
Abstract
Developing CRISPR/Cas9-mediated non-transgenic mutants in asexually propagated perennial crop plants is challenging but highly desirable. Here, we report a highly useful method using an Agrobacterium-mediated transient CRISPR/Cas9 gene expression system to create non-transgenic mutant plants without the need for sexual segregation. We have also developed a rapid, cost-effective, and high-throughput mutant screening protocol based on Illumina sequencing followed by high-resolution melting (HRM) analysis. Using tetraploid tobacco as a model species and the phytoene desaturase (PDS) gene as a target, we successfully created and expediently identified mutant plants, which were verified as tetra-allelic mutants. We produced pds mutant shoots at a rate of 47.5% from tobacco leaf explants, without the use of antibiotic selection. Among these pds plants, 17.2% were confirmed to be non-transgenic, for an overall non-transgenic mutation rate of 8.2%. Our method is reliable and effective in creating non-transgenic mutant plants without the need to segregate out transgenes through sexual reproduction. This method should be applicable to many economically important, heterozygous, perennial crop species that are more difficult to regenerate.
Similar content being viewed by others

Introduction
Transgenic technologies provide powerful tools for crop improvement. However, the application of these technologies has been hampered by public apprehension toward potential food safety and gene flow concerns, resulting from the presence and/or expression of transgenes1,2. Recent development of CRISPR/Cas9-mediated genome editing has made targeted mutagenesis an attractive alternative to traditional transgenic technologies3,4. In plants, the most widespread application of CRISPR/Cas9 entails the stable integration of Cas9 endonuclease and single-guide RNA (sgRNA) genes into host genomes5,6,7,8,9,10,11,12,13,14,15,16. When CRISPR/Cas9-mediated gene editing is used in sexually propagated annual crop plants, transgenes (Cas9, sgRNA, and so on) can be eliminated from host genomes following sexual reproduction and screening of segregating populations. This segregation of CRISPR/Cas9 transgenes from mutations of interest can result in non-transgenic mutant plant progeny17,18,19,20. However, this strategy is rarely feasible or practical for vegetatively propagated perennial plants. These plants generally require years to reach sexual maturity; thus, multiple years are needed before sexual reproduction is feasible21. Additionally, these plants are highly heterozygous for genes controlling many important traits, and these traits will segregate and recombine following sexual reproduction, resulting in non-transgenic mutant progeny likely lacking a combination of desirable traits22.
Developing a method to generate CRISPR/Cas9-mediated non-transgenic mutants is highly desirable for many applications of genome editing, particularly for asexually propagated, heterozygous, perennial crop plants. It has been reported that pre-assembled CRISPR/Cas9 ribonucleoproteins can be delivered into protoplasts to induce mutations, without the need for stable integration of CRISPR/Cas9 genes into the host-plant genome23,24,25. Particle bombardment has also been used to deliver CRISPR/Cas9 ribonucleoproteins to wheat and maize cells, producing non-transgenic mutants26,27,28,29. However, working with protoplasts, as well as utilizing biolistics, limits the potential for full-plant regeneration to some species and tissue types. Therefore, it is important to also develop alternative methods to produce non-transgenic CRISPR mutants of perennial crop plant species.
In contrast to the limited success of plant regeneration from protoplasts30,31, plant regeneration from leaf, hypocotyl, epicotyl, shoot, root, cotyledon, or callus explants has been well established for the majority of crop plant species32, including many that are recalcitrant to regeneration from protoplasts33,34. It has also been shown that proteins can be produced following transient expression of Agrobacterium T-DNA genes35,36. In addition, Agrobacterium inoculation protocols have been developed for many perennial crop species37. To circumvent the limited regeneration potential when using CRISPR ribonucleoproteins to produce non-transgenic mutants, we report a method for using Agrobacterium to transiently express the Cas9 and sgRNA genes in plant cells, using tobacco as a model plant and PDS as a model target gene. We have also developed a high-throughput screening protocol utilizing next-generation sequencing in combination with high-resolution DNA melting (HRM) analysis to efficiently identify mutants from a population of shoots regenerated in the absence of selection pressure. We demonstrate that the combination of Agrobacterium-mediated transient CRISPR/Cas9 expression with a highly efficient screening protocol makes it possible to efficiently obtain non-transgenic mutant plants, a method that should be applicable to heterozygous perennial crop species.
Results
Production of mutants via Agrobacterium-mediated expression without antibiotic selection
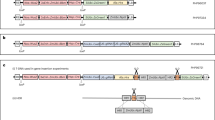
Using tobacco (Nicotiana tabacum Xanthi) as a model plant and an intron-containing GUS gene as a marker (Fig. 1a), we observed that transient expression of T-DNA genes in inoculated leaf discs peaked 3–4 days following Agrobacterium infection in the absence of kanamycin selection. Figure 1d shows the GUS activity in tobacco leaf discs 2–6 days post infection (dpi), and Fig. 1e shows the GUS activity in leaf discs after 5 additional days in timentin-containing media. The antibiotic timentin was used to suppress Agrobacterium growth following an initial 2–6 day co-incubation; thus, the GUS activities shown in Fig. 1e should result from stable integration of the GUS gene into the tobacco genome. The difference in GUS expression between explants in Fig. 1d and those in Fig. 1e is indicative of transient GUS expression, demonstrating that there are high levels of transient expression of the genes in the T-DNA region. The results in Fig. 1d, e indicated that a 3-day or 4-day co-incubation was optimal for Agrobacterium-mediated transient expression of the GUS gene. Three days of Agrobacterium co-incubation was subsequently used for transient expression of CRISPR/Cas9 genes.

