
Abstract
Spatial scale partly explains the differentiated effects of habitat fragmentation on plant biodiversity, but the mechanisms remain unclear. To investigate the effects of habitat fragmentation on genetic diversity at different scales, we sampled Actinidia chinensis Planch. at broad and fine scales, China. The broad-scale sampling included five mountain populations and one oceanic island population (Zhoushan Archipelago), and the fine-scale sampling covered 11 lake islands and three neighboring land populations in Thousand-Island Lake (TIL). These populations were genotyped at 30 microsatellite loci, and genetic diversity, gene flow, and genetic differentiation were evaluated. Genetic differentiation was positively related to geographical distance at the broad scale, indicating an isolation-by-distance effect of habitat fragmentation on genetic diversity. The oceanic population differed from the mainland populations and experienced recent bottleneck events, but it showed high gene flow with low genetic differentiation from a mountain population connected by the Yangtze River. At the fine scale, no negative genetic effects of habitat fragmentation were found because seed dispersal with water facilitates gene flow between islands. The population size of A. chinensis was positively correlated with the area of TIL islands, supporting island biogeography theory, but no correlation was found between genetic diversity and island area. Our results highlight the scale-dependent effects of habitat fragmentation on genetic diversity and the importance of connectivity between island-like isolated habitats at both the broad and fine scales.
Similar content being viewed by others
Introduction
Habitat fragmentation caused by anthropogenic activities is considered a key factor threatening biodiversity1,2. Habitat fragmentation affects biodiversity by increasing isolation between populations and decreasing effective population size, which alters inbreeding and genetic drift within populations, as well as gene-flow frequency between populations3,4,5. This mechanism can be explained by island biogeography theory6,7.
Habitat fragmentation changes continuous habitats to isolated patches and results in the formation of island-like habitats. Guo defined island-like habitats as island entities or areas, such as mountains, oases, or discontinuous forests, isolated by farmlands8. Populations in island-like habitats are usually thought to have low genetic diversity9,10, which is explained by the species–area relationship from island biogeography theory11. Population size is positively related to island area, so small islands are likely influenced by genetic drift, which usually leads to low genetic diversity12,13. Habitat fragmentation can decrease gene flow among populations because low connectivity between habitats limits seed dispersal and insect pollination14,15,16. If gene flow is too low to buffer the negative effects of inbreeding and genetic drift in fragmented populations17, then habitat fragmentation may decrease population fitness and increase the risk of extinction18,19. Toczydlowski and Waller found high levels of inbreeding and genetic differentiation between isolated populations of Impatiens capensis20.
However, some studies did not find negative effects of habitat fragmentation on genetic diversity21,22. The differentiated effects of habitat fragmentation on genetic diversity may be associated with species characteristics (e.g., mating pattern), environmental features (e.g., landscape attributes)23,24,25, or the scale of habitat fragmentation26,27. Compared to monoecious plants, dioecious plants have higher potential for adaptation and higher genetic diversity because they have higher selection efficacy both against deleterious mutations and for beneficial mutations28. Long-lived species have higher potential against genetic erosion from habitat fragmentation than short-lived species because of the occurrence of gene flow among populations29. In a broad-scale study across Tibet, Sichuan, and Yunnan provinces in China (ca. 1 × 106 km2), habitat fragmentation was found to weaken gene flow between populations and cause isolation-by-distance effects30, while there was a lack of evidence of isolation-by-distance effects in a fine-scale-fragmented habitat (ca. 1.5 km2) on Stephens Island, New Zealand31. Long-distance dispersal of seeds can promote gene flow and reduce the genetic distance between populations on a broad scale32. Thus, spatial scale and species characteristics, such as dioecy, a long lifespan, and long-distance dispersal ability, should be considered when studying the effects of habitat fragmentation on genetic diversity.
Here, we investigated the genetic diversity of Actinidia chinensis in China across its national distribution range, considering geographic distance at a broad scale (ca. 1 × 106 km2) and a fine scale (ca. 30 km2). This study aimed to investigate (1) whether island-like isolated habitats limit gene flow and cause differentiation between populations, (2) whether the correlation between genetic diversity and island area in fragmented habitats supports island biogeography theory, and (3) whether the genetic effect of habitat fragmentation is scale-dependent. We employed microsatellites known as simple sequence repeats (SSRs) for genetic diversity analysis because they are codominant loci and have the advantages of a low cost, high polymorphism, high reliability, and a low requirement for DNA quality33,34, which facilitates the analysis of population demography in fine-scale regions35. These findings are expected to provide useful information for the conservation of genetic diversity.
