
Abstract
Transformation in grass species has traditionally relied on immature embryos and has therefore been limited to a few major Poaceae crops. Other transformation explants, including leaf tissue, have been explored but with low success rates, which is one of the major factors hindering the broad application of genome editing for crop improvement. Recently, leaf transformation using morphogenic genes Wuschel2 (Wus2) and Babyboom (Bbm) has been successfully used for Cas9-mediated mutagenesis, but complex genome editing applications, requiring large numbers of regenerated plants to be screened, remain elusive. Here we demonstrate that enhanced Wus2/Bbm expression substantially improves leaf transformation in maize and sorghum, allowing the recovery of plants with Cas9-mediated gene dropouts and targeted gene insertion. Moreover, using a maize-optimized Wus2/Bbm construct, embryogenic callus and regenerated plantlets were successfully produced in eight species spanning four grass subfamilies, suggesting that this may lead to a universal family-wide method for transformation and genome editing across the Poaceae.
Similar content being viewed by others
Main
The genetic transformation and regeneration of plants with new phenotypes was a breakthrough in early plant biotechnology and generated high expectations among plant scientists. However, soon after the first report of successful Nicotiana tabacum transformation using Agrobacterium infection1, it became apparent that tobacco is the exception and harnessing the potential of this technology in major crops would be prevented by poor transformation and/or regeneration2. Stepwise improvements in transformation have continued for various plant species, but for many of them this process has been limited by recalcitrance to in vitro manipulation. Finding the best combination of transformation method and culture system for each crop has required the evaluation of different explants ranging from single cells such as protoplasts; to embryonic tissues such as meristem, scutellum or cotyledons; to seedling-derived tissues such as apical/axillary meristems, hypocotyl or leaves; and finally to in planta alternatives.
Recent development of genome editing technologies based on CRISPR (clustered regularly interspaced short palindromic repeats)–Cas (CRISPR-associated) nucleases further exacerbates the need for improved and efficient transformation processes3. RNA-guided Cas nucleases generate targeted double-stranded breaks in the genome, which are repaired either through non-homologous end joining (NHEJ) or homology-directed repair (HDR) pathways. NHEJ is prone to imperfect repair and often results in small insertions or deletions, while HDR can be used to introduce predefined modifications by providing a donor DNA template with regions of homology to the double-stranded break site.
Since the first reports of Cas9-mediated genome editing in tobacco, Arabidopsis and rice, the list of edited plant species has continued to expand4,5,6,7. However, while NHEJ-mediated mutagenesis and gene knockouts are now common practice, other types of genome edits (such as HDR-mediated gene insertions, large-scale deletions, inversions and translocations) occur with much lower frequencies, requiring a high number of plants to be regenerated and screened to find one with the desired modification. This limitation is true for most species in the Poaceae (for example, indica rice, maize, wheat, barley, sorghum and pearl millet), where transformation methods rely on immature embryos and in general remain very species- and/or genotype-dependent, limiting their large-scale deployment. In addition, a consistent year-round supply of immature embryos as a transformation explant requires greenhouse infrastructure that makes it resource- and cost-prohibitive for most academic institutions. Consequently, transformation service facilities have become a necessity8. Improved transformation processes with high frequencies of regeneration therefore become a prerequisite for these complex genome editing applications9.
A major breakthrough in immature-embryo-based transformation came with the use of the morphogenic genes Wuschel2 (Wus2) and Babyboom (Bbm), allowing plant regeneration at high frequencies from recalcitrant maize genotypes10,11. The use of Wus2/Bbm also improved transformation in three sorghum varieties, Tx430, Tx623 and P898012 (refs. 12,13), and two public maize lines, FFMM14 and B104 (ref.15). In addition, the expression of Wus2 alone has been shown to be effective for improving transformation in maize16 and genome editing in sorghum17. Following these reports, other genes stimulating cell division and the formation of embryogenic tissue have been identified and used with similar outcomes. For example, Grf4/Gif1 (ref. 18) and Wox5 (ref.19) have improved immature embryo transformation and regeneration in a wide range of wheat cultivars. However, for some grass species (such as bamboos, Setaria, teff and the small millets), seed and immature embryos are simply too small to efficiently isolate, while other crops (such as sugarcane and banana) are vegetatively propagated.
