
Abstract
The center–periphery hypothesis (CPH) states that the genetic diversity, genetic flow, and population abundance of a species are highest at the center of the species’ geographic distribution. However, most CPH studies have focused on the geographic distance and have ignored ecological and historical effects. Studies using niche models to define the center and periphery of a distribution and the interactions among geographical, ecological, and historical gradients have rarely been done in the framework of the CPH, especially in biogeographical studies of animal species. Here, we examined the CPH for a widely distributed arthropod, Tetranychus truncatus (Acari: Tetranychidae), in eastern China using three measurements: geographic distance to the center of the distribution (geography), ecological suitability based on current climate data (ecology), and historical climate data from the last glacial maximum (history). We found that the relative abundances of different populations were more strongly related to ecology than to geography and history. Genetic diversity within populations and genetic differentiation among populations based on mitochondrial marker were only significantly related to history. However, the genetic diversity and population differentiation based on microsatellites were significantly related to all three CPH measurements. Overall, population abundance and genetic pattern cannot be explained very well by geography alone. Our results show that ecological gradients explain the variation in population abundance better than geographic gradients and historical factors, and that current and historical factors strongly influence the spatial patterns of genetic variation. This study highlights the importance of examining more than just geography when assessing the CPH.
Similar content being viewed by others


Introduction
Disentangling the mechanistic processes and ecological factors that shape patterns of spatial variation in species’ population structures has long been a focus in ecology and evolution (Hengeveld and Haeck 1982; Sagarin and Gaines 2002; Pironon et al. 2017). One frequently observed pattern that explains the variation in demographic, genetic, and ecological characteristics of species across their ranges is the ‘center–periphery’ hypothesis (CPH) (Pironon et al. 2017), which is also known as the ‘abundant-center’ (Sagarin and Gaines 2002) or ‘central–marginal’ hypothesis (Eckert et al. 2008). This hypothesis assumes that populations are under optimal ecological (abiotic and/or biotic) conditions near the center of a species’ distribution range, whereas harsher conditions exist at the periphery of their range (Sagarin and Gaines 2002). Unfavorable conditions would lead to declines in population density and fitness (Hengeveld and Haeck 1982; Brown 1984; Brown et al. 1995) at the geographic range periphery, and populations then become smaller and more isolated. Because of density-dependent migration, gene flow should be asymmetric, with a higher number of migrants moving from populations at the center to the range edge (Hansson 1991; Hanski et al. 1994; Kubisch et al. 2011). High isolation and small population size are then predicted to cause decreased genetic diversity within populations and increased genetic differentiation among populations (Excoffier et al. 2009; Dixon et al. 2013; Swaegers et al. 2013).
Lower level of the genetic diversity in the periphery populations, in turn, may constrain their adaptation to changeable environment and influence their demographic performance (Takahashi et al. 2016). Highly fragmented populations with limited gene flow may again limit the availability of locally beneficial alleles and prevent adaptation (Holt and Keitt 2000). In addition, allee effects induced by a small population size could impede population settlement beyond species range limits, because low density could result in reproductive failure, or a breakdown in biotic interactions (Keitt et al. 2001). CPH has long been considered to be one of explanations for understating species’ range limits.
One important presupposition of the CPH is that the center of the distribution tends to have the most optimal climate conditions (Brown 1984; Pironon et al. 2017). However, species interactions (e.g. inter-specific competition, predation) may be more important in regulating the population abundance than the abiotic environment (Dallas et al. 2017). Moreover, habitat suitability does not necessarily linearly decline as geographic distance from the center of a species’ range increases (Sagarin and Gaines 2002; Rivadeneira et al. 2010). Hence, species demographic performance does not have similar responses to geographic and climatic conditions (Pironon et al. 2015). Population abundance may be more related to environmental quality (Pironon et al. 2017), and gene flow patterns may be correlated to environment gradients but not the geographic distance (Sexton et al. 2014).
Moreover, environmental conditions change over time, and species distributions change over time under the influence of changing demographic and dispersal parameters (Travis et al. 2013). Populations currently considered to be ecologically or geographically central may have been peripheral in the past, and vice versa (Pironon et al. 2017). Therefore, the population genetic and abundance patterns across the range could be strongly influenced by historical range shifts (Micheletti and Storfer 2015). For example, climatic reconstructions of species distributions during the Last Glacial Maximum (LGM) showed that older populations at the center of the historical distribution (refugia) tend to have higher genetic diversity, whereas younger populations (recolonizing populations relative to older populations) have lower genetic diversity in some species (Duncan et al. 2015; Pironon et al. 2017). Consequently, these issues may produce mixed population genetic and demographic patterns in the context of the CPH (Eckert et al. 2008; Duncan et al. 2015; Pironon et al. 2017). For example, only 6% (1 of 16) of tests conducted on inset provide population genetic support for the CPH (Pironon et al. 2017). Whereas, historical or current ecological factors often better explain the geographically genetic patterns in insects (e.g. European Coenagrionid damselfly species) (Johansson et al. 2013).
One way to address these issues is by evaluating ecological and historical effects on population genetic and abundance patterns (Eckert et al. 2008; Sexton et al. 2014; Suárez-Atilano et al. 2017; Jiang et al. 2018). Several influential studies have noted the importance of incorporating a historical dimension and current climate in the CPH framework (Hampe and Petit 2005; Eckert et al. 2008; Duncan et al. 2015). In fact, many studies demonstrated that post-glacial recolonization and current climate may shape the population structure (Pérez-Collazos et al. 2009; Sexton et al. 2014; Wei et al. 2016). However, only a few studies have examined the spatial population structure using ecological niche models, which can be used to depict historical and contemporary ecological suitability, to define the central and peripheral populations in the context of the CPH. In addition, the interactions among geographical, ecological, and historical gradients have rarely been studied (Pironon et al. 2017).
In this study, we investigated geographical patterns of the population structure (relative abundance and genetic variation) and the underlying ecological and evolutionary drivers for a widely distributed arthropod, Tetranychus truncatus (Acari: Tetranychidae), across its distribution in China. The effect of historical glaciation in China has been debated for a long time, because no unified ice sheet developed during the Quaternary period (especially in east China) (Liu et al. 2012). Until now, very few empirical studies tested whether and how historical processes affect occurrence and genetic variation in the context of the CPH in China (Wei et al. 2016). First, we defined the CPH in terms of geography (distance from center of the organism’s range), ecology (current environmental suitability, 1950–2000), and history (historical environmental suitability, LGM) based on occurrence point data and ENMs. Second, we tested the CPH by examining patterns of the relative abundance, genetic diversity, and genetic differentiation along geographic, ecological, and historical gradient. Third, we used Akaike’s information criterion (AICc) to determine the model that best explained patterns of population genetic variation and relative abundance. Last, by examining gene flow patterns, we analyzed whether historical migration rate was asymmetrical, which would be consistent with post-glacial expansion, and whether contemporary gene flow moves from central to peripheral populations. Taken together, these results allow us to evaluate geographic, ecological, and historical assumptions of the CPH and help us understand how ecology, history, and geography influence biogeographic patterns that ultimately structure species’ ranges.