Materials and methods
Plant materials and sampling regions
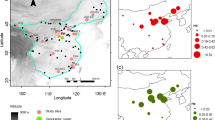
A. chinensis (2n = 2X = 58) is a perennial, dioecious, and deciduous liana36 with a wide distribution in China (Fig. 1). A. chinensis plants are cross-pollinated by insects or wind and disperse through seeds37,38. One-year-old shoots are gray-green brown and glabrous or sparsely covered with white downy hairs, while 2-year-old stems are dark brown and glabrous39. A. chinensis plants generally start to flower 3 years after seed germination.

The green shading in the left subfigures indicates the distribution range of wild A. chinensis populations in China. Lower-left subfigure, XHZ and TPZ (Henan Province in the north), SYH (Hunan Province in the west), and YAC (Guangxi Province in the south) were sampling points around the distribution edge of A. chinensis in China; TIL, Thousand-Island Lake in Zhejiang Province; ZSA, the Zhoushan Archipelago. Right subfigure, sampling points on 11 TIL islands and the neighboring mainland (the SOU site is not marked in the subfigure because it is located over 30 km away from the lake)
Thousand-Island Lake (TIL, Fig. 1), located in eastern China (Chun’an County, Zhejiang Province), is an artificial lake constructed for impoundment water40. The islands of TIL were originally a range of continuous mountains near neighboring mainland and then became fragmented due to the construction of the Xin-An Jiang Reservoir in 19585. The Zhoushan Archipelago (ZSA), located in the East China Sea, was originally a part of mainland China and formed by rising sea levels ca. 7000–9000 years ago27 (Fig. 1).
For the TIL fine-scale analysis, to test island biogeography theory, we investigated the abundance of A. chinensis on all TIL islands, except for the largest (JSI and MGI) islands, where population size was estimated by multiplying the average number of plants in three randomly sampled bays by the number of bays (Supplementary Table S1 and Fig. 1). At the same time, we measured the diameter at breast height (DBH) of A. chinensis plants. To infer plant age on the islands, we counted the tree rings of six A. chinensis plants and constructed a linear regression between tree rings and DBH. In addition, three neighboring land populations (north, east, and south) were sampled around the TIL islands (Fig. 1), which were used and labeled as one (TIL-M) population in the next analysis at a broad scale.
For the broad-scale analysis, we sampled A. chinensis on four mountains (TPZ, XHZ, YAC, and SYH) and one oceanic island (ZSA) in China (Fig. 1), with 15–40 plants per population. We sampled individuals at least 5 m from each other whenever possible.
A total of four to six fresh leaves of A. chinensis were sampled per plant. Half of the leaves per plant were dried by allochroic silica gel for DNA extraction, and the other half was kept fresh for flow cytometry.
Ploidy detection
To determine the genetic background of A. chinensis plants, we detected the ploidy of samples with flow cytometry. Thirty milligrams of fresh leaves per plant were chopped and dissociated by 1 mL of HEPES buffer. After filtration and centrifugation, the dissociated products were dyed with 500 µl of PI (propidium iodide) solution for 10–15 min. Ploidy analysis was conducted by flow cytometry (BD LSRFortessa) with FSC 488, SSC 290, and PI 411–432 (adjusted based on the peak value of the control sample)41.
DNA extraction and microsatellite genotyping
Whole-genomic DNA of A. chinensis was extracted from 25 mg of dry leaves using a Hi-DNA Secure Plant Kit (TIANGEN Biotech Co., Ltd.), using a modified version of the SDS method5. The genetic diversity of A. chinensis populations was detected using 30 microsatellite markers described by Huang et al.42 (Supplementary Table S2). All forward primers were labeled with four kinds of 5′-fluorescein bases (FAM, HEX, TRAMA, or ROX). The multiplex PCR procedure derived from the method of Huang et al.42 was run on a BIO-RAD T100TM Thermal Cycler. Detection of multiplex PCR products was carried out with an ABI 3730xl DNA Analyzer by Sangon Biotech Co., Ltd., setting a default range of standard length ± 40 bp. The detection bands of 30 markers were scored using GeneMarker version 1.91 (size standard: GS500).
Statistical analysis
We employed MICRO-CHECKER 2.2 to detect null alleles, scoring errors, and allele dropout43. Linkage disequilibrium and deviation from Hardy–Weinberg equilibrium were tested in Arlequin v. 3.544. No scoring errors or allele dropout was detected by MICRO-CHECKER. Five of 30 loci were null alleles, and no pattern of linkage disequilibrium was found among any loci (P > 0.05) (Supplementary Table S3).
Genetic diversity indices, including the number of alleles (A), effective number of alleles (Ae), observed heterozygosity (Ho), expected heterozygosity (He), and fixation index (FIS), were calculated for the 278 plants sampled from 19 populations by GenAlEx version 6.50345. Analysis of variance (ANOVA) was employed to investigate the influence of isolation on genetic diversity, with Tukey’s HSD test and 95% confidence intervals, by using SPSS46.