a, b T-DNA constructs for (a) GUS and (b) CRISPR/Cas9 (Cas9 and a PDS-targeted sgRNA) expression. The GUS construct consists of a GUSPlus gene interrupted by a cas1 intron (in), to prevent bacterial expression, under the control of a CaMV 35S promoter, and a kanamycin resistance gene (KanR). b The CRISPR construct (hCas9-NtPDS) consists of a Cas9 gene interrupted with an IV2 intron (in), an sgRNA targeting the tobacco PDS gene under the control of a U6 promoter, and a kanamycin (KanR) resistance gene. Primer sets 1, 2, and 3 were used for PCR analysis to determine the presence of T-DNA fragments stably integrated into the tobacco genome. c The fourth exon of the tobacco PDS gene was selected as the target site for the sgRNA. d, e Histochemical staining of GUS activity in tobacco leaf discs (d) without or (e) with timentin to suppress Agrobacterium growth. d GUS activity in tobacco leaf discs 2–6 days following infection by Agrobacterium. e Reduction in GUS activity in tobacco explants transferred to timentin-containing media, to suppress Agrobacterium growth, for an additional 5 days following the initial 2–6 day co-incubation. The difference in GUS expression activities between explants in d and those in e indicated transient GUS expression demonstrating that transient GUS expression peaked at 3–4 days post infection
The sgRNA used in these experiments (Fig. 1b) targets the beginning of the fourth exon of the endogenous tobacco phytoene desaturase gene (PDS), as shown in Figure 1c. Previous studies have shown that disruption of this gene leads to an albino phenotype38. We used this phenotype as a visual marker to identify tobacco mutants whose PDS gene had been edited following the expression of Cas9 and PDS-targeting sgRNA genes. We infected 415 tobacco leaf-disc explants in three independent experiments using 3 days of Agrobacterium co-incubation without any selection for transgenic cells or shoots (Table 1). A total of 197 shoots regenerated from infected explants exhibited the albino phenotype, indicating a mutation in PDS, demonstrating a mutation rate of 0.475 pds mutants per explant. However, due to the lack of chemical selection, the total number of shoots regenerated from each explant was very high, and therefore, the mutation rate per total regenerated shoots was quite low (2.57%, Supplementary Table 1). These results indicate that pds mutant plants can be produced via Agrobacterium-mediated expression of Cas9 and sgRNA genes without using antibiotic selection. Ten independent pds mutant plants were randomly chosen for further analysis, and six are shown in Table 2. The specific genetic mutations in these plant lines were identified via high-throughput sequencing, which demonstrated that all plants contained tetra-allelic mutations, meaning that all four alleles were mutagenized and no wild-type alleles could be detected. Microscopic analysis of plant tissues was unable to uncover the presence of green cells in albino pds mutants, suggesting that all PDS genes in all cells were mutated in these plants.
The albinism resulting from the disrupted PDS gene enabled us to conveniently identify pds mutant shoots at early stages of shoot development in this study. However, the vast majority of desirable mutations for crop improvement are unlikely to display any visually identifiable phenotypes at the early stages of shoot development. When no selection pressure is applied during callus and shoot regeneration following Agrobacterium infection, the vast majority of regenerated shoots or plantlets should be non-mutant, as demonstrated above. Therefore, the ability to efficiently identify mutants lacking any visually identifiable phenotype from a population of regenerated shoots is essential for using the above-described Agrobacterium-mediated transient mutagenesis system. Toward this end, we tested the effectiveness and efficiency of a two-step screening method using the newly produced pds mutants. The first step, an initial identification of mutants, takes advantage of the high-throughput nature of Illumina sequencing, and the second step, a fine identification of mutants, makes use of the high resolution of HRM analysis.
Initial screening of CRISPR-mediated mutants using high-throughput DNA sequencing analysis
Although our mutagenesis rate was relatively high per explant, without a visible phenotype, it would be difficult to identify mutant plants due to the high number of regenerated shoots in the absence of chemical selection. Additionally, other CRISPR mutagenesis projects could have an even lower mutation rate than the one reported here. Therefore, we have developed a two-step method for high-throughput screening of shoots to identify the presence of targeted mutations. We first mixed leaf tissue from an albino pds mutant (MT), pds-12, with leaf tissues from independently derived non-mutant shoots (WT), regenerated from Agrobacterium-infected explants, at MT-to-WT ratios of 1:20, 1:41, and 1:83. We isolated genomic DNA from these pooled tissue samples and performed PCR reactions to amplify a 186-bp fragment that contained the sgRNA-target region on the fourth PDS exon (Fig. 1c). PCR products were sequenced on an Illumina platform to ~×60,000 to ×100,000 coverage. Next, we measured the amount of PCR product derived from the MT-to-WT ratios of 1:20, 1:41, and 1:83 mixed tissues and diluted it with ×6 the amount of PCR product (ng) derived from WT plant tissue. The diluted PCR product was also used for Illumina sequencing analysis.