Materials and methods
Study system
Tetranychus truncatus (TTR) is a herbivorous arthropod that is primarily distributed in China and spans ~40° in latitude (18°–50°) (Sun et al. 2018). This species was also occasionally found in several other East Asian countries (especially Japan) (Ehara 1999). It was reported to be the most abundant spider mite in a warm temperate zone (center China), but was less frequent in cold temperate (northern China) and warmer subtropical (southern China) zones in mainland China (Jin et al. 2018). Dispersal of spider mites is generally achieved by walking and drifting through the aerial currents, and the dispersal distance were usually <1 km within plants or fields (Smitley and Kennedy 1985). Besides, making use of aerial currents and transportation of mite-infested plant material, the spider mites also can engage in long-distance dispersal (Yaninek 1988; van Petegem et al. 2016).
Identifying the center–periphery throughout species’ distribution range
Geographic center–periphery
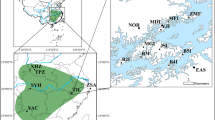
TTR locality information (113 records) was obtained from a survey conducted from 2008–2017 in China (Jin et al. 2018) and published literature (Table S1), which covered most of TTR’s distribution range. Sampling bias due to the different sources may affect the accuracy of presence-only modeling (Phillips et al. 2006). Spider mites are very small and the species identifications were often problematic (Jin et al. 2018), so we only chose the papers with detailed identification and sufficient geographical information. Occurrence localities from different studies may be biased (Reddy and Dávalos 2003). TTR is a common species that feed on many crops; they are often highly correlated with the nearby presence of agricultural system. So the sampling intensity may vary widely across the study area or different ecological system (Reddy and Dávalos 2003). To avoid the effect of nonuniform sampling of species localities, auto-correlated occurrence points were eliminated by the ‘Spatially Rarefy Occurrence Data Tool’ of SDMtoolbox v2.2 following a default protocol (Brown 2014) in ArcGIS v10.5 (ESRI, Redlands, CA, USA). After eliminating the auto-correlated occurrence points, a total of 84 occurrence records were used for ecological niche modeling (Fig. 1). We drew the polygons based on the proximity of the geographical records, which represented the accessibility area considering the known geographical distribution. The presence of geographical barriers like basins, ocean, and physiographic depressions were also taken into account (Suárez-Atilano et al. 2017). The geographic range center was determined by finding the center point of a polygon around all filtered occurrence points using ‘Mean Center’ tools in the ArcGIS v10.5 (McMinn et al. 2017).

a Sample localities (N = 22) across the major distribution range of Tetranychus truncatus. Locations are represented by red points with the population name nearby. Dark points represent the recorded sites of TTR from 2008 to 2018 and published research (Table S1). The green point represents the geographic center that was calculated using the ‘Mean Center’ tool in ArcGIS v10.5. Distribution patterns of haplotype diversity (Hd) (b) and expected heterozygosity (c) on the map
Ecological (current climate) and historical (paleo-climate) center–periphery
To construct current [Current conditions (~1960–1990)] and palaeo-climate niche models [System Model (CCSM4) and Max-Planck-Institute Earth System Mode P (MPI-ESM-P)], we carried out ecological niche modeling using the program MaxEnt v3.3e (Phillips et al. 2006; Franklin 2013; Micheletti and Storfer 2015). Palaeo-distribution models are generated using current climate conditions with the addition of palaeo-climate estimates on which to project the species distribution (Cane et al. 2006; Duncan et al. 2015; Wei et al. 2016). It should be mentioned that the projections into past climatic conditions rely on the assumption of niche conservatism. At this moment, historical occurrence data or other data are not available to make more accurate predictions.
Nineteen bioclim variables of current climate were obtained from the WorldClim 1.4 database (Hijmans et al. 2005; http://worldclim.org/) at 2.5 arc-min resolution. To reduce the impact of highly auto-correlated (r > 0.9) input environmental data, we only used eight bioclimatic variables (annual mean temperature, mean diurnal range, isothermality, temperature seasonality, mean temperature of warmest quarter, annual precipitation, precipitation of driest month, and precipitation seasonality) using the ‘Remove Highly Correlated Variables’ tool in SDMtoolbox (Brown 2014). For paleo-climate reconstructions, we used LGM climate information in CCSM4 and MPI-ESM-P at a spatial resolution of 2.5 arc-min from the WorldClim database (http://worldclim.org).
Ecological niche modeling was performed in MaxEnt using the logistic output format (Phillips and Dudík 2008). MaxEnt was run with 50 bootstrap replications with a random test percentage of 25% and regularization multiplier of 1 (Micheletti and Storfer 2015). Default parameters achieve very good performance based on a series of tests using diverse dataset of 226 species from six regions (Phillips and Dudík 2008). Therefore, default settings were used for all other parameters. Model performance was evaluated using the area under the curve (AUC) of the receiving operator characteristics plot (Phillips et al. 2006). An AUC value closer to one indicates a good fit and AUC < 0.5 indicates a poor fit (Swets 1988). Predicted habitat suitability values of current and historical climate (as a logistic value) ranged from 0 to 1 (unsuitable to highly suitable).
Meanwhile, principal components analysis (PCA) method was also used to define the current and historical climate centre (Dallas et al. 2017). To do this, we created a data matrix including 5000 points sampled at random from polygons used in the geographic anaylsis (Lira-Noriega and Manthey 2014). We calculated the first two principal components of the set of eight BioClim/ variables which also were used for MaxEnt (Hijmans et al. 2005), translating geographic points into climatic niche space (Dallas et al. 2017) for current climate. Niche center position was calculated as the mean values of first two principal components (PC1 and PC2). Climate distance to the center was calculated using Euclidean distance between points in niche space created by the two PCA niche axes.