We separately calculated genetic differentiation and gene flow at the broad and fine scales using GenAlEx45. The genetic differentiation coefficient (FST) was calculated by analysis of molecular variance (AMOVA)47. The significance of FST values was tested with 999 permutations, followed by Bonferroni correction. Gene flow (Nm) was estimated based on the formula \(Nm = \left( {1 - F_{ST}} \right)/4F_{ST}\). Hierarchical AMOVA with 999 permutations was constructed to determine the genetic differentiation between populations by GenAlEx45,47.
We performed a Mantel test to detect the correlation between the genetic differentiation matrix and geographic distance matrix, using 999 permutations, FST/(1 − FST) and logarithmic geographic distance48. Since oceanic isolation is the main reason for the genetic divergence of the ZSA population from the mountain populations, we excluded the ZSA population from the Mantel test to remove the interference of oceanic isolation on IBD analysis. The pairwise matrix of geographic distances was calculated based on geographic coordinates, which were identified by a global positioning system (GPS) device (Garmin Oregon 450). All calculations above were performed by GenAlEx45.
To infer population structure, Bayesian clustering analysis was conducted in STRUCTURE version 2.3.449. We set the number of groups (K) from 1 to 19 with ten independent runs using an admixture ancestry model and 100,000-step Markov chain Monte Carlo (MCMC) replicates after a 10,000-step burn-in for each run. The best K value was inferred by delta K in STRUCTURE HARVESTER50,51. A cluster analysis based on genetic distance was performed by the unweighted pair-group method with arithmetic mean (UPGMA) approach in MEGA 6.052. We used the TREEMIX program to detect historical migration among populations53.
BOTTLENECK was used to examine recent bottleneck events54 under the TPM (two-phase model), which allows multiple-step mutations55, with a 10% infinite allele mutation and 90% stepwise mutation model and 1000 replicates56. The significance of heterozygote excess was determined by Wilcoxon’s signed-rank test (P < 0.05). Departures from mutation-drift equilibrium were detected by a mode-shift test57.
We applied the program DIYABC to infer the divergence history of A. chinensis populations58. According to the phylogenetic relationships reconstructed by STRUCTURE and TREEMIX, we set a demographic scenario to infer the effective population size and divergence time of four clusters, i.e., TIL islands, neighboring land populations, mountain populations, and the ZSA population. We set the number of simulated data sets to 100,000, and the number of selected data sets to 1000. The historical model, genetic data, and summary statistics were set to default values.
To identify the relationships between genetic diversity and population size in A. chinensis and features of TIL islands (Supplementary Table S1), we conducted linear regression analysis with the Pearson correlation coefficient46. Features of 11 TIL islands, including area, perimeter, the shape index (SI), distance to the nearest island (DTI), and distance to the nearest mainland (DTL), were measured with a geographic information system (GIS). The island-shape index (SI) was calculated as follows: \(SI = P/\left[ {2 \times \left( {\pi \times A} \right)^{0.5}} \right]\)59.
Results
Habitat fragmentation due to geographic isolation on a broad scale
Actinidia chinensis plants in the TPZ, XHZ, SYH, YAC, and TIL-M populations were diploid, while 18 plants in the ZSA population were tetraploid (Supplementary Fig. S1). The genetic diversity of A. chinensis in TPZ, XHZ, and TIL-M was significantly greater than that in YAC, SYH, and ZSA, and the ZSA population significantly differed from the TPZ, XHZ, and TIL-M populations but not from YAC and SYH (Table 1).
In the TPZ, XHZ, SYH, YAC, and ZSA populations, >50% of the tested loci deviated from Hardy–Weinberg equilibrium (Supplementary Table S4), indicating frequent inbreeding. Mountain populations (XHZ, TPZ, SYH, and YAC) showed significant heterozygote excess but no evidence of bottleneck events (Supplementary Table S5), while the ZSA population experienced recent bottleneck events according to departures from mutation-drift equilibrium in the allele frequency distribution (Supplementary Table S5).
The pairwise-estimated FST between populations was moderate (0.012–0.123), with the lowest FST index in the comparison of ZSA and SYH populations (Table 2). The gene flow (Nm) between populations ranged from 1.786 to 20.833, with the highest Nm in the comparison of the ZSA and SYH populations (Table 2). The Mantel test showed that genetic differentiation was positively related to geographical distance (log-transformed) on a broad scale (R2 = 0.383, P = 0.033) (Fig. 2), indicating an isolation-by-distance effect.

Genetic differentiation was calculated as [FST/(1 − FST)], and geographical distance was log-transformed. A, five mountain populations on a broad scale; B, TIL populations
Structure analysis showed that the ZSA population differed from other mainland populations (K = 4) (Fig. 3), and UPGMA cluster analysis confirmed these structure results (Supplementary Fig. S2). Analysis of molecular variance (AMOVA) was performed between ZSA and the mountain populations, and only 3% (P < 0.05) of the total genetic variance was attributed to the differences between the two groups. The vast majority (90%, P < 0.05) of the variance occurred among individuals within populations, whereas only 7% of variance occurred among populations (Supplementary Table S6).

