCS patients. Sanchez et al.5 demonstrated that POLR1B mutations located on 2q14.1 were also found in patients with TCS. TCOF1 gene mutations are responsible for ~86% of TCS cases, POLR1C: 1.2%, POLR1D: 6%, and POLR1B: 1.3%4,5,6. TCOF1 encodes a 144 kDa nuclear phosphoprotein called Treacle, which is composed of 1488 amino acids7.
Mutations in TCOF1 may lead to haploinsufficiency due to the formation of a shortened protein. Treacle contains three domains, of which the central domain is the largest, embodying ten repeats of serine clusters subdivided by stretches rich in the amino acids alanine, proline, lysine, and glutamic acid8. Ciccia et al.9 studied the interaction between the DNA damage response factor (DDR) and TCOF1. This gene plays a regulatory role as a DDR factor and Treacle forms part of the NBS1 protein complex. The N and C termini of Treacle contain nuclear export and import signals, which is evidence of its dynamic localization10. Protein localization is related to the LisH motif, which exists in the N-terminal domain11. Treacle plays an meaningful role in proper rDNA transcription during ribosome biogenesis7 and mitosis12.
More than 200 mutations have been reported in TCOF113,14,15. The majority of these mutations are small deletions that result in a premature termination codon, which produces a truncated protein. Bowman et al.16 identified multiple pathogenic TCOF1 gene rearrangements of different sizes at a rate of 5% in examined unrelated individuals with TCS. Beygo et al.17 used multiplex ligation‐dependent probe amplification to identify a large 3.367 kb deletion eliminating the entire exon 3 in one patient, suggesting that the rate of large deletions was low at <1%. Vincent et al.6 described large 1 Mb and 262 kb mutations resulting in complete gene deletions of TCOF1 and CAMK2A. No correlation was observed between the type of mutation and patient phenotype. The management of patients requires a multidisciplinary treatment team involving pediatricians, otolaryngologists, clinical geneticists, orthodontists, audiologists, and surgeons.
In this study, we presented three novel mutations (two in exon 16 and one in exon 24) of the TCOF1 gene in MFD patients.
The described examinations were conducted according to guidance from an ethics committee and in accordance with the Declaration of Helsinki. Blood from affected individuals was collected after obtaining informed consent from the patients or their legal guardians.
Genetic material from three different, unrelated patients of Caucasian ethnicity was examined. The patients had commonly observed clinical features characteristic of TCS, such as downward-slanting palpebral fissures, mandibular hypoplasia, delayed speech development, and atresia of the external ear canal. Typically, TCS patients do not demonstrate cognitive disability. The phenotypic features of the examined patients are summarized in Table 1.
In all three cases, DNA was isolated from the peripheral blood leukocytes of patients and parents. In addition, blood was collected from the siblings of one patient. PCR reaction of exons 1–27 with adjacent intron fragments was then performed. PCR products were sequenced after the sample purification phase. Using appropriate primers for each exon, samples were analyzed by capillary gel electrophoresis based on the ABI 310 DNA System. The nomenclature for sequence variants used in our report is in accordance with Human Genome Variation Society (HGVS) guidelines. The National Center for Biotechnology Information (NCBI) database was used for verification of the mutations (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/). The sequencing results were compared with Homo sapiens chromosome 5, referring to the NCBI reference sequence NM_001135243.2.gb transcript variant 4 for TCOF1. The exon position of the mutation, kind on amino acid involved in the change and predicted effect on the protein, were indicated by https://www.mutalyzer.nl/ and confirmed by http://www.mutationtaster.org/. The results were submitted to dbSNP (https://www.ncbi.nlm.nih.gov/snp/).
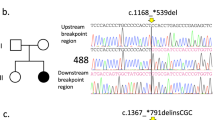
Sequence analysis allowed us to identify three mutations, one frameshift and two nonsense mutations, located in TCOF1 hotspots. In patient 1, we found a heterozygous 2 bp deletion in exon 16 (NM_001135243.2:c.2604_2605delAG). In this case, an in-frame deletion caused the premature termination of translation at amino acid 871 (p.Gly869Glufs*3). This resulted in the formation of a protein product lacking a nuclear localization signal. Direct sequencing of the DNA from the patient’s siblings and parents did not show similar sequences. Compared with a wild-type sequence, the mutation in the proband was considered de novo. Sequence analysis of material from the second patient revealed a novel transition (NM_001135243.2:c.2575C>T) at (p. Gln859*) in exon 16. The predicted effect of this substitution is shortening of the Treacle protein. In the case of individual three, the substitution NM_001135243.2:c.4111G>T was found in exon 24 and resulted in a stop codon at (p.Glu1371*). Neither substitution was found in the parents of the respective probands, indicating de novo mutations. All three recognized sequence variants are heterozygous. The examination results are presented as chromatograms comparing patient and control sequences in Fig. 1.

Sequence analysis of the amplified fragments of the TCOF1 gene: 1, patient with c.2604_2605delAG in exon 16; 2, patient with c.2575C>T in exon 16; 3, patient with c.4111G>T in exon 24; and C, controls. Arrows indicate the location of mutations and stop codons are marked by *.