Relative abundance estimation
Spider mites are very small arthropods and have a very short generation time (10–20 generations per year) (Gotoh et al. 2015). Different species with similar morphological characters often co-occur in agriculture (Ehara 1999; Jin et al. 2018). The time-consuming nature of identification and potentially huge population size make it difficult to quantify population density or abundance by direct counting. In this research, we used frequency of occurrence to represent the relative abundance in each location. At each location, 2–6 (average, 5) sampling area (1000 × 10 m) was established to collect mites. To avoid sampling full siblings which could affect estimates of population genetic diversity, a maximum of three mites from each individual plant were collected, and each plant had a minimum distance of 1 m to other plants. The frequency of occurrence in each sample area was calculated as the proportion of sampled TTR individuals, and mean frequency of occurrence across different sample areas for each location were used for demographic tests. In total, 22 locations in different radial transects (center, northeastern, northwestern, and southern) were surveyed from 2013–2015 (Fig. 1).
Population genetic characters
DNA extraction and genotyping
In each location, spider mite samples for genetic analysis were collected and soaked in 95% ethanol for DNA extraction in the lab. DNA was extracted using Qiagen DNeasy Blood and Tissue Kits following the standard protocol (Qiagen Valencia, CA, USA). Individual DNA samples were amplified, and a 741-bp fragment of the mitochondrial cytochrome c oxidase subunit I gene (mtCOI) was sequenced following the protocol described by Chen et al. (2016). Microsatellite variation was genotyped at 14 loci developed by Ge et al. (2013) (TUFG6, TUFG12, TUFG33, TUFG18, TUFG169, TUFG130N) and Zhang et al. (2016) (TUGZ155, TUFZ4, TUFZ3, TUFZ 34, TUFZ30, TUFZ60, TUFZ23). PCR was performed using the protocol described by Zhang et al. (2016). Fragment lengths were sequenced in an ABI 3170 automated sequencer using LIZ-500 size standard. All allele scores were manually checked for quality and consistency.
Genetic diversity and differentiation
Mitochondrial sequences were aligned using the default parameters of CLUSTAL W (Thompson et al. 1994) in Mega 7 (Kumar et al. 2016) and refined by eye. We assessed genetic variation within mtCOI by calculating the nucleotide polymorphisms, number of haplotypes and haplotype diversity (Hd) using DnaSP v5 (Librado and Rozas 2009). Genealogical relationships among mtCOI haplotypes were reconstructed by a haplotype network analysis by the minimum spanning method implemented in PopART version 1.7 (http://popart.otago.ac.nz, Leigh and Bryant 2015). The level of genetic differentiation between pairs of populations was estimated using pairwise Fst values computed from 1000 permutations in DnaSP v5 (Librado and Rozas 2009).
For microsatellites, deviation from Hardy–Weinberg equilibrium (HWE) was tested using GENEPOP on the web (http://genepop.curtin.edu.au/) (Raymond and Rousset 1995). Significance values of HWE were corrected for multiple comparisons by applying standard Bonferroni correction. We estimated the microsatellite polymorphisms based on the number of effective alleles, observed and expected (He) heterozygosity using Genalex v6.5 (Peakall and Smouse 2012). Genetic differences among populations were examined using Fst in Genalex v6.5(Peakall and Smouse 2012). The presence of null alleles (NULL) was assessed with MICRO-CHECKER v2.2.3 (van Oosterhout et al. 2004), and the estimate of null allele frequency was performed in FreeNA (Chapuis and Estoup 2007).
Population structure and gene flow
Population genetic structure was determined by the Bayesian model-based clustering analysis in STRUCTURE v2.3 (Pritchard et al. 2000). A Markov chain Monte Carlo (MCMC) approach was used to cluster individuals into K panmictic groups by minimizing deviations from HWE. We performed ten replicates for each simulation from K = 1–16 under the admixed model. Each run was performed with a burn-in of 20,000 iterations and 100,000 MCMC iterations. The number of clusters (K) was selected based on the Delta K method (Evanno et al. 2005) using Structure Harvester (Earl and vonHoldt 2012). Cluster matching was conducted in CLUMPP v1.1.2 (Jakobsson and Rosenberg 2007). Distruct 1.1 (Rosenberg 2004) was used to create bar charts for the output data derived from CLUMPP.
The asymmetric migration rate among populations was calculated using Fst and Nm statistics with the divMigrate function in the R package diveRsity (Keenan et al. 2013). This approach facilitates estimation of directional gene flow between pairs of populations with low computational effort using any classical or modern measures of genetic differentiation. Recent gene flow was estimated using BayesAss + v1.3 (Wilson and Rannala 2003). This software package uses a Bayesian MCMC resampling method to estimate the posterior probability distribution of the proportion of migrants between populations (Islam et al. 2014).
Population abundance and genetic tests of the CPH
Linear regression was used to test whether our three center–periphery measurements (geography, current and historical ecological suitability) can predict frequency of population occurrence and population genetic diversity (mitochondrial, Hd, and microsatellites, He). Model validation was performed using the function “gvlma” in R packages gvlma. This method performs a single global test to assess the linear model assumptions, as well as perform specific directional tests designed to detect skewness, kurtosis, a nonlinear link function, and heteroscedasticity (Peña and Slate 2006). To determine the model that best fitted the occurrence patterns and genetic diversity, we calculated AICc corrected for small sample size (Hurvich and Tsai 1989) for each respective model. The model with the smallest AICc value was considered as the best among all models (Burnham and Anderson 2004). The CPH predicts that genetic differentiation increases at periphery (geographic and climate) of populations. Therefore, we corrected the Fst values for each population using the averages of all pairwise population differentiations (Wei et al. 2016). Then, we carried out linear regression analyses of corrected Fst values against each population’s geographic distance to the center and their habitat suitability. AICc was used to determine the model that best fitted the observed genetic pattern.
Results
Species range and demographic tests of the CPH
The geographic centroid of TTR’s range (35.440N, 114.591E; Fig. 1) was located in the western edge of the North China Plain. Two models of LGM climate showed highly similar patterns (Fig. S4, Pearson’s correlation, r = 0.9552, p < 0.0001). Distance to the niche center Only CCSM4 was used for subsequent analysis. MaxEnt models performed fairly well in predicting current and historical niche model of TTR (AUC training = 0.889, AUC test = 0.851). Higher current ecological suitability was primarily located in the center region and parts of the southern regions (Fig. 2a) and there are three peaks of current habitat suitability along the latitude gradient (Fig. 2b). Compared with the current ENM, the historical ENM showed higher levels of ecological suitability in the center regions with a slight southward trend (Fig. 2c). There is one only peak of historical habitat suitability along the latitude gradient (Fig. 2d).