Population structure bar plots show the clustering of samples into two (best delta K) and four clusters. Each vertical bar indicates an individual, and the height of each colored bar represents the proportion of assignment to that cluster. Abbreviations are population names; see Fig. 1
Approximate Bayesian Computation (DIYABC) revealed a smaller effective population size (Ne) in ZSA than in the mountain populations (Supplementary Table S7 and Supplementary Fig. S3). The differentiation time of the ZSA population (median t2 = 1920 generations ≈11,520 years, mode t2 = 1290 generations ≈7740 years) coincided with the formation of ZSA islands (ca. 7000–9000 years ago). The ZSA population differentiated a mean of 2450 generations ago, later than the formation time of the mountain populations (8880 generations; Supplementary Table S7). TREEMIX analysis revealed historic migration from the northern TPZ population to the eastern TIL-M and ZSA populations, but not to the southern SYH and YAC populations (Fig. 4).

Top subfigure, A. chinensis populations at the broad scale; bottom subfigure, the A. chinensis TIL populations at the fine scale
Habitat fragmentation from water isolation on a fine scale in TIL
All 480 sampled plants in TIL were diploid (Supplementary Fig. S1). The population size of A. chinensis per island was positively correlated with island area (r = 0.963, P < 0.001) and perimeter (r = 0.943, P < 0.001) (Supplementary Tables S1 and S8). More than one-half of the A. chinensis plants were seedlings on the XJS, B3I, and B4I islands, and most of the seedlings appeared at the edge of the islands.
With a hypothesis of plants migrating from old to young populations, we can detect possible dispersal pathways of A. chinensis by measuring tree rings to define the age of plants in populations. Linear regression analysis revealed a significant positive correlation between tree rings and DBH (Supplementary Fig. S4) (R2 = 0.669, P = 0.023). The maximum DBH of sampled A. chinensis plants on the TIL islands was 30.8 cm in the MGI population (Supplementary Table S1), indicating that the oldest plant was ~23 years old. The maximum DBHs of plants on TIL islands were similar and did not show dispersal pathways, indicating the simultaneous existence of A. chinensis plants on these islands. However, the TREEMIX results showed historic migration from MDI to YJI islands (Fig. 4). The differentiation time between TIL island populations and neighboring mainland populations was 62 generations (6 years/generation; Supplementary Table S7), indicating that the present A. chinensis plants emerged before the formation of the TIL reservoir (ca. 60 years ago).
The genetic diversity of A. chinensis was not related to TIL island features or geographic distance between islands (Supplementary Table S8). The genetic diversity of A. chinensis populations in the neighboring mainland populations and on large islands (JSI and MGI) was higher than that on other small islands (Table 3). The absolute value of FIS of all populations was close to zero (Table 3), indicating random mating and a limited isolation effect in these populations. The TIL populations showed significantly higher Ho and lower FIS values than the other populations (Table 1).
The pairwise-estimated FST between populations in TIL ranged from 0 to 0.163, with a high level of gene flow between these populations (Nm from 1.285 to 82.245) (Supplementary Table S9). Recent bottleneck events were not detected in TIL island populations, except for the YJI population with a small population size and small island area (Supplementary Table S5). Structure analysis revealed that the TIL populations differed from other mountain populations (K = 2), and the nearby B2I, B3I, B4I, and B5I islands exhibited frequent gene flow (Fig. 3).
The AMOVA results attributed a low percentage of the total genetic variance (1%) to differences between TIL island and neighboring land populations, with 5% among populations and 94% within populations (Supplementary Table S6). Genetic differentiation of island populations was not related to geographic distance between islands (R2 = 0.001, P = 0.835) (Fig. 2).
Discussion
Habitat fragmentation has been found to reduce genetic diversity56,60,61, while some studies have found no negative effects21,22. To investigate whether the differentiated genetic effects of habitat fragmentation are associated with the scale of habitat fragmentation, we measured the genetic diversity of A. chinensis populations at a broad and a fine scale. Our results showed scale-dependent effects of habitat fragmentation on A. chinensis populations, with an isolation-by-distance effect at the broad scale, but no negative effects on genetic diversity at the TIL (fine) scale.
The positive correlation between A. chinensis population size and island area in TIL supported island biogeography theory62,63,64, while the relationship between the genetic diversity of A. chinensis and island area did not. Landscape attributes are key factors determining the genetic diversity effects of habitat fragmentation65,66,67. Small habitat area and island isolation have been found to decrease the genetic diversity of plant species68. Landscape connectivity is beneficial to the conservation of genetic diversity and helps reduce demographic bottlenecks in natural populations66. We found most seedlings on the edges of TIL islands, and the ripe fruits of A. chinensis are able to float in freshwater. The upstream Xin’an River is located west of the TIL and leads to northern and southern flow from west to east, which likely explains the northern and southern clusters (Fig. 3 and Supplementary Fig. S2). The results showed low pairwise-estimated FST, low FIS, and no bottleneck events (except in YJI). These results indicate that seed dispersal with lake water flow facilitates gene flow between islands.