Various research groups have explored alternative starting explants for the transformation of grass species. Except for japonica rice, readily transformed via Agrobacterium infection of mature seeds20, these groups have used either mature seed-derived embryos21,22 or seedling-derived leaf bases23,24,25,26 to first establish callus cultures, which are then used as the target for Agrobacterium infection. The use of Wus2 and Bbm expanded the list of alternative explants for transformation, as demonstrated by the regeneration of fertile transgenic plants using mature embryo slices and leaf-base tissue segments in maize10. Using constructs developed by Lowe et al.10, recent reports have shown leaf-base transformation in switchgrass27 and successful gene mutagenesis in Eragrostis tef28. However, further improvements are needed to increase transformation frequency, to expand the method within the grass family and to eventually enable more complex genome editing applications.
Here we describe a new combination of promoters regulating the expression of Wus2 and Bbm that stimulate direct rapid formation of somatic embryos and regeneration of T0 plants after Agrobacterium infection of maize and sorghum seedling-derived early leaf tissue. We also demonstrate that this improved leaf transformation method enables Cas9-mediated genome modifications, including gene dropout and targeted gene insertion in maize. Finally, using a vector design, media and leaf transformation protocol optimized for maize, we demonstrate leaf transformation in a variety of grass species, including rice (both indica and japonica), teff, switchgrass, pearl millet, foxtail millet, barley and rye.
Results
Our previous work using Wus2/Bbm established both the starting point and our new objective for the optimization of leaf transformation. Lowe et al.10 determined that the use of Wus2 regulated by the Nopaline synthase (Nos) promoter and Bbm regulated by Ubiquitin (Ubi) produced embryogenic callus that after 10–12 weeks could produce transgenic plants for recalcitrant maize genotypes (our starting point). Later, Lowe et al.11 demonstrated that by providing a morphogenic pulse using a different set of promoters regulating Wus2 and Bbm expression (Axig1 and Pltp, respectively), immature embryos would rapidly (in 7–14 days) produce somatic embryos that could be regenerated into T0 plants. Our new objective for leaf transformation was therefore the identification and evaluation of promoters and tissue culture conditions that allow rapid leaf transformation and plant regeneration with efficiencies that allow complex genome editing applications, such as targeted gene insertion. The genetic components used in expression cassettes within the transfer DNA (T-DNA) in different plasmids are listed in Supplementary Table 1.
Identification of promoters that stimulate rapid somatic embryo formation in leaf tissue
To evaluate promoter combinations with the objective of improving and accelerating the embryogenic response in leaves, we used Agrobacterium tumefaciens strain LBA4404 TD THY- harbouring helper plasmid PHP71539 (pVIR9; ref. 29) and a binary donor vector (see PHP96037 as a general example; Fig. 1a) to transform leaf tissue from the lower portion of seedlings of Pioneer maize line PH1V69. The amount of callus or direct somatic embryos produced from leaf fragments was used as the criterion to evaluate various combinations of promoters regulating the expression of Wus2 and Bbm (the scoring is described in Fig. 2). The results of these experiments are summarized for maize inbred PH1V69 in Table 1. In general, weaker expression of Wus2/Bbm resulted in no embryogenesis in the leaf tissue, while different combinations of stronger and/or constitutive promoters produced an increasing embryogenesis response.

a, T-DNA vectors used in random integration experiments: PHP86491 is a control vector containing only visible (ZsGreen1) and selectable (Hra) marker genes; PHP96037 and PHP97334 are vectors containing morphogenic genes (Wus2 and Bbm), Cre recombinase, a visible marker gene (ZsGreen1) and either Hra or NptII as a selectable marker gene, respectively. b, T-DNA vector used for Cas9-mediated Wx1 dropout containing morphogenic genes (Wus2 and Bbm), Cre recombinase, Cas9 and two gRNA expression cassettes, visible (ZsGreen1) and selectable (NptII) marker genes. c, Depiction of Cas9-mediated targeted gene insertion experiment, including (i) the T-DNA vector containing the morphogenic genes (Wus2 and Bbm), Cre recombinase, Cas9 and gRNA expression cassettes, a selectable marker gene (Hra), the donor cassette comprising the second selectable marker gene (NptII), homology arms (HR1 and HR2) flanked by Cas9 cut sites (or target sites (TS)) and a visible marker gene (ZsGreen1); (ii) targeted gene insertion via HDR upon Cas9 cleavage of the genomic target site and both target sites within the T-DNA to release the donor sequence and (iii) the targeted gene insertion locus with the positions of PCR primer pairs used for detecting the HR1 (1) and HR2 (2) junctions, and the long-PCR product across the entire insertion (3). RB and LB = the right and left border sequences of the Agrobacterium T-DNA.