Ecological habitat suitability estimated with MaxEnt based on 84 point localities for Tetranychus truncatus in a the current climate and b projected climate during the Last Glacial Maximum based on Community Climate System Model 4 and scatter plots between latitude and habitat suitability (c, d) with smooth curve to its points. Suitability values indicate logistic probability of presence
Frequency of occurrence (Table S2) of TTR populations was negatively related to population distance from the geographical center (Fig. 3a, F1,18 = 7.464, r2 = 0.293, p = 0.014) and positively related to the population’s current ecological suitability (Fig. 3b, F1,18 = 8.320, r2 = 0.320, p = 0.009). A weak but significantly positive relationship was detected between frequency of occurrence and historical ecological suitability (Fig. 3c, F1,18 = 4.696, r2 = 0.207, p = 0.044). AICc analyses showed that the current habitat suitability best fitted the difference in frequency of occurrence (AICc = −4.256), followed by current habitat suitability + geography (AICc = −4.009) and geography alone (AICc = −3.595) (Table 1). Climate distance calculated based on PCA got similar results (Table S3). First two components explained more than 80% variation for both current and historical environment conditions (Fig. S3). Frequency of occurrence of TTR populations was negatively related to climate distance from current climate center of (Table S3, F1,18 = 10.151, r2 = 0.396, p = 0.002).

Linear model fit of frequency of occurrence with geography (distance from range centroid in km; log-transformed), ecology (current ecological suitability) and history (historical ecological suitability)
Population genetic tests of the CPH
Twenty-two populations across TTR’s distribution were sampled for population genetic analyses. In the final dataset, 740 individuals from 22 populations were evaluated for mitochondrial polymorphisms. After removing samples with more than six microsatellites missing, 602 individuals from 20 populations were evaluated for microsatellite polymorphisms. Deviations from HWE were detected at 48 of the 280 total locus/population combinations and were largely (50% of instances) limited to the loci TUFG12 and TUFZ30 after standard Bonferroni correction. These loci showed a significant heterozygote deficit. Potential null alleles were identified at 87 of the 280 locus/population combinations using the MICRO-CHECKER, and high null allele frequencies (>0.02) were again largely limited (56%) to TUFG12 and TUFZ30. The present HWE deviation induced by heterozygote deficiency and null alleles was commonly observed in prior genetic studies of Tetranychus species (Boubou et al. 2012; Sun et al. 2012; Zhang et al. 2016); nevertheless, we also used the datasets that excluded one or two loci to calculate population genetic diversity and differentiation. Biogeographical conclusions from these analyses were consistent with those drawn from the full 14-locus dataset. The results below reflect analysis of this full dataset.
No significant relationships were detected between mitochondrial Hd ecology (Fig. 4b, F1,18 = 2.396, r2 = 0.118, p = 0.139), but Hd significantly increased with the historical suitability gradients (Fig. 4c, F1,18 = 13.800, r2 = 0.434, p = 0.002) and decreased with the distance to the center (Fig. 4a, F1,18 = 4.459, r2 = 0.198, p = 0.049) Expected heterozygosity significantly declined with increasing distance from the center of the distribution (Fig. 4d, F1,18 = 7.931, r2 = 0.331, p = 0.012) and significantly increased with ecological suitability (Fig. 4e, F1,16 = 11.640, r2 = 0.421, p = 0.004 and historical suitability gradients (Fig. 4f, F1,16 = 9.880, r2 = 0.382, p = 0.006). AICc analyses showed that the model that best described differences in expected heterozygosity was current habitat suitability + geography (Table 1, AICc = −25.155). None of the three measurements of center and periphery alone explained expected heterozygosity well (Table 1).

Linear model fit of genetic diversity (expected heterozygosity = He; haplotype diversity = Hd) with geography (distance from range centroid in km; log-transformed), ecology (current ecological suitability) and history (historical ecological suitability)
Fst calculated by mtCOI showed no significant relationships with geography (Fig. 5a) and ecology (Fig. 5b), but showed significant relationships with historical ecological suitability.(Fig. 5c, F1,18 = 16.31, r2 = 0.475, p < 0.001). Fst calculated from 14 microsatellites was significantly correlated with the three measurements of center and periphery (Fig. 5d–f); Fst values significantly increased with increasing distance from the center of the distribution (F1,16 = 16.310, r2 = 0.387, p = 0.005), and declined with historical (F1,16 = 11.1901, r2 = 0.412, p = 0.004) and current suitability gradients (F1,16 = 10.12, r2 = 0.418, p = 0.004). AICc analyses showed that the model that best described differences in expected heterozygosity was current habitat suitability + geography (AICc = −75.991). However, none of the three measurements of center and periphery alone explained expected heterozygosity well (Table 1).

Linear model fit of genetic differentiation (corrected Fst value for both mitochondrial and nuclear makers) with geography (distance from range centroid in km; log-transformed), ecology (current ecological suitability) and history (historical ecological suitability)
Using the Delta K method based on structure analyses, K = 2 was the optimal K value. Populations were divided into two major groups (northern and southern, see Fig. 6a). Within the northern group, K = 5 was the optimal number of genetic clusters (Fig. 6b), which indicated geographical structure in the northern group. To detect gene flow among subgroups, northern populations were roughly subdivided into six subgroups (N1–N6) based on genetic structure and geographic position. Within the southern group, K = 3 was the optimal number of genetic clusters (Fig. 6c). The haplotype network indicated a star-like structure with only two haplotypes from southern populations (Fig. S1).

Genetic structure of Tetranychus truncatus (TTR) and gene flow among population groups. a Individual specimens from the 22 populations were grouped into two main clusters (K = 2): north and south. b, c Hierarchical clustering analyses split the north and south clusters into five and three subclusters (K = 5 and 3), respectively; Estimated migration rates among population groups were calculated by diveRsity in R (d) and BayesAss (e), respectively. Thickness of arrows indicates relative strength of migration. f A map of TTR populations showing genetic composition inferred by structure for northern group (b) and south group (c). See Table S2 for explanation of population IDs
The relative migration rate estimated by diveRsity was greater into the northern group from the southern group (Nm = 1) than vice versa (Nm = 0.35) (Fig. 6d). Within the northern group, asymmetrical gene flow between group N4 (near the center regions) and other groups (except N1) were detected, with higher emigration than immigration rates (Fig. 6d). Within the southern, asymmetrical gene flow was detected between groups S2 and S1. The contemporary gene flow between the south and north calculated by BayesAss was relatively low (south to north: 0.0025, north to south: 0.0031). Within the northern group, a higher migration rate was observed among populations in the central regions (N4, N5, and N6). Again, asymmetrical gene flow between group N4 and most other groups was detected, with higher emigration than immigration rates (Fig. 6e). There was no apparent asymmetrical gene flow within the southern group.