In addition, the mating pattern, dioecy, and relatively long lifespan of A. chinensis may buffer the area, isolation, and edge effects resulting from the fine scale of habitat fragmentation in TIL29,69,70. Dioecy can decrease the inbreeding rate and counteract the negative effects of isolation due to forced outcrossing71. Dioecious species experience lower frequencies of genetic drift than monoecious species28. Species with short lifespans in highly fragmented landscapes may experience genetic degradation due to the loss and fixation of alleles29, while long-lived species could resist the genetic erosion caused by habitat fragmentation29,60. The age of A. chinensis plants was greater than 20 years (Supplementary Fig. S4), and A. chinensis plants almost simultaneously emerged on these islands before the formation of the TIL reservoir (Supplementary Table S7), except for individual plants in the YJI island population that migrated from the MDI island (Fig. 4).
However, the buffering mechanisms discussed above were limited at the broad scale because geographic isolation resulted in low gene flow and high differentiation between mountain populations, and led to an isolation-by-distance effect. This was consistent with the findings of other studies on a broad scale of habitat fragmentation26,30. These mountain populations are located in isolated island-like habitats that lack connectivity between populations, which limits seed dispersal and insect pollination. Inbreeding caused by isolation leads to the fixation and loss of alleles and then the loss of genetic variance17,18. We found loci deviating from HWE and high fixation indices (FIS > 0.7) in ZSA island and mountain populations.
The ZSA population, on an oceanic island, significantly differed from the mainland populations (Fig. 3 and Supplementary Fig. S3), with high FIS values and bottleneck events (Table 1 and Supplementary Table S5). Founder effects could not explain this phenomenon. The divergence time between mainland and the ZSA population coincides with the formation time of ZSA, but ZSA is genetically closer to the far-away SYH population than to the nearby TIL population. The results showed high gene flow and low differentiation between the ZSA and SYH populations, resulting in similar levels of genetic diversity between them. The Yangtze River connects the two populations, which might facilitate the seed dispersal of A. chinensis. As discussed above, in TIL, seed dispersal via floating on water is the main dispersal method of A. chinensis. Gene flow through long-distance pollen or seed dispersal could disrupt isolation and weaken the negative effects of habitat fragmentation15,16,69. In fact, some studies revealed gene flow and introgression between diploid and tetraploid populations of A. chinensis72. Breeding experiments showed that diploid A. chinensis could crossbreed with tetraploid A. chinensis and produce fertile offspring73. The existence of transitional phenotypes of A. chinensis in the wild proves the occurrence of gene flow and introgression between the two cytotypes in nature74. This phenomenon suggests that the design of corridors is very important for the conservation of genetic diversity.
A. chinensis plants in ZSA were tetraploid, and their origin was unclear. Diploid A. chinensis is thought to be the progenitor of tetraploid A. chinensis75,76. We detected historical migration events from TPZ to ZSA, indicating that the ZSA population may have come from northern populations, but the mechanism of polyploidization is still unknown. Generally, polyploidization can increase biological diversity, but the genetic diversity of the tetraploid ZSA population was lower than that of the diploid populations. This could be explained by treating tetraploid plants as diploid plants to score bands, the high inbreeding rate of the ZSA population, and bottleneck events resulting from the small population size. Llorens et al. reported bottleneck events and decreased genetic diversity in the shrubs Grevillea caleyi and G. longifolia, indicating that they had experienced long-term habitat isolation77. Moreover, the detrimental effects of long-term local adaptation likely decrease genetic diversity78. ZSA is an oceanic island that has undergone long-term isolation of ca. 7000–9000 years27. Long-term oceanic isolation decreased the genetic diversity and increased the genetic differentiation of insular populations, while there were small genetic effects on the TIL islands that experienced short-term isolation by water. This is likely to support a time-delayed effect of habitat fragmentation. Such time delays tend to mask the genetic effects of habitat fragmentation79.
In conclusion, we found a scale-dependent effect of habitat fragmentation on the genetic diversity of A. chinensis populations, with an isolation-by-distance effect at the broad scale. The connectivity between populations explains the scale-dependent effect because it facilitates the seed dispersal of A. chinensis and buffers against the negative effects of habitat fragmentation, not only at the fine scale but also at the broad scale. Therefore, our results highlight the importance of constructing corridors for biodiversity conservation, particularly in broad-scale island-like fragmented habitats and on long-term isolated islands.