We observed that the PCR products derived from a 42-plant (1MT: 41WT) pooled tissue sample containing the pds-12 mutant showed a drastically elevated nucleotide variant frequency (NVF) at positions 45–51 (red triangles, Fig. 2, showing a 1MT: 41WT pooled tissue sample), which is consistent with verified mutations at these nucleotide positions (Table 2). NVF is a measure of the frequency of abnormal (compared to WT reference) nucleotides detected by DNA sequencing at a given position due to mutations or sequencing error. When we diluted the same PCR products with 6× WT PCR products (blue squares, Fig. 2), we observed significant reductions in NVF at positions 45–51 relative to the undiluted PCR product. The observed elevations and reductions of NVF before and after a 6× WT DNA dilution further verified the presence of mutations at nucleotide positions 45–51, as NVF resulting from sequencing error would be unaffected by dilution. As shown in Table 2, high-throughput sequencing uncovered four types of mutations in the pds-12 mutant line: a 1-bp deletion at position 51, a 5-bp deletion at positions 45–49, a 2-bp deletion at positions 49–50, and a 4-bp deletion at positions 46–49 (Table 2). Similar results were observed using the 1MT: 20WT and 1MT: 83WT pooled tissue samples, with a more drastic elevation of NVF for the 1MT: 20WT samples and reduced elevated NVF for the 1MT: 83WT samples compared to the 1MT: 41WT samples (data not shown).

The red-marked TGG sequence on the x axis represents the three PAM nucleotides. Black circles represent nucleotide variant frequencies (NVF) of PCR product amplified from wild-type (WT) DNA. Red triangles represent the NVF of PCR product amplified from the genomic DNA of a 42-plant pool containing the pds-12 mutant line, displaying much higher NVF at positions 45–51. Blue squares represent the NVF of the same PCR products as the red triangles, but diluted six times with the WT PCR product, demonstrating that the NVF at nucleotide positions 45–51 are significantly reduced following the dilution. Using the WT PCR product dilution method, mutations at positions 45–51 were identified, consistent with the DNA sequencing results of the pds-12 mutant plant (Table 2)
Using a single-blind approach (the researchers who conducted the experiment did not know which 42-plant groups contained pds mutants), we tested the accuracy of the mutant screening method based on elevations and reductions of NVF before and after a 6× WT DNA dilution as described above. We created eight 42-plant pools, five of which contained plant tissue from a single pds mutant plant line and three of which contained 100% WT plants. The five pds mutants used were pds-9, pds-10, pds-11, pds-13, and pds-14. We confirmed that the screening method was reliable for identifying all 42-plant pools that contained pds mutant plants at a ratio of 1MT: 41WT (Supplementary Figure 1), with 100% accuracy (Table 3). Thus, the elevations and reductions of NVF before and after a 6× WT DNA dilution were excellent indicators of 42-plant pools that contained mutant plants.
Fine identification of mutants using DNA high-resolution melting analysis
After identification of mutant-containing 42-plant pools, HRM analysis was used to identify individual mutant plant lines within each of the 42-plant pools (Supplementary Figure 2). To determine the sensitivity of HRM analysis, we performed HRM analysis on PCR products amplified from various DNA templates combined at different ratios. These template mixes were created using one-part pds-12 plant tissue combined with different parts independently regenerated non-mutant (WT) tissues in the following ratios: 1:1, 1:6, 1:19, and 1:29. Figure 3a shows that mutant-containing PCR products at ratios of 1:1, 1:6, and 1:19 (MT:WT) could be distinguished from a wild-type DNA reference. The plant pool size we chose for subsequent HRM analysis was 7; thus, each 42-plant pool containing DNA from mutants could be divided into six pools of seven plants each.

a HRM analysis of DNA from pooled samples containing pds-12 with various ratios of mutant (MT) to wild-type (WT) plant tissue. The pooled samples were as follows: a 100% WT plant pool, a 2-plant pool (1MT + 1WT), a 7-plant pool (1MT + 6WT), a 20-plant pool (1MT + 19WT), and a 30-plant pool (1MT + 29WT). Melt curves demonstrate that the 20-plant pool sample can be clearly distinguished from those of the 100% WT pool. b HRM analysis of the 7-plant pool containing different pds mutants. HRM curves of the 100% WT pool (negative control) or 7-plant pools containing a single pds-9, pds-10, pds-11, pds-12, pds-13, or pds-14 mutant demonstrate that HRM analysis is effective to identify single mutants from pooled samples of 7-plants. The WT curve is representative of the three negative control pools used in this single-blind screen. Difference RFU values in b represent the average of three replicates at each temperature point. Error bars show variance for each temperature point for each mutant line
We also used a single-blind experiment approach to test the accuracy of HRM analysis to identify mutant plants. We created eight additional 7-plant pools, five containing a single pds mutant each (pds-9, pds-10, pds-11, pds-13, or pds-14), and the remaining three negative control pools containing only wild-type plants. The HRM analysis results are shown in Fig. 3b and demonstrate that all pooled samples containing pds mutant plants could be identified with 100% accuracy. Finally, upon the identification of mutant-containing 7-plant pools, individual mutant(s) within each pool were identified via HRM based on a 1:1 mix between each putative mutant and a WT plant. Through this method, we successfully identified all pds mutant plants (Table 2).
Determination of non-transgenic pds mutants
To distinguish transgenic from non-transgenic mutants, we performed PCR on 29 randomly selected pds mutant lines using primers targeted to three regions in the T-DNA fragment (Fig. 1b). Mutant plants were considered to be non-transgenic if they lacked a PCR product for all three primer sets (Fig. 4). Approximately 17.2% of the tested pds mutant lines were determined to be non-transgenic following PCR analysis (Table 4). As shown in Supplementary Figure 3, a non-transgenic plant (pds-7), along with a transgenic plant (pds-9), was cultured on MS media containing 100 mg/L kanamycin. The non-transgenic pds-7 plant died under kanamycin selection, while the transgenic pds-9 plant grew normally.