Discussion
The key finding of this study is that geography alone cannot explain population abundance and genetic diversity of TTR, which is a widely distributed arthropod species in eastern China, in the context of the CPH. This was particularly reflected in the distribution patterns of relative population abundance and mitochondrial genetic diversity. The CPH has remained a central hypothesis in biogeography for more than a century (Pironon et al. 2017), although mixed evidence exists. An inappropriate definition of the center may be responsible for the inconsistent patterns relative to the CPH (Wagner et al. 2011; Duncan et al. 2015). Ecological and historical definitions of the CPH should be applied and distinguished with geography (Pfenninger et al. 2011; Pironon et al. 2017). However, such studies were especially lacking in biogeographic research of animal species. (Sagarin and Gaines 2002; Micheletti and Storfer 2015; Pironon et al. 2017). Our study represents one of a few studies that have attempted to reveal the relative effects of current and historical ecological conditions on species population structure within the context of the CPH.
In this study, we defined center and periphery using geography (distance to center) and habitat suitability (ecological and historical suitability based on ENMs). Our geographically defined center and periphery revealed that frequency of occurrence and genetic diversity were significantly related to geography. Lower genetic differentiation in the center region and asymmetrical gene flow were detected in this study. However, population abundance was more related to ecology than geography or history. AICc analyses also showed that the best explanation of occurrence variation among populations was ecology alone (AICc = −4.256). Mitochondrial genetic diversity was only significantly related to history (Fig. 4). In most cases, geography alone could not explain population structure well. These results provided mixed evidence for the CPH and, more importantly, highlight the effects of ecology and history on population variation across a species’ distribution range.
The CPH assumes that geographic distance from the center is associated with environmental suitability, such that a population will be more abundant at the center of the species’ range, where there are stable and favorable conditions (Brown et al. 1995). In this research, TTR relative abundance was more significantly associated with ecological suitability than geographic distance from the center. There is a potentially complex occurrence pattern for this species, because the frequency of occurrence in the eastern coastal area was also relatively high, even though these populations are far away from the geographic center (Table S2). This may occur because the ecological conditions do not precisely coincide with the geographic center and demographic parameters (e.g., vital rates, stochastic population growth rates) may be more related to abiotic environmental quality (Pironon et al. 2017).
In this study, we found that current habitat suitability best fitted to the relative abundance (AICc = −4.256). But the difference in AICc are minimal (AICc ranges between 4.3 and 3) and AICc values were very high compared to the AICc values in population generic tests. This suggests that unmeasured variation (e.g. biotic interactions) may be important in regulating population abundance than the environment (Santini et al. 2019). Although large-scale patterns of distribution or abundance were often related to geographic factors, there were evidences that abundance of such species were strongly associated with the agricultural intensification at a coarse scale (Thomas et al. 2008). TTR is a common species in agriculture. It is possible that abundance and species type of the plant are susceptible to modify the biology of the TTR. The theoretical basis of the CPH (Brown et al. 1995) clearly oversimplifies the reality of spatial demographic variation (Pironon et al. 2015).
Aside from demographic properties, population genetic patterns are also often influenced by ecological conditions (Eckert et al. 2008). Sexton et al. (2014) found that gene flow in natural populations is often ecologically structured (i.e., isolation-by-ecology). In such scenarios, gene flow is stronger among similar environments and greater than predicted under isolation-by-distance (Wang et al. 2013), and the genetic differentiation among populations would thus differ along and across gradients (similar and different environments, respectively) within dispersal limits (Sexton et al. 2014). We studied the population genetic pattern of TTR in eastern China using 14 microsatellite markers. Genetic diversity was significantly higher at both the geographic and ecological centers. Genetic differentiation among populations was also significantly correlated with geography and ecology. Climatic conditions are often strongly related to geography (Wang et al. 2013). Distinguishing the relative effects of geographic and ecological factors on the population genetic patterns has thus emerged in recent years. In our study, we used AICc analyses to choose models that could suitably explain the population patterns. We found that the geography and ecology together was the best explanation of genetic diversity and population divergence. In population genetic studies of invertebrates, the genetic diversity and differentiation patterns have not typically supported the CPH (Pironon et al. 2017). We suggest that the lack of a meaningful definition of the centers in CPH may be one of the reasons for the undetectable CPH patterns.
Recently, several important reviews suggested that studies that test the CPH should incorporate geography and ecology in addition to historical measurements of center and periphery (Sagarin and Gaines 2002; Eckert et al. 2008; Pironon et al. 2017). Because genetic diversity is often a reflection of glacial and post-glacial (re)colonization cycles rather than current conditions (Hewitt 1999; Provan and Bennett 2008). For example, Duncan et al. (2015) found that history matters when explaining genetic diversity within the context of the CPH hypothesis in a widely distributed amphibian (Lithobates sylvaticus) in North America. A similar pattern is also found in plants (Pironon et al. 2015). However, very few studies have explicitly tested the CPH by examining historical vs. contemporary ecological suitability in arthropods (Duncan et al. 2015; Pironon et al. 2017). Our results confirmed that historical suitability was significantly related to TTR population genetic diversity and differentiation using 14 microsatellite markers, although these correlations were weaker than those with ecology and geography in AICc analyses. The mitochondrial genetic patterns seemed to be more associated with history than geography or ecology. Together, these results demonstrated a potential effect of history on shaping population genetic variation.