References
Wu, J. Key concepts and research topics in landscape ecology revisited: 30 years after the Allerton Park workshop. Landsc. Ecol. 28, 1–11 (2013).
Wilson, M. C. et al. Habitat fragmentation and biodiversity conservation: key findings and future challenges. Landsc. Ecol. 31, 219–227 (2016).
Leimu, R., Vergeer, P., Angeloni, F. & Ouborg, N. J. Habitat fragmentation, climate change, and inbreeding in plants. Ann. NY Acad. Sci. 1195, 84–98 (2010).
Young, A., Boyle, T. & Brown, T. The population genetic consequences of habitat fragmentation for plants. Trends Ecol. Evolution 11, 413–418 (1996).
Yuan, N., Comes, H. P., Mao, Y., Qi, X. & Qiu, Y. Genetic effects of recent habitat fragmentation in the Thousand-Island Lake region of southeast China on the distylous herb Hedyotis chrysotricha (Rubiaceae). Am. J. Bot. 99, 1715–1725 (2012).
MacArthur, R. H. & Wilson, E. O. An equilibrium theory of insular zoogeography. Evolution 17, 373–387 (1963).
MacArthur, R. H. & Wilson, E. O. The Theory of Island Biogeography (Princeton University Press, Princeton, NJ, 1967).
Guo, Q. Island biogeography theory: emerging patterns and human effects. Earth Syst. Environ. Sci. 32, 1–5 (2015).
Wroblewska, A. High genetic diversity within island-like peripheral populations of Pedicularis sceptrum-carolinum, a species with a northern geographic distribution. Ann. Bot. Fenn. 50, 289–299 (2013).
Csergo, A.-M. et al. Genetic structure of peripheral, island-like populations: a case study of Saponaria bellidifolia Sm. (Caryophyllaceae) from the Southeastern Carpathians. Plant Syst. Evol. 278, 33–41 (2009).
Doyle, J. M., Hacking, C. C., Willoughby, J. R., Sundaram, M. & DeWoody, J. A. Mammalian genetic diversity as a function of habitat, body size, trophic class, and conservation status. J. Mammal. 96, 564–572 (2015).
Reynolds, R. G. et al. Archipelagic genetics in a widespread Caribbean anole. J. Biogeogr. 44, 2631–2647 (2017).
Costanzi, J.-M. & Steifetten, Ø. Island biogeography theory explains the genetic diversity of a fragmented rock ptarmigan (Lagopus muta) population. Ecol. Evol. 9, 3837–3849 (2019).
Hermansen, T., Minchinton, T. & Ayre, D. Habitat fragmentation leads to reduced pollinator visitation, fruit production and recruitment in urban mangrove forests. Oecologia 185, 221–231 (2017).
Broeck, A. et al. Dispersal constraints for the conservation of the grassland herb Thymus pulegioides L. in a highly fragmented agricultural landscape. Conserv Genet. 16, 765–776 (2015).
Browne, L. & Karubian, J. Habitat loss and fragmentation reduce effective gene flow by disrupting seed dispersal in a neotropical palm. Mol. Ecol. 27, 3055–3069 (2018).
Bijlsma, R. & Loeschcke, V. Genetic erosion impedes adaptive responses to stressful environments. Evol. Appl. 5, 117–129 (2012).
Lienert, J. Habitat fragmentation effects on fitness of plant populations-a review. J. Nat. Conserv. 12, 53–72 (2004).
Luquet, E. et al. Genetic erosion in wild populations makes resistance to a pathogen more costly. Evolution 66, 1942–1952 (2012).
Toczydlowski, R. H. & Waller, D. M. Drift happens: molecular genetic diversity and differentiation among populations of jewelweed (Impatiens capensis Meerb.) reflect fragmentation of floodplain forests. Mol. Ecol. 28, 2459–2475 (2019).
Jimenez, J. F., Sanchez-Gomez, P., Canovas, J. L., Hensen, I. & Aouissat, M. Influence of natural habitat fragmentation on the genetic structure of Canarian populations of Juniperus turbinata. Silva Fenn. 51, 1–14 (2017).
Garciaverdugo, C. et al. Do island plant populations really have lower genetic variation than mainland populations? Effects of selection and distribution range on genetic diversity estimates. Mol. Ecol. 24, 726–741 (2015).
Vandepitte, K., Jacquemyn, H., Roldan-Ruiz, I. & Honnay, O. Landscape genetics of the self-compatible forest herb Geum urbanum: effects of habitat age, fragmentation and local environment. Mol. Ecol. 16, 4171–4179 (2007).
Krauss, J. et al. Habitat fragmentation causes immediate and time-delayed biodiversity loss at different trophic levels. Ecol. Lett. 13, 597–605 (2010).
Heinken, T. & Weber, E. Consequences of habitat fragmentation for plant species: do we know enough? Perspect. Plant Ecol. Syst. 15, 205–216 (2013).