Primer set 1 was used to amplify the T-DNA region between the left T-DNA border and the 5′ end of the CaMV 35S promoter. Primer set 2 was used to amplify the 3′ end of the Cas9 coding sequence. Primer set 3 was used to amplify a region of the kanamycin resistance gene (KanR). The absence of all three T-DNA PCR fragments was indicative of non-transgenic plants. WT wild type
Discussion
Producing non-transgenic mutants of heterozygous perennial crop plants using CRISPR/Cas9 technology is highly desirable but challenging. We developed an effective method for producing and identifying CRISPR/Cas9-mediated non-transgenic mutant plants, which should be applicable to many perennial heterozygous crop plants. We have demonstrated that we can use Agrobacterium to transiently express CRISPR/Cas9 genes, and such expression can lead to the production of tetra-allelic, non-transgenic mutant plants. We have also demonstrated that the first step of our mutant identification, based on elevated and reduced nucleotide variance frequencies before and after a WT DNA dilution, using high-throughput DNA sequencing analysis, is reliable and highly efficient. Furthermore, the second step of mutant identification, using HRM analysis, is simple and effective.
With one sgRNA targeting the tobacco PDS gene, we achieved a 47.5% mutation rate (i.e., 0.475 pds mutants per explant) using no selective pressure during callus or shoot regeneration. At least 17.2% of the pds mutant plants produced this way were non-transgenic, for an overall non-transgenic mutation rate of 8.2% (i.e., 0.082 pds mutants per explant). We expect that the rate of recovery for non-transgenic mutant plants following Agrobacterium-mediated transient expression of CRISPR/Cas9 genes could be much higher than we reported here. One reason is that the albino phenotype caused by pds mutations used in this study can result in cell- or shoot-growth disadvantages, which may have contributed to lower rates of mutant shoot production. Additionally, multiple sgRNA sequences may be used to target the same gene39 to increase the efficiency of mutant production.
Protoplast-mediated delivery of CRISPR/Cas9 ribonucleoproteins offers advantages for creating non-transgenic mutant plants23,24,25,26,27,28,40. However, regenerating plants from protoplasts can be difficult and has not been demonstrated to be possible for many important crop species. Low efficiency of plant regeneration from protoplasts has been reported in economically important crops such as avocado (Persea americana)41, grape (Vitis vinifera)42, and apple (Malus domestica)43. Furthermore, regeneration protocols have not been successfully demonstrated in many other plant species31,44. In the case of citrus, for instance, a globally important fruit crop that is often heterozygous for important trait genes, protoplast regeneration can be achieved only if protoplasts are derived from juvenile tissues45. However, plants generated from juvenile citrus tissues require years to reach a mature fruit production stage, and this delay is an impediment to the efficient evaluation of gene functions in fruit and for other genes pertinent to citrus breeding programs46,47. On the other hand, tissues from mature citrus trees, such as shoot segments, can be transformed with Agrobacterium48.
Jacobs et al.49 discussed the possibility of using Agrobacterium-mediated transient expression of Cas9 and sgRNA genes to produce non-transgenic mutant plants. Iaffaldano et al.50 reported the production of CRISPR/Cas9-mediated mutant plants using Agrobacterium without any selection of stable transgenic plants. However, Iaffaldano et al.50 did not characterize whether their mutant plants were transgenic or non-transgenic, nor did they calculate the percentage of each type. The non-transgenic mutation rate reported in this study is lower than mutation rates for the previously reported protoplast microinjection methods25 but higher than for particle bombardment methods (per explant)26,27,28. However, Agrobacterium can be a more versatile tool because of its ability to efficiently infect many different tissues types across most plant species51,52, and it is the most widely used method for plant transformation. Thus, protocols for Agrobacterium infection and subsequent callus and shoot regeneration are readily available for many crop plant species. Furthermore, even when other methods are applicable, Agrobacterium infection procedures are relatively simple and easy to perform for many plant species. Thus, the method presented here provides an alternative method for the production of non-transgenic mutant plants.
Our method of using Agrobacterium to transiently express CRISPR/Cas9 genes, without the use of chemical selection such as kanamycin, also offers some other advantages. The lack of a chemical selection agent following Agrobacterium infection allows for even greater rates of plant regeneration compared to when chemical selection is used and therefore likely enhances mutant callus/shoot production. This is because selection agents, such as kanamycin, hygromycin, and various herbicides, can suppress shoot regeneration for many plant species, even when those shoots express relevant resistance genes53. Furthermore, our high-throughput screening method, based on a novel DNA sequencing strategy in combination with HRM analysis, makes mutant identification fast and easy to perform. The drawback of our method is that there is a risk of creating transgenic mutant plants; however, identifying and discarding these plants does not add any significant time to the screening process.
We have observed that the threshold of mutant plant detection using the Illumina sequencing method can be much more sensitive than the detection of one mutant out of 42 plants (the 1MT: 41WT) described in this study. The use of the 42-plant sample pools, therefore, assures that the presence of a mutant plant can be readily detected in pooled samples. Additionally, the threshold of HRM analysis to detect mutant plants can be as low as 1 of 19 plants, but we chose one out seven (1MT: 6WT). Again, this pooling choice provides a safe margin for error by reducing the chance of missing mutant plants due to false negatives. Furthermore, a small number of false positives from the sequencing analysis of 42-plant pools are not a concern because they can be easily identified in the subsequent step of screening. Thus, our method, which combines a unique DNA sequencing strategy with HRM analysis, provides a reliable protocol to screen for CRISPR/Cas9-mediated mutant shoots from a population of shoots regenerated in the absence of selection pressure. The overall protocol for mutant identification is schematically represented in Supplementary Figure 4. Using this protocol, it would take ~14 days to identify all mutants from 1000 independent shoots that were subjected to Agrobacterium-mediated transient expression of CRISPR/Cas9 genes.