The majority of TCOF1 mutations are deletions that range in size from 1 to 40 nucleotides, resulting in a truncated protein13,18,19. Most commonly recognized sequence variants are small insertions, splice site variants, and missense and nonsense variants (19–23% of mutations). All three variations detected here were in the coding sequence of the TCOF1 gene. Exons 16 and 24 contain a portion of the coding sequence and encode the central domain of the Treacle protein. The central domain plays a significant role in protein structure, as it contains kinase C and casein kinase 2-phosphorylation sites. Therefore, the identified mutations may affect the abnormal conformation and dysfunction of the Treacle protein. Mutational hotspots in TCOF1 include exons 10, 15, 16, 23, and 24. In this study, we present one deletion, NM_001135243.2:c.2604_2605delAG, and two substitutions, NM_001135243.2:c.2575C>T and NM_001135243.2:c.4111G>T. All three reported mutations are in hotspot regions (exons 16 and 24) and result in a shortened protein. These pathogenic variants are not observed in relatives of the examined probands, which indicates that all are de novo mutations. All genetic alterations identified within our work are presented in Table 2.
The first descriptions of TCS date back to the early twentieth century; however, a full understanding of its genetic origins requires further study. Research has confirmed that in addition to TCOF1, mutations in POLR1C, POLR1D, and POLR1B4,5 can lead to TCS. However, for 8–11% of patients, the pathogenesis of the disorder remains uncertain18. This may indicate the involvement of other genes or other mechanisms leading to TCS. Duan et al.20 presented the hypothesis that oxidative stress may be one of the causes of TCS.
Molecular diagnosis is important in rare disorders and is used in prenatal and postnatal screening. Other disorders, such as Oculoauriculovertebral spectrum, Miller syndrome and Nager syndrome, possess phenotypic features similar to those of TCS, making the diagnosis of TCS difficult. The most effective tool to further explore mutations and genes associated with the origin of TCS would be whole-exome sequencing18.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3086, https://doi.org/10.6084/m9.figshare.hgv.3089, and https://doi.org/10.6084/m9.figshare.hgv.3092.
References
Gorlin R. J., Cohen M. M. & Levin L. S. Syndromes of the Head and Neck (Oxford Univ. Press, 1990).
Treacher Collins Syndrome Collaborative Group. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat. Genet. 12, 130–136 (1996).
So, R. B. et al. Another face of the Treacher Collins syndrome (TCOF1) gene: identification of additional exons. Anotheene 328, 49–57 (2004).
Dauwerse, J. G. et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat. Genet. 43, 20–22 (2011).
Sanchez, E. et al. POLR1B and neural crest cell anomalies in Treacher Collins syndrome type 4. Genet. Med. 22, 547–556 (2020).
Vincent, M. et al. Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genet. Med. 18, 49–56 (2016).
Valdez, B. C., Henning, D., So, R. B., Dixon, J. & Dixon, M. J. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc. Natl Acad. Sci. USA 101, 10709–10714 (2004).
Isaac, C. et al. Characterization of the nucleolar gene product, treacle, in Treacher Collins syndrome. Mol. Biol. Cell 11, 3061–3071 (2000).
Ciccia, A. et al. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl Acad. Sci. USA 111, 18631–18636 (2014).
Winokur, S. T. & Shiang, R. The Treacher Collins syndrome (TCOF1) gene product, treacle, is targeted to the nucleolus by signals in its C-terminus. Hum. Mol. Genet. 7, 1947–1952 (1998).
Wise, C. A. et al. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc. Natl Acad. Sci. USA 94, 3110–3115 (1997).
Sakai, D. & Trainor, P. A. Face off against ROS: Tcof1/Treacle safeguards neuroepithelial cells and progenitor neural crest cells from oxidative stress during craniofacial development. Dev. Growth Differ. 58, 577–585 (2016).
Conte, C. H. et al. Novel mutations of TCOF1 gene in European patients with Treacher Collins syndrome. BMC Med. Genet. 12, 125 (2011).
Marszałek-Kruk, B. A., Wójcicki, P., Śmigiel, R. & Trzeciak, W. H. Novel insertion in exon 5 of the TCOF1 gene in twin sisters with Treacher Collins syndrome. J. Appl. Genet. 53, 279–282 (2012).
Zhang, C. H. et al. Mutation analysis of TCOF1 gene in Chinese Treacher Collins syndrome patients. J. Clin. Lab Anal. 35, e23567 (2021).
Bowman, M. et al. Gross deletions in TCOF1 are a cause of Treacher–Collins–Franceschetti syndrome. Eur. J. Hum. Genet. 20, 769–777 (2012).
Beygo, J. et al. First report of a single exon deletion in TCOF1 causing Treacher Collins Syndrome. Mol. Syndromol. 2, 53–59 (2012).
Fan, X. et al. TCOF1 pathogenic variants identified by whole-exome sequencing in Chinese Treacher Collins syndrome families and hearing rehabilitation effect. Orphanet J. Rare Dis. 14, 178 (2019).
Zeng, H., Xie, M., Li, J., Xie, H. & Lu, X. A novel nonsense mutation in the TCOF1 gene in one Chinese newborn with Treacher Collins syndrome. Int. J. Pediatr. Otorhinolaryngol. 141, 110561 (2021).
Duan, X., Kelsen, S. G., Clarkson, A. B. Jr, Ji, R. & Merali, S. SILAC analysis of oxidative stress-mediated proteins in human pneumocytes: new role for treacle. Proteomics 10, 2165–2174 (2010).
Author information
Authors and Affiliations
Contributions
All contributors have read this manuscript and approved its submission for consideration, for publication as a Data Report in the Human Genome Variation. B.A.M.-K. led the manuscript writing, defined the article structure, collected and interpreted the data, performed research on previous literature, implemented research methods, interpreted the results, and submitted variation data to dbSNP. P.W. described the clinical features, and collected and interpreted patient data. NCBI database was used for the verification of mutations: https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marszałek-Kruk, B.A., Wójcicki, P. Identification of three novel TCOF1 mutations in patients with Treacher Collins Syndrome. Hum Genome Var 8, 36 (2021). https://doi.org/10.1038/s41439-021-00168-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-021-00168-4