It was known from previous work that the combination of the Axig1 and Pltp promoters produces a pulse of Wus2 and Bbm expression, respectively, which was sufficiently strong in the immature embryo scutellar epithelium to stimulate somatic embryos within 7–14 days11. However, in leaf tissue this combination (PHP83652) only produced small clusters of fluorescing cells without further growth. Likewise, two constructs using diurnally regulated promoters (Sweet 11 or Diurnal 10) driving Wus2 expression along with Ubi::Bbm did not produce a growth response (PHP96730 and PHP96731, respectively). A weak growth response (+) was observed with a variety of promoter combinations, which included an auxin-inducible promoter (8xDR5 in PHP81855 and PHP94636); photosynthetic promoters such as PepC1(PHP95073), Rubisco Ssu (PHP95205) and Cab (PHP95394); diurnal promoters such as Diurnal 12 (PHP95074) and Diurnal 11 (PHP96032); or two viral promoters, Cassava Vein Mosaic Virus (CSVMV) (PHP95393) and Sugarcane Bacilliform Virus (SCBV) (PHP95987), in general being too transient or too weak to elicit rapid embryogenic growth. A more consistent callus growth response (++) was observed with (1) constitutively expressed Wus2 with transient Bbm expression (PHP81857), (2) transient Wus2 with constitutive Bbm (PHP94715, PHP98564 and PHP98565) or (3) our original Nos::Wus2/Ubi::Bbm combination (PHP81858 or PHP97978). Three promoter combinations to drive the respective expression of Wus2 and Bbm (Actin/Ubi in PHP95385, Gos2/Ubi in PHP96031 or Ubi/3xEnh–Ubi in PHP97417; 3xEnh, three consecutive viral enhancers from Figwart Mosaic Virus (FMV), Peanut Chlorotic Streak Virus (PCSV) and Mirabilis Mosaic Virus (MMV); Supplementary Table 1) resulted in more rapid embryogenic growth with some somatic embryo formation within 14 days. Finally, various constitutive promoters for Wus2 (Ubi, Actin and Nos) along with 3xEnh–Ubi::Bbm (PHP93925 to PHP103735) (Table 1) produced rapid somatic embryo formation in 60–80% of leaf fragments. The presence of 3xEnh–Ubi promoter driving Bbm expression (PHP97334 and PHP96277) significantly increased transcript levels not only of Bbm but also of Wus2 compared with PHP97978 and PHP95385 with Ubi::Bbm (Supplementary Table 2). We also observed that having any expression cassette upstream of Wus2 reduced the growth response, as noted in two specific examples. First, PHP97335 with Ubi::NptII (neomycin phosphotransferase II) upstream of Nos::Wus2 produced a moderate growth response, while PHP97334 (where Ubi::NptII is downstream) generated a strong rapid somatic embryo response; second, PHP93738 with a Heat Shock Protein17 promoter (Hsp17) driving Cre upstream of Actin::Wus2 resulted in a moderate callus response, while PHP95385 (where Hsp17::Cre is downstream) produced a rapid somatic embryo response. Of all constructs tested, PHP96037 and PHP97334, both with Nos::Wus2 and 3xEnh–Ubi::Bbm but two different selectable markers, consistently demonstrated a strong and rapid growth response and were chosen for further experiments.

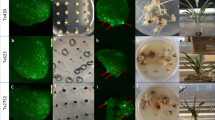
The images show examples of scoring the growth of fluorescent cells and somatic embryos 14 days after Agrobacterium infection. The images are representative of three independent experiments. Scale bar = 1 mm. NA = not applicable since no resulting tissue grew.
Agrobacterium-mediated transformation of seedling-derived leaf tissue in maize
To evaluate the overall leaf tissue transformation process (including T0 plant regeneration), the binary vectors containing Wus2, Bbm, Cre, a fluorescent marker gene (ZsGreen1) and a selectable marker gene (Highly-Resistant ZmAls (Hra) (PHP96037) or NptII (PHP97334)) were compared with a control vector (PHP86491) lacking Wus2/Bbm (Fig. 1a). LoxP sites were introduced, allowing the excision of Wus2/Bbm and Cre expression cassettes prior to T0 plant regeneration. Seedling preparation and the transformation process are shown in Fig. 3. Surface-sterilized seed was germinated on agar-solidified medium for 14 days (Fig. 3a–c), and then approximately 3 cm of leaf tissue above the mesocotyl (Fig. 3d) was bisected longitudinally and mechanically chopped in a food processor while immersed in the Agrobacterium suspension (Fig. 3e). The leaf fragments were distributed on filter papers and placed on co-cultivation medium for 24 hours and then resting medium (RM) for 7 days (Fig. 3f). Green-fluorescent somatic embryo formation was observed from the leaf fragments approximately 7–10 days after infection (Fig. 3g). After three weeks of selection on the filter papers (Fig. 3h), the plates containing the tissue were moved into a 45 °C incubator for two hours to induce Cre-mediated excision, and then individual pieces of healthy embryogenic tissue were transferred onto maturation medium (Fig. 3i) and finally onto rooting medium (Fig. 3j), where plantlet development was completed before transfer to soil in the greenhouse.