Duminil, J. et al. Large-scale pattern of genetic differentiation within African rainforest trees: insights on the roles of ecological gradients and past climate changes on the evolution of Erythrophleum spp (Fabaceae). BMC Evol. Biol. 13, 195- (2013).
Yuan, N. et al. A comparative study on genetic effects of artificial and natural habitat fragmentation on Loropetalum chinense (Hamamelidaceae) in Southeast China. Heredity 114, 544 (2014).
Muyle, A. et al. Dioecy in plants: an evolutionary dead end? Insights from a population genomics study in the Silene genus. Preprint at https://www.biorxiv.org/content/10.1101/414771v1.full (2018).
Fuller, M. R. & Doyle, M. W. Gene flow simulations demonstrate resistance of long-lived species to genetic erosion from habitat fragmentation. Conserv Genet. 19, 1439–1448 (2018).
Hu, Y. et al. Genetic structuring and recent demographic history of red pandas (Ailurus fulgens) inferred from microsatellite and mitochondrial DNA. Mol. Ecol. 20, 2662–2675 (2011).
Moore, J. A., Miller, H. C., Daugherty, C. H. & Nelson, N. J. Fine-scale genetic structure of a long-lived reptile reflects recent habitat modification. Mol. Ecol. 17, 4630–4641 (2008).
Martínez-López, V., García, C., Zapata, V., Robledano, F. & De la Rúa, P. Intercontinental long-distance seed dispersal across the Mediterranean Basin explains population genetic structure of a bird-dispersed shrub. Mol. Ecol. 29, 1408–1420 (2020).
Grover, A. & Sharma, P. C. Development and use of molecular markers: past and present. Crit. Rev. Biotechnol. 36, 290–302 (2016).
Vieira, M. L. C., Santini, L., Diniz, A. L. & Munhoz, C. D. F. Microsatellite markers: what they mean and why they are so useful. Genet Mol. Biol. 39, 312–328 (2016).
Selkoe, K. A. & Toonen, R. J. Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol. Lett. 9, 615–629 (2006).
Huang, H. et al. Genetic diversity in the genus Actinidia (in Chinese). Chin. Biodivers. 8, 1–12 (2000).
Logan, D. P. & Xu, X. Germination of kiwifruit, Actinidia chinensis, after passage through Silvereyes, Zosterops lateralis. New Zeal. J. Ecol. 30, 407–411 (2006).
Costa, G., Testolin, R. & Vizzotto, G. Kiwifruit pollination: an unbiased estimate of wind and bee contribution. N. Zeal J. Crop Hort. 21, 189–195 (1993).
Huang, H. The Genus Actinidia, A World Monograph (Science Press, Beijing, 2014).
Lv, K. et al. Habitat fragmentation influences gene structure and gene differentiation among the Loxoblemmus aomoriensis populations in the Thousand Island Lake. Mitochondrial DNA A 29, 222–227 (2017).
Liu, Y. F. et al. Rapid radiations of both kiwifruit hybrid lineages and their parents shed light on a two-layer mode of species diversification. N. Phytol. 215, 877–890 (2017).
Huang, W. G., Cipriani, G., Morgante, M. & Testolin, R. Microsatellite DNA in Actinidia chinensis: isolation, characterisation, and homology in related species. Theor. Appl. Genet. 97, 1269–1278 (1998).
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P. M. & Shipley, P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538 (2004).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Peakall, R. & Smouse, P. E. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295 (2006).
Pallant, J. SPSS survival manual: a step by step guide to data analysis using SPSS. Aust. N.Z. J. Public Health 37, 597–598 (2013).
Excoffier, L., Smouse, P. E. & Quattro, J. M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491 (1992).
Smouse, P. E., Long, J. C. & Sokal, R. R. Multiple regression and correlation extensions of the mantel test of matrix correspondence. Syst. Biol. 35, 627–632 (1986).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. & Vonholdt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012).
Cornuet, J. M. & Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144, 2001–2014 (1996).
Di Rienzo, A. et al. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl Acad. Sci. USA 91, 3166–3170 (1994).
Wang, S. et al. Population size and time since island isolation determine genetic diversity loss in insular frog populations. Mol. Ecol. 23, 637–648 (2014).
Luikart, G., Allendorf, F., Cornuet, J. & Sherwin, W. Distortion of allele frequency distributions provides a test for recent population bottlenecks. J. Hered. 89, 238–247 (1998).
Cornuet, J.-M. et al. DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30, 1187–1189 (2014).
Ewers, R. M. & Didham, R. K. The effect of fragment shape and species’ sensitivity to habitat edges on animal population size. Conserv Biol. 21, 926–936 (2007).
Ortego, J., Bonal, R. & Munoz, A. Genetic consequences of habitat fragmentation in long-lived tree species: the case of the Mediterranean holm oak (Quercus ilex, L.). J. Hered. 101, 717–726 (2010).