In conclusion, we have developed a highly useful method utilizing Agrobacterium-mediated transient expression of Cas9 and sgRNA genes combined with multi-step pooled screening, enabling the reliable production and efficient identification of non-transgenic mutants regenerated in the absence of selection pressure. Due to the versatility of CRISPR/Cas9 and Agrobacterium-mediated infection, as well as the ease of plant regeneration from leaf, shoot, or root explants, this method is applicable to many economically important plant species, particularly heterozygous perennial plant species that are recalcitrant to regeneration from protoplasts or following biolistic bombardment.
Materials and methods
Gene constructs
To determine the optimum experimental conditions for maximum transient expression of T-DNA in plant cells, we used pCAMBIA1305.1, a plasmid containing an intron-containing GUS gene under the control of a CaMV 35S promoter and a NOS terminator. We modified this construct by adding a kanamycin resistance gene (NPTII) under the control of a CaMV 35S promoter and CaMV 35S terminator using the KpnI and XbaI restriction enzymes (Fig. 1a). For CRISPR/Cas9-based disruption of the tobacco PDS gene, we made a number of modifications to the pK7WGF2::hCas9 plasmid (Addgene plasmid #46965). First, we added an IV2 intron to a Cas9 gene to prevent Cas9 expression in Agrobacterium, this gene was used to replace the original Cas9 DNA in the vector using the SpeI and ApaI restriction enzyme sites. Second, we added an sgRNA targeting the fourth exon of the PDS gene in Nicotiana benthamiana54 under the control of the Arabidopsis thaliana U6–26 gene promoter (accession # At3G13855), using the XbaI and KpnI restriction enzymes. All of the added polynucleotides were synthesized by Genscript Corporation (Piscataway, NJ, USA). The final CRISPR/Cas9-containing plasmid was named hCas9-NtPDS (Fig. 1b).
Agrobacterium-mediated transformation for transient expression
A. tumefaciens (EHA105) was used to deliver Ti-plasmid DNA into tobacco leaf-disc explants. Agrobacterium cells were cultured overnight (16 h, 200 r.p.m., 28 °C) in 5 mL of liquid LB medium containing 100 mg/L spectinomycin. The overnight culture was diluted with 50 mL of fresh liquid LB media (1:10 dilution) and then grown for 6–8 h (200 r.p.m., 28 °C). Agrobacterium cells were collected at an OD600 of 0.6–0.8, centrifuged at 5000 r.p.m. for 15 min, and finally re-suspended in 50 mL of liquid MS medium containing 100 µM acetosyringone (AS). Re-suspended bacterial cells were shaken (180 r.p.m., 28 °C) for 1 h before use. Tobacco leaf discs (0.5 cm2, from vegetatively propagated clonal plants) were incubated in Agrobacterium cell solution for 20 min. Explants were blotted dry on sterile filter paper and transferred onto solid MS media containing 100 µM AS and no antibiotics.
Histochemical GUS activity assay
To optimize the time needed for robust transient T-DNA expression, tobacco leaf explants inoculated with Agrobacterium (containing the pCAMBIA1305.1 vector) were incubated in the dark at 25 °C for 2, 3, 4, 5, or 6 days. Half of the leaf discs (six) from each treatment were stained for GUS activity55. The remaining explants were transferred to solid MS medium containing 150 mg/L timentin to suppress Agrobacterium growth. These explants were stored in the dark for an additional 5 days. Leaf discs from each treatment were histochemically stained to estimate stable versus transient expression levels of the GUS gene.
Callus and shoot regeneration
For transient expression of the CRISPR/Cas9 genes, tobacco leaf discs were incubated in the dark at 25 °C for 3 days before being transferred to solid media containing MS salts, 2.0 mg/L benzylaminopurine (to promote shoot initiation), and 150 mg/L timentin (to repress Agrobacterium growth). No antibiotics for the selection of transgenic plant cells were included in the culture media. The plates were stored at 25 °C under a 16-h photoperiod. After 4 weeks, visual screening was performed to identify white-colored mutant shoots. The white shoots were then transferred to solid MS media containing 150 mg/L timentin for rooting. Genomic DNA was isolated using the NucleoSpin plant II kit (Macherey-Nagel, Dueren, Germany). The primer pair PDS-F (5′-CTGAAGCAGTCACCAAGA-3′) and PDS-R (5′-AGTACGCATTCTTGAGGAGTC-3′) was used to amplify the sgRNA-target region. A second round of PCR was performed, with the primer pair PDS-FA (5′–ACACTCTTTCCCTACACGACGCTCTTCCGATCTCTGAAGCAGTCACCAAGA-3′) and PDS-RA (5′–GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTAGTACGCATTCTTGAGGAGTC-3′), using the PCR product of the first round as the template to add general adapter sequences to the PCR products. A final round of PCR was performed to add a barcode tag to the PCR products, with the primer pair PDS-FAI (5′-AATGATACGGCGACCACCGAGATCTACAC-index-ACACTCTTTCCCTACACGA-3′) and PDS-RAI (5′-CAAGCAGAAGACGGCATACGAGAT-index-GTGACTGGAGTTCAGACGTG-3′). Sequencing reactions were performed using the barcoded PCR products and the Illumina MiSeq platform (paired-end ×2 150 bp read length). The raw Illumina reads were mapped to the 186-bp reference sequence in the tobacco genome using bwa mem (-c 300000 –v 2) of BWA v0.5.956 to determine the nature of mutations.