The complex relationships between population genetic patterns and historical suitability revealed in this study may be a reflection of no unified ice sheet in China during the Quaternary period (Liu et al. 2012). The effect of the LGM on biogeographic patterns in China may not be very strong (Chen et al. 2012). The TTR distribution only slightly contracted southward during the LGM, and historical suitability was very similar to that of the current distribution except in the eastern coastal area. A similar pattern in mainland China was also found for a tree species, Euptelea pleiospermum (Wei et al. 2016), and reported in a region without glaciation (Gugger et al. 2013). Especially, geographic patterns of the mitochondrial and nuclear genetic diversity were different in TTR. Some population maintained higher nuclear genetic diversity but very low mitochondrial genetic diversity. Several process (e.g. adaptive, sex-biased dispersal, demographic expansion) were reported could induce Mito-nuclear discordance (Toews and Brelsford 2012). Male-biased dispersal may drive the Mito-nuclear discordance because of the matrilineal inheritance of mtDNA (Excoffier et al. 2009). However, adult males didn’t manifest the aerial-dispersal behavior (Smitley and Kennedy 1985). This suggested that Mito-nuclear discordance in TTR may be not induced by male-biased dispersal. In our study, we found that TTR seems to have experienced a recent range expansion (Fig. 2). New colonization populations associated with genetic drift effects are expected to have a more pronounced impact in the mitochondrial than the nuclear genome, since mtDNA is haploid and maternally inherited, and therefore has a fourfold smaller effective population size (Sun et al. 2015). Other process like adaptive introgression also promotes this patterns, Gathering more genetic, phenotypic, and environmental data from natural populations will greatly aid in testing alternative explanations (Toews and Brelsford 2012).
High Fst values (average Fst = 0.146) in this research revealed high levels of genetic structure across the TTR distribution range. As expected, we detected asymmetrical gene flow between the south and north (Fig. 6d, f). Contemporary gene flow calculated by BayesAss was greater into south group from north group (0.0031) than vice versa (0.0025). Alternatively, the gene flow calculated by diveRsity was greater into north group from south (relative Nm = 1) than vice versa (relative Nm = 0.35). This pattern was also detected in some other species from China (Wei et al. 2016). Wei et al. (2016) suggested that southward gene flow was linked to population expansion after the LGM. However, the lower genetic diversity and lower historical suitability in the southern TTR populations indicated that the southern regions may not represent suitable refugia for this species. There are multiple glacial refugia across the different regions of China (Liu et al. 2012). The geographic center for TTR in each subregion (north center: group N4; south center: group C2) was very close to the predicted refugia (Qiu et al. 2011). It is possible that ancestral TTR populations survived in glacial refugia in both the north and south. After the LGM, some individuals dispersed northward following host plant range expansion. TTR is a phytophagous arthropod and many of their hosts are domesticated plants (Jin et al. 2018). Many food crops were first domesticated or introduced in the North China Plain (e.g., wheat, sorghum). We inferred that TTR benefitted from agricultural expansion, and the population size may have increased faster than in the south. In addition, the dispersal direction may have recently become inverse from north to south.
The effects of limited gene flow and loss of genetic variation on local adaptation and range limits have received considerable theoretical attention, but the empirical support for the same is lacking (Takahashi et al. 2016). In this study, we found higher Fst values, lower genetic diversity, and limited gene flow in the periphery populations (geographic and ecological TTR populations). This suggested a potentially fragmented distribution pattern near the periphery of their range. Limited gene flow may disrupt their metapopulation network and thus hinder recovery of genetic diversity around their distribution limit. Lower genetic diversity then may prevent adaptation, and decrease the population performance and abundance at the range margin in TTR. This may also could explain the positively significant relationship between microsatellite genetic diversity and relative abundance (Pearson’s correlation, r = 0.584, p = 0.011). However, several other factors (e.g. biotic interactions and geographic barrier) may play important roles on the population genetic and abundance pattern (Ayre et al. 2011; Hansen and Moran 2014; Sexton et al. 2014). The actual causes of geographical patterns of population genetic and abundance thus need more explorations.
Limited and geographically biased sampling may affect the result of CPH studies (McMinn et al. 2017). Although the conclusions with limited sites were statistic reliable (Table S5), there is a sampling bias in our dataset, with only six of 22 sites were from southern regions. We had surveyed more than 180 sites (Jin et al. 2018), TTR was rare in south regions. This is the major reason why only six sites were available for estimation of population genetic diversity and abundance. Limited sites number (22 populations) majorly resulted from the inevitable trade-off between within-population effort (for population abundance and genetic analysis) and the number of populations examined (for CPH Identification). More data will give more accurate tests of CPH if possible. In population genetic research of Tetranychus species, HWE deviations and presence of null alleles were frequently reported (Sun et al. 2012; Ge et al. 2013; Sauné et al. 2015). These problems may inflate genetic differentiation, but it is usually difficult to avoid these issues from a technical standpoint (Zhang et al. 2016). In our research, around 17% of the locus–population combinations significantly deviated from HWE and one-third of the locus–population combinations had null alleles. The proportion of deviation from HWE (17%) is relatively lower than that observed in other population genetic research of spider mites (e.g., 20% in Ge et al. 2013; 27% in Boubou et al. 2012). We also used datasets that excluded one or two loci that significantly deviated from HWE and had a high null allele frequency. Biogeographical conclusions from these analyses were similar to those drawn from the full 14-locus dataset. Therefore, we believe that the microsatellite loci used in this study were reliable and had little impact on the examination of genetic diversity and differentiation.
Conclusion
The CPH is a long-standing hypothesis in ecological and biogeographic research. We found statistical support for decreased relative abundance and genetic diversity, and increased genetic differentiation from the geographic center to the periphery, which supports the CPH. However, we found that ecological and historical factors also strongly influenced the abundance and population genetic patterns. Overall, this study demonstrates that geography alone cannot sufficiently explain the genetic and abundances pattern and highlights the effects of ecological suitability and historical events, such as post-glacial recolonization, on population variation across the species’ distribution range.
Data availability
Genotype data of the Tetranychus truncatus populations is available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.80gb5mkn3. Sampling information was uploaded as online supplemental material (Table S1-S2).