Fletcher, J. R. J. et al. Is habitat fragmentation good for biodiversity? Biol. Conserv 226, 9–15 (2018).
Fattorini, S., Borges, P. A. V., Dapporto, L. & Strona, G. What can the parameters of the species-area relationship (SAR) tell us? Insights from Mediterranean islands. J. Biogeogr. 44, 1018–1028 (2017).
Matthews, T. J. et al. Island species-area relationships and species accumulation curves are not equivalent: an analysis of habitat island datasets. Global Ecol. Biogeogr. 25, 607–618 (2016).
Matthews, T., Cottee-Jones, H., Whittaker, R. & Brotons, L. Habitat fragmentation and the species-area relationship: a focus on total species richness obscures the impact of habitat loss on habitat specialists. Divers Distrib. 20, 1136–1146 (2014).
McGlaughlin, M. E. et al. Do the island biogeography predictions of MacArthur and Wilson hold when examining genetic diversity on the near mainland California Channel Islands? Examples from endemic Acmispon (Fabaceae). Bot. J. Linn. Soc. 174, 289–304 (2014).
Jangjoo, M., Matter, S. F., Roland, J. & Keyghobadi, N. Connectivity rescues genetic diversity after a demographic bottleneck in a butterfly population network. Proc. Natl Acad. Sci. USA 113, 10914–10919 (2016).
Reisch, C. et al. Genetic diversity of calcareous grassland plant species depends on historical landscape configuration. BMC Ecol. 17, 1–13 (2017).
Bottin, L. et al. Genetic diversity and population structure of an insular tree, Santalum austrocaledonicum in New Caledonian archipelago. Mol. Ecol. 14, 1979–1989 (2005).
Breed, M. F. et al. Mating patterns and pollinator mobility are critical traits in forest fragmentation genetics. Heredity 115, 108–114 (2015).
Llorens, T. M., Byrne, M., Yates, C. J., Nistelberger, H. M. & Coates, D. J. Evaluating the influence of different aspects of habitat fragmentation on mating patterns and pollen dispersal in the bird-pollinated Banksia sphaerocarpa var.caesia. Mol. Ecol. 21, 314–328 (2012).
Rosche, C. et al. Sex ratio rather than population size affects genetic diversity in Antennaria dioica. Plant Biol. 20, 789–796 (2018).
Liu, Y., Li, D., Yan, L. & Huang, H. The microgeographical patterns of morphological and molecular variation of a mixed ploidy population in the species complex Actinidia chinensis. PLoS ONE 10, e0117596 (2015).
Guijun, Y., Ferguson, A. R. & McNeilage, M. A. Ploidy races in Actinidia chinensis. Euphytica 78, 175–183 (1994).
Chat, J., Jáuregui, B., Petit, R. J. & Nadot, S. Reticulate evolution in kiwifruit (Actinidia, Actinidiaceae) identified by comparing their maternal and paternal phylogenies. Am. J. Bot. 91, 736–747 (2004).
Crowhurst, R. N. et al. Analysis of expressed sequence tags from Actinidia: applications of a cross species EST database for gene discovery in the areas of flavor, health, color and ripening. BMC Genomics 9, 351 (2008).
Zheng, Y. Q., Li, Z. Z. & Huang, H. W. Preliminary study on SSR analysis in kiwifruit cultivars. J. WH Bot. Res. 21, 444–448 (2003).
Llorens, T. M., Ayre, D. J. & Whelan, R. J. Anthropogenic fragmentation may not alter pre-existing patterns of genetic diversity and differentiation in perennial shrubs. Mol. Ecol. 27, 1541–1555 (2018).
Van de Peer, Y., Mizrachi, E. & Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424 (2017).
Bommarco, R., Lindborg, R., Marini, L. & Ockinger, E. Extinction debt for plants and flower-visiting insects in landscapes with contrasting land use history. Divers. Distrib. 20, 591–599 (2014).
Acknowledgements
We thank Mr. Dun Zhang, Ms Fang Liu, Ms. Yuan Liu, and local farmers for their assistance in field sampling. This work was financially supported by the Biodiversity Survey, Observation and Assessment of the Ministry of Ecology and Environment, China (2019HJ2096001006), and the General Research Project of CRAES, China (No. 2016YSKY-08).
Author information
Authors and Affiliations
Contributions
Y.B.L. designed the study. W.Y., B.W., X.W., Z.Y., Y.H.L., and Y.B.L. performed the experiments. W.Y., B.W., and Y.B.L. analyzed the data. W.Y. and Y.B.L. wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, W., Wu, B., Wang, X. et al. Scale-dependent effects of habitat fragmentation on the genetic diversity of Actinidia chinensis populations in China. Hortic Res 7, 172 (2020). https://doi.org/10.1038/s41438-020-00401-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-00401-1