High-throughput sequencing
For the 21-, 42-, and 84-plant pools used in the high-throughput sequencing screen, leaf tissue from pds-12 mutant plants was combined with an equal amount of tissue from each of 20, 41, or 83 independently regenerated non-mutant shoots, respectively. One control pool was created using the tissue from 84 independently regenerated non-mutant shoots. Genomic DNA was isolated, amplified, tagged, and barcoded as described previously. For dilution, the PCR product amplified from the 42-plant pool was quantified using a NanoDrop and was then diluted with ×6 the amount of PCR product (ng) amplified from a pool composed of 84 independently regenerated non-mutant shoots. Sequencing reactions were performed in triplicate using the barcoded PCR products and the Illumina MiSeq platform (paired-end ×2 150 bp read length). The raw Illumina reads were mapped to the 186-bp reference sequence in the tobacco genome using bwa mem (-c 300000 –v 2) of BWA v0.5.956. NVFs were calculated over a 77-bp region. To confirm the high-throughput screening method, five additional mutant-containing 42-plant pools were created, each comprising tissue from a single mutant plant (pds-9, pds-10, pds-11, pds-13, or pds-14) in combination with tissue from 41 independently regenerated non-mutant shoots. Three WT shoot control pools were created using the tissue from 42 independently regenerated non-mutant shoots. All eight pools were given randomly generated labels to assure that high-throughput sequencing screening could be performed in a single-blind manner.
HRM analysis
For 2-, 7-, 20-, and 30-plant pools used in the high-resolution melting analysis screen, leaf tissue from pds-12 mutant plants was combined with an equal amount of tissue from each of 1, 6, 19, or 29 independently regenerated non-mutant shoots, respectively. One control pool was created using the tissue from 30 independently regenerated non-mutant shoots. Genomic DNA was isolated using the NucleoSpin plant II kit (Macherey-Nagel). We used primers PDS-F and PDS-R (described previously) to amplify the sgRNA-target region. We also tested 85 bp (F primer: 5′-ATCTGTTCTGCACCTGAATAC-3′, R primer: 5′-AACCCAGTCTCATACCAA-3′) and 146 bp (F primer: 5′-CTGAAGCAGTCACCAAGA-3′, R primer: 5′-AACCCAGTCTCATACCAA-3′) PCR products for HRM analysis. The 186-bp fragments were used for our experiments, as shown in our results. PCR amplifications were performed in 10 µL volumes containing 5 µL of Precision Melt Supermix (Bio-Rad, USA), 0.5 µL of each 2 µM primer, and 50 ng of genomic DNA. PCR was carried out in triplicate using 96-well white-walled PCR plates and a CFX96™ Real-Time PCR Detection System (Bio-Rad). The amplification started with an initial denaturation step at 95 °C for 5 min, followed by 50 cycles of 95 °C for 10 s, 60 °C for 30 s, and 72 °C extension for 1 min. High-resolution melting analyses of the PCR amplicons were carried out in the same plate immediately following PCR amplification, using the following cycle: 95 °C for 30 s and 60 °C for 1 min. Melting curves were generated over a 65–95 °C range with 0.2 °C increment. Melting curves were analyzed using Precision Melt Analysis software (Bio-Rad). To confirm our HRM screening method, five mutant-containing 7-plant pools were created, each comprised tissue from a single mutant plant (pds-9, pds-10, pds-11, pds-13, or pds-14) in combination with tissue from six independently regenerated non-mutant shoots. Three control pools were created using the tissue from seven independently regenerated non-mutant shoots. The eight pools were labeled randomly to assure that HRM screening was performed in a single-blind manner.
Determination of non-transgenic mutant plants
Stable integration of the CRISPR/Cas9 T-DNA was determined by PCR analysis of the tobacco genomic DNA. Primers 1F (5′-AGGTGGCGAAGTCATCTGC-3′) and 1R (5′-TGTCGTTTCCCGCCTTCAG-3′) were used to amplify a 701-bp region of the T-DNA fragment. Primers 2F (5′-GCCTGTTTGGTAATCTTATCGC-3′) and 2R (5′-TCTTTCCACTCTGCTTGTCTCG-3′) were used to amplify a 1326-bp fragment of the Cas9 gene. Primers 3F (5′-ACTGGGCACAACAGACAATC-3′) and 3R (5′-ACCGTAAAGCACGAGGAA-3′) were used to amplify a 668-bp fragment of the kanamycin resistance gene. The absence of PCR products from all three primer sets was deemed to be indicative of a lack of stable transgene integration into the tobacco genome, thus indicating a non-transgenic mutant plant. Shoots from pds mutant plants were cultured on MS media containing 100 mg/L kanamycin and allowed to grow for 35 days, producing further evidence for the lack of transgene insertion into the genome.
References
Li, W. et al. Elevated auxin and reduced cytokinin contents in rootstocks improve their performance and grafting success. Plant Biotechnol. J. 15, 1556–1565 (2017).