References
Ayre DJ, Coulson LA, Perrin C, Roberts DG, Minchinton TE (2011) Can limited dispersal or biotic interaction explain the declining abundance of the whelk, Morula marginalba, at the edge of its range? Biol J Linn Soc 103:849–862
Boubou A, Migeon A, Roderick GK, Auger P, Cornuet JM, Magalhães S et al. (2012) Test of colonisation scenarios reveals complex invasion history of the red tomato spider mite Tetranychus evansi. PLoS ONE 7:e35601
Brown JH (1984) On the relationship between abundance and distribution of species. Am Nat 124:255–279
Brown JL (2014) SDMtoolbox: a python-based GIS toolkit for landscape genetic, biogeographic and species distribution model analyses. Methods Ecol Evol 5:694–700
Brown JH, Mehlman DW, Stevens GC (1995) Spatial variation in abundance. Ecology 76:2028–2043
Burnham KP, Anderson DR (2004) Multimodel inference: understanding AIC and BIC in model selection. Socio Methods Res 33:261–304
Cane MA, Braconnot P, Clement A, Gildor H, Joussaume S, Kageyama M et al. (2006) Progress in paleoclimate modeling. J Clim 19:5031–5057
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Chen Y, Compton SG, Liu M, Chen XY (2012) Fig trees at the northern limit of their range: the distributions of cryptic pollinators indicate multiple glacial refugia. Mol Ecol 21:1687–1701
Chen Y-T, Zhang Y-K, Du W-X, Jin P-Y, Hong X-Y (2016) Geography has a greater effect than Wolbachia infection on population genetic structure in the spider mite, Tetranychus pueraricola. Bull Entomol Res 106:685–694
Dallas T, Decker RR, Hastings A (2017) Species are not most abundant in the centre of their geographic range or climatic niche. Ecol Lett 20:1526–1533
Dixon AL, Herlihy CR, Busch JW (2013) Demographic and population-genetic tests provide mixed support for the abundant centre hypothesis in the endemic plant Leavenworthia stylosa. Mol Ecol 22:1777–1791
Duncan SI, Crespi EJ, Mattheus NM, Rissler LJ (2015) History matters more when explaining genetic diversity within the context of the core-periphery hypothesis. Mol Ecol 24:4323–4336
Earl DA, vonHoldt BM (2012) Structure Harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Eckert CG, Samis KE, Lougheed SC (2008) Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Mol Ecol 17:1170–1188
Ehara S (1999) Revision of the spider mite family Tetranychidae of Japan (Acari, Prostigmata). Species Divers 4:63–141
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481–501
Franklin J (2013) Species distribution models in conservation biogeography: developments and challenges. Divers Distrib 19:1217–1223
Ge C, Sun JT, Cui YN, Hong XY (2013) Rapid development of 36 polymorphic microsatellite markers for Tetranychus truncatus by transferring from Tetranychus urticae. Exp Appl Acarol 61:195–212
Gotoh T, Moriya D, Nachman G (2015) Development and reproduction of five Tetranychus species (Acari: Tetranychidae): do they all have the potential to become major pests? Exp Appl Acarol 66:453–479
Gugger PF, Ikegami M, Sork VL (2013) Influence of late Quaternary climate change on present patterns of genetic variation in valley oak, Quercus lobata Née. Mol Ecol 22:3598–3612
Hampe A, Petit RJ (2005) Conserving biodiversity under climate change: the rear edge matters. Ecol Lett 8:461–467
Hansen AK, Moran NA (2014) The impact of microbial symbionts on host plant utilization by herbivorous insects. Mol Ecol 23:1473–1496
Hanski I, Kuussaari M, Nieminen M (1994) Metapopulation structure and migration in the butterfly Melitaea Cinxia. Ecology 75:747–762
Hansson L (1991) Dispersal and connectivity in metapopulations. Biol J Linn Soc 42:89–103
Hengeveld R, Haeck J (1982) The distribution of abundance. I. Measurements. J Biogeogr 9:303
Hewitt GM (1999) Post-glacial re-colonization of European biota. Biol J Linn Soc 68:87–112
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978
Holt RD, Keitt TH (2000) Alternative causes for range limits: a metapopulation perspective. Ecol Lett 3:41–47
Hurvich CM, Tsai CL (1989) Regression and time series model selection in small samples. Biometrika 76:297–307
Islam MS, Lian C, Kameyama N, Hogetsu T (2014) Low genetic diversity and limited gene flow in a dominant mangrove tree species (Rhizophora stylosa) at its northern biogeographical limit across the chain of three Sakishima islands of the Japanese archipelago as revealed by chloroplast and nuclear SSR. Plant Syst Evol 300:1123–1136
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jiang XL, An M, Zheng SS, Deng M, Su ZH (2018) Geographical isolation and environmental heterogeneity contribute to the spatial genetic patterns of Quercus kerrii (Fagaceae). Heredity 120:219–233
Jin P-Y, Tian L, Chen L, Hong X-Y (2018) Spider mites of agricultural importance in China, with focus on species composition during the last decade (2008–2017). Syst Appl Acarol 23:2087–2098
Johansson H, Stoks R, Nilsson-Örtman V, Ingvarsson PK, Johansson F (2013) Large-scale patterns in genetic variation, gene flow and differentiation in five species of European Coenagrionid damselfly provide mixed support for the central-marginal hypothesis. Ecography 36:744–755
Keenan K, Mcginnity P, Cross TF, Crozier WW, Prodöhl PA (2013) DiveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788
Keitt TH, Lewis MA, Holt RD (2001) Allee effects, invasion pinning, and species’ borders. Am Nat 157:203–216
Reddy S, Dávalos LM (2003) Geographical sampling bias and its implications for conservation priorities in Africa. J Biogeogr 30:1719–1727
Kubisch A, Poethke HJ, Hovestadt T (2011) Density-dependent dispersal and the formation of range borders. Ecography 34:1002–1008
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis Version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lira-Noriega A, Manthey JD (2014) Relationship of genetic diversity and niche centrality: a survey and analysis. Evolution 68:1082–1093
Liu JQ, Sun YS, Ge XJ, Gao LM, Qiu YX (2012) Phylogeographic studies of plants in China: advances in the past and directions in the future. J Syst Evol 50:267–275
McMinn RL, Russell FL, Beck JB (2017) Demographic structure and genetic variability throughout the distribution of Platte thistle (Cirsium canescens Asteraceae). J Biogeogr 44:375–385
Micheletti SJ, Storfer A (2015) A test of the central-marginal hypothesis using population genetics and ecological niche modelling in an endemic salamander (Ambystoma barbouri). Mol Ecol 24:967–979
Peakall R, Smouse PE (2012) GenALEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539
Peña EA, Slate EH (2006) Global validation of linear model assumptions. J Am Stat Assoc 101:341–354
Pérez-Collazos E, Sanchez-Gómez P, Jiménez F, Catalán P (2009) The phylogeographical history of the Iberian steppe plant Ferula loscosii (Apiaceae): a test of the abundant-centre hypothesis. Mol Ecol 18:848–61