Tuteja, N., Verma, S., Sahoo, R. K., Raveendar, S. & Reddy, I. B. Recent advances in development of marker-free transgenic plants: regulation and biosafety concern. J. Biosci. 37, 167–197 (2012).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Bortesi, L. & Fischer, R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol. Adv. 33, 41–52 (2015).
Čermák, T. et al. A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 29, 1196–1217 (2017).
Feng, Z. et al. Efficient genome editing in plants using a CRISPR/Cas system. Cell Res. 23, 1229–1232 (2013).
Gil-Humanes, J. et al. High‐efficiency gene targeting in hexaploid wheat using DNA replicons and CRISPR/Cas9. Plant J. 89, 1251–1262 (2017).
Lawrenson, T. et al. Induction of targeted, heritable mutations in barley and Brassica oleracea using RNA-guided Cas9 nuclease. Genome Biol. 16, 258–270 (2015).
Lu, Y. & Zhu, J. K. Precise editing of a target base in the rice genome using a modified CRISPR/Cas9 system. Mol. Plant 10, 523–525 (2017).
Mao, Y. et al. Application of the CRISPR–Cas system for efficient genome engineering in plants. Mol. Plant 6, 2008–2011 (2013).
Xie, K. & Yang, Y. RNA-guided genome editing in plants using a CRISPR–Cas system. Mol. Plant. 6, 1975–1983 (2013).
Xiong, J. S., Ding, J. & Li, Y. Genome-editing technologies and their potential application in horticultural crop breeding. Hortic. Res. 2, 15019 (2015).
Gao, Y. et al. Auxin binding protein 1 (ABP1) is not required for either auxin signaling or Arabidopsis development. Proc. Natl Acad. Sci. USA 112, 2275–2280 (2015).
Gao, Y. & Zhao, Y. Specific and heritable gene editing in Arabidopsis. Proc. Natl Acad. Sci. USA 111, 4357–4358 (2014).
Ma, X. et al. A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol. Plant 8, 1274–1284 (2015).
Char, S. N. et al. An Agrobacterium‐delivered CRISPR/Cas9 system for high‐frequency targeted mutagenesis in maize. Plant Biotechnol. J. 15, 257–268 (2017).
Zhang, H. et al. The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol. J. 12, 797–807 (2014).
Zhou, H., Liu, B., Weeks, D. P., Spalding, M. H. & Yang, B. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. 42, 10903–10914 (2014).
Gao, X., Chen, J., Dai, X., Zhang, D. & Zhao, Y. An effective strategy for reliably isolating heritable and Cas9-free Arabidopsis mutants generated by CRISPR/Cas9-mediated genome editing. Plant Physiol. 171, 1794–1800 (2016).
Artlip, T. S., Wisniewski, M. E., Arora, R. & Norelli, J. L. An apple rootstock overexpressing a peach CBF gene alters growth and flowering in the scion but does not impact cold hardiness or dormancy. Hortic. Res. 3, 16006 (2016).
Norelli, J. L. et al. Genotyping-by-sequencing markers facilitate the identification of quantitative trait loci controlling resistance to Penicillium expansum in Malus sieversii. PLoS ONE 12, e0172949 (2017).
Malnoy, M. et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Front. Plant Sci. 7, 1904 (2016).
Subburaj, S. et al. Site-directed mutagenesis in Petunia×hybrida protoplast system using direct delivery of purified recombinant Cas9 ribonucleoproteins. Plant Cell Rep. 35, 1535–1544 (2016).
Woo, J. W. et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat. Biotechnol. 33, 1162–1164 (2015).
Liang, Z. et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nat. Commun. 8, 14261 (2017).
Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K. & Cigan, A. M. Genome editing in maize directed by CRISPR–Cas9 ribonucleoprotein complexes. Nat. Commun. 7, 13274 (2016).
Zhang, Y. et al. Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nat. Commun. 7, 12617 (2016).
Kim, H. et al. CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat. Commun. 8, 14406 (2017).
Davey, M. R., Anthony, P., Power, J. B. & Lowe, K. C. Plant protoplasts: status and biotechnological perspectives. Biotechnol. Adv. 23, 131–171 (2005).
Eeckhaut, T., Lakshmanan, P. S., Deryckere, D., Van Bockstaele, E. & Van Huylenbroeck, J. Progress in plant protoplast research. Planta 238, 991–1003 (2013).
Gaj, M. D. Factors influencing somatic embryogenesis induction and plant regeneration with particular reference to Arabidopsis thaliana (L.) Heynh. Plant Growth Regul. 43, 27–47 (2004).
Carimi, F., De Pasquale, F. & Crescimanno, F. G. Somatic embryogenesis and plant regeneration from pistil thin cell layers of citrus. Plant Cell Rep. 18, 935–940 (1999).
Paul, H., Belaizi, M. & Sangwan-Norreel, B. S. Somatic embryogenesis in apple. J. Plant Physiol. 143, 78–86 (1994).
Fischer, R. & Emans, N. Molecular farming of pharmaceutical proteins. Transgenic Res. 9, 279–299 (2000).
Pogue, G. P. et al. Production of pharmaceutical‐grade recombinant aprotinin and a monoclonal antibody product using plant‐based transient expression systems. Plant Biotechnol. J. 8, 638–654 (2010).
Krenek, P. et al. Transient plant transformation mediated by Agrobacterium tumefaciens: principles, methods and applications. Biotechnol. Adv. 33, 1024–1042 (2015).
Wang, M., Wang, G., Ji, J. & Wang, J. The effect of pds gene silencing on chloroplast pigment composition, thylakoid membrane structure and photosynthesis efficiency in tobacco plants. Plant Sci. 177, 222–226 (2009).
Xie, K., Minkenberg, B. & Yang, Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl Acad. Sci. USA 112, 3570–3575 (2015).
Andersson, M. et al. Efficient targeted multiallelic mutagenesis in tetraploid potato (Solanum tuberosum) by transient CRISPR-Cas9 expression in protoplasts. Plant Cell Rep. 36, 117–128 (2017).
Litz, R. E. & Grosser, J. W. Isolation, culture and regeneration of avocado (Persea americana Mill.) protoplasts. Plant Cell Rep. 18, 235–242 (1998).
Jardak R., Mliki A., Ghorbel A. & Reustle G. L. Transfer expression of UIDA gene in grapevine protoplasts after PEG-mediated transformation. Int. J. Vine Wine Sci. (2002).
Saito, A. & Suzuki, M. Plant regeneration from meristem-derived callus protoplasts of apple (Malus domestica cv.Fuji’). Plant Cell Rep. 18, 549–553 (1999).
Maćkowska, K., Jarosz, A. & Grzebelus, E. Plant regeneration from leaf-derived protoplasts within the Daucus genus: effect of different conditions in alginate embedding and phytosulfokine application. Plant Cell Tissue Organ Cult. 117, 241–252 (2014).
Bonga J. M. Cell and Tissue Culture in Forestry (Springer, 1987)
Peña, L. et al. Constitutive expression of Arabidopsis LEAFY or APETALA1 genes in citrus reduces their generation time. Nat. Biotechnol. 19, 263–267 (2001).
van Nocker, S. & Gardiner, S. E. Breeding better cultivars, faster: applications of new technologies for the rapid deployment of superior horticultural tree crops. Hortic. Res. 1, 14022 (2014).
Wu, H. et al. Genetic transformation of commercially important mature citrus scions. Crop Sci. 55, 2786–2797 (2015).
Jacobs, T. B., Zhang, N., Patel, D. & Martin, G. B. Generation of a collection of mutant tomato lines using pooled CRISPR libraries. Plant Physiol. 174, 2033–2037 (2017).
Iaffaldano, B., Zhang, Y. & Cornish, K. CRISPR/Cas9 genome editing of rubber producing dandelion Taraxacum kok-saghyz using Agrobacterium rhizogenes without selection. Ind. Crops Prod. 89, 356–362 (2016).
Altpeter, F. et al. Advancing crop transformation in the era of genome editing. Plant Cell 28, 1510–1520 (2016).
Sood, P., Bhattacharya, A. & Sood, A. Problems and possibilities of monocot transformation. Biol. Plant 55, 1–5 (2011).
Wilmink, A. & Dons, J. J. Selective agents and marker genes for use in transformation of monocotyledonous plants. Plant Mol. Biol. Rep. 11, 165–185 (1993).
Gao, J. et al. CRISPR/Cas9-mediated targeted mutagenesis in Nicotiana tabacum. Plant Mol. Biol. 87, 99–110 (2015).
Li, W. et al. An AGAMOUS intron‐driven cytotoxin leads to flowerless tobacco and produces no detrimental effects on vegetative growth of either tobacco or poplar. Plant Biotechnol. J. 14, 2276–2287 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Acknowledgements
We thank the financial support from the USDA National Institute of Food and Agriculture SCRI (grant no. 2015-70016-23027), the Florida Citrus Development Foundation (2016-001) and the Genetically Modified Organisms Breeding Major Projects of China (2014ZX0801008B-001). The Connecticut-Storrs Agriculture Experimental Station, the Priority Academic Program Development of Jiangsu Higher Education Institutions and the Innovative Research Project of JAAS (ZX-17-2006) also contributed financially to some experiments presented in this manuscript. Y.L. holds a 2-month/-year-visiting professor position at Nanjing Agricultural University, China.
Authors’ contributions
Y.L. and L.C. designed the experiments; L.C., W.L., X.G., Y.L., T.G., R.W., J.D., and L.K.-G. conducted transformation and molecular characterization of pds mutants; J.D., W.L., and T.G. conducted DNA sequencing data analysis; W.L., X.G., L.C., and Y.L. performed HRM analysis; L.K.-G., W.L., J.D., T.G., X.L., Z.D., R.J.M., F.G.G., and Z.D. provided suggestions for the experiments; and Y.L., L.K.-G., W.L., J.D., T.G., R.M., F.G.G., Z.D., and Y.Z. wrote/edited the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, L., Li, W., Katin-Grazzini, L. et al. A method for the production and expedient screening of CRISPR/Cas9-mediated non-transgenic mutant plants. Hortic Res 5, 13 (2018). https://doi.org/10.1038/s41438-018-0023-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-018-0023-4
This article is cited by
-
A new biotechnology for in-planta gene editing and its application in promoting flavonoid biosynthesis in bamboo leaves
Plant Methods (2023)
-
Heritable transgene-free genome editing in plants by grafting of wild-type shoots to transgenic donor rootstocks
Nature Biotechnology (2023)
-
Transgene-free genome editing of vegetatively propagated and perennial plant species in the T0 generation via a co-editing strategy
Nature Plants (2023)
-
Recent advances and challenges in potato improvement using CRISPR/Cas genome editing
Planta (2023)
-
Strategic transgene-free approaches of CRISPR-based genome editing in plants
Molecular Genetics and Genomics (2023)