Pfenninger M, Salinger M, Haun T, Feldmeyer B (2011) Factors and processes shaping the population structure and distribution of genetic variation across the species range of the freshwater snail Radix balthica (Pulmonata, Basommatophora). BMC Evol Biol 11:135
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecol Model 190:231–259
Phillips SJ, Dudík M (2008) Modeling of species distributions with MaxEnt: new extensions and a comprehensive evaluation. Ecography 31:161–175
Pironon S, Papuga G, Villellas J, Angert AL, García MB, Thompson JD (2017) Geographic variation in genetic and demographic performance: new insights from an old biogeographical paradigm. Biol Rev 92:1877–1909
Pironon S, Villellas J, Morris WF, Doak DF, García MB (2015) Do geographic, climatic or historical ranges differentiate the performance of central versus peripheral populations? Glob Ecol Biogeogr 24:611–620
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–59
Provan J, Bennett K (2008) Phylogeographic insights into cryptic glacial refugia. Trends Ecol Evol 23:564–71
Qiu YX, Fu CX, Comes HP (2011) Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol Phylogenet Evol 59:225–244
Raymond M, Rousset F (1995) GENEPOP (Version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rivadeneira MM, Hernáez P, Antonio Baeza J, Boltaña S, Cifuentes M, Correa C et al. (2010) Testing the abundant-centre hypothesis using intertidal porcelain crabs along the Chilean coast: linking abundance and life-history variation. J Biogeogr 37:486–498
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Sagarin RD, Gaines SD (2002) The ‘abundant centre’ distribution: to what extent is it a biogeographical rule? Ecol Lett 5:137–147
Santini L, Butchart SHM, Rondinini C, Benítez-López A, Hilbers JP, Schipper AM et al. (2019) Applying habitat and population-density models to land-cover time series to inform IUCN Red List assessments. Conserv Biol 33:1084–1093
Sauné L, Auger P, Migeon A, Longueville J-E, Fellous S, Navajas M (2015) Isolation, characterization and PCR multiplexing of microsatellite loci for a mite crop pest, Tetranychus urticae (Acari: Tetranychidae). BMC Res Notes 8:247
Smitley DR, Kennedy GG (1985) Photo-oriented Aerial-dispersal Behavior of Tetranychus urticae (Acari: Tetranychidae) enhances escape from the leaf surface. Ann Entomol Soc Am 78:609–614
Sexton JP, Hangartner SB, Hoffmann AA (2014) Genetic isolation by environment or distance: which pattern of gene flow is most common? Evolution 68:1–15
Suárez-Atilano M, Rojas-Soto O, Parra JL, Vázquez-Domínguez E (2017) The role of the environment on the genetic divergence between two Boa imperator lineages. J Biogeogr 44:2045–2056
Sun J-T, Jin P-Y, Hoffmann AA, Duan X-Z, Dai J, Hu G et al. (2018) Evolutionary divergence of mitochondrial genomes in two Tetranychus species distributed across different climates. Insect Mol Biol 27:698–709
Sun J-T, Lian C, Navajas M, Hong X-Y (2012) Microsatellites reveal a strong subdivision of genetic structure in Chinese populations of the mite Tetranychus urticae Koch (Acari: Tetranychidae). BMC Genet 13:8
Sun JT, Wang MM, Zhang YK, Chapuis MP, Jiang XY, Hu G et al. (2015) Evidence for high dispersal ability and mito-nuclear discordance in the small brown planthopper, Laodelphax striatellus. Sci Rep 5:8045
Swaegers J, Mergeay J, Therry L, Larmuseau MHD, Bonte D, Stoks R (2013) Rapid range expansion increases genetic differentiation while causing limited reduction in genetic diversity in a damselfly. Heredity 111:422–429
Swets J (1988) Measuring the accuracy of diagnostic systems. Science 240:1285–1293
Takahashi Y, Suyama Y, Matsuki Y, Funayama R, Nakayama K, Kawata M (2016) Lack of genetic variation prevents adaptation at the geographic range margin in a damselfly. Mol Ecol 25:4450–4460
Toews DPL, Brelsford A (2012) The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol 21:3907–3930
Thomas CD, Bulman CR, Wilson RJ (2008) Where within a geographical range do species survive best? A matter of scale. Insect Conserv Divers 1:2–8
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Travis JMJ, Delgado M, Bocedi G, Baguette M, Bartoń K, Bonte D et al. (2013) Dispersal and species’ responses to climate change. Oikos 122:1532–1540
van Oosterhout C, Hutchinson WF, Wills PM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
van Petegem KHP, Boeye J, Stoks R, Bonte D (2016) Spatial selection and local adaptation jointly shape life-history evolution during range expansion. Am Nat 188:485–498
Wagner V, Durka W, Hensen I (2011) Increased genetic differentiation but no reduced genetic diversity in peripheral vs. central populations of a steppe grass. Am J Bot 98:1173–1179
Wang IJ, Glor RE, Losos JB (2013) Quantifying the roles of ecology and geography in spatial genetic divergence. Ecol Lett 16:175–182
Wei X, Sork VL, Meng H, Jiang M (2016) Genetic evidence for central-marginal hypothesis in a Cenozoic relict tree species across its distribution in China. J Biogeogr 43:2173–2185
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–91
Yaninek JS (1988) Continental dispersal of the cassava green mite, an exotic pest in Africa, and implications for biological control. Exp Appl Acarol 4:211–224
Zhang J, Sun JT, Jin PY, Hong XY (2016) Development of microsatellite markers for six Tetranychus species by transfer from Tetranychus urticae genome. Exp Appl Acarol 70:17–34
Acknowledgements
We sincerely thank Jia Zhang, Jia-Fei Ju, Jian-Xin Sun, Jing-Feng Guo, Xiao Han, Ya-Ting Chen and Yu-Xi Zhu of Nanjing Agricultural University, China for their help with the sample collection and species identification. We thank Feng Cheng from Chinese Academy of Sciences for advice on data analyses. We also thank Dr. Xiao-Li Bing of Nanjing Agricultural University, China for his helpful comments on the manuscript. This study was supported in part by a grant-in-aid for Scientific Research (31672035, 31871976) from the National Natural Science Foundation of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Jin, PY., Sun, JT., Chen, L. et al. Geography alone cannot explain Tetranychus truncatus (Acari: Tetranychidae) population abundance and genetic diversity in the context of the center–periphery hypothesis. Heredity 124, 383–396 (2020). https://doi.org/10.1038/s41437-019-0280-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-019-0280-5
This article is cited by
-
A multiplex direct PCR method for the rapid and accurate discrimination of three species of spider mites (Acari: Tetranychidae) in fruit orchards in Beijing
Experimental and Applied Acarology (2024)
-
Suitable climate space and genetic diversity of the mountain-affiliated moth Cosmosoma maishei (Erebidae: Arctiinae: Arctiini: Euchromiina) in cloud forests of Chiapas, Mexico
Journal of Insect Conservation (2023)