a, Surface sterilization of maize seeds. b, Maize seeds on germination medium. c, Fourteen-day-old maize seedlings used as the explant source. d, Preparation of 3 cm segments of the leaf whorl directly above the mesocotyl followed by slicing the tissue longitudinally (not shown). e, The tissue was placed in 100 ml of Agrobacterium suspension in the mini-Cuisinart blender and subjected to ten brief pulses to further slice up the leaf tissue. After a 20-minute incubation in the Agrobacterium suspension, the liquid was poured through a metal sieve to collect the leaf fragments, which were briefly blotted onto dry filter paper to wick away excess Agrobacterium (not shown). f, Leaf fragments distributed onto filter paper on the solid RM. g, Fluorescent somatic embryo growth observed 7–10 days after Agrobacterium infection (the image is representative of over 100 independent experiments; scale bar, 1 mm). h, After culturing on the RM, leaf tissue was transferred to selection medium. i, Following three weeks on filter papers on selection medium, the tissue was subjected to 45 °C/70% RH for two hours (not shown), transferred to maturation medium and cultured for an additional three weeks. j, Formation of T0 regenerants after two weeks on rooting medium.
The origin of somatic embryos within transformed leaf pieces (Extended Data Fig. 1a,b) was inferred on the basis of histological observations of the tissue, which clearly showed multicellular clusters appearing to initiate from internal leaf cells (Extended Data Fig. 1d–f). These clusters continued to grow as somatic embryos finally pushed out through the surface of the leaf (Extended Data Fig. 1c,f).
The results of leaf tissue transformation for maize stiff-stalk (SS) inbred PH1V69 from four experiments using vector PHP96037 are shown in Table 2a. In four replicates, each starting with ten seedlings used to generate the leaf fragments, the number of transgenic T0 plants recovered ranged from six (Experiments 1 and 4) to 14 (Experiment 2), resulting in a mean transformation frequency of 98 ± 43% (calculated on the basis of the number of T0 regenerants per starting seedling). In three replicate treatments with the control vector PHP86491 (Fig. 1a), T-DNA delivery comparable to other Ubi::ZsGreen1-containing plasmids was observed, but no green-fluorescent somatic embryo formation occurred in these leaf pieces, and no transgenic T0 plants were recovered using the same transformation protocol.
These leaf transformation experiments produced a high percentage (from 0% to 57% with a mean of 36 ± 25%) of high-quality T0 regenerants with a single copy of the integrated T-DNA (containing selectable and fluorescent marker genes) with no backbone sequences inserted elsewhere, as determined by quantitative PCR (qPCR). Comparing this number of high-quality transgenic events with the number of starting seedlings used in these experiments provided a clear measure of overall efficiency for producing single-copy T0 plants, with a mean frequency of 40 ± 29%.
Single-copy transgenic regenerants were grown to maturity in the greenhouse. The T0 plant phenotype was comparable to that of wild-type (non-transgenic) seed-derived plants. T0 plants were then either self-pollinated or crossed with wild-type plants and produced on average 172 (11 ears) and 195 (17 ears) seeds per ear, respectively (Supplementary Table 3). Thirty-six single-copy T0 plants were also analysed using Southern-by-Sequencing (SbS) technology30 (Supplementary Table 4). Twenty-seven plants (75%) were ‘SbS-pass’, indicating that a single intact copy of the integrated T-DNA with Wus2, Bbm and Cre excised was present, with no unlinked, independently segregating small DNA fragments detected. The remaining 25% failed due to an assortment of issues including single nucleotide polymorphisms, linked or unlinked fragments, plasmid backbone or mismatched border sequences (Supplementary Table 4).
Ancymidol during maize seedling growth and heat treatment improves leaf transformation frequency
During rapid seedling growth, leaves often contacted the lid of the growth container, resulting in tissue necrosis and phenolic production. To mitigate this problem, we added ancymidol, an antagonist of gibberellin-regulated elongation31, to the germination and seedling growth medium to produce shorter, more compact seedlings. In addition, we observed that ancymidol pretreatment of seedlings improved transformation frequency as demonstrated using PHP97334 vector, which contained a T-DNA with the same Wus2, Bbm, Cre and ZsGreen1 expression cassettes as in PHP96037, except for an SiUbi::NptII replacing the SbAls::ZmHra as a selectable marker gene (Fig. 1a).
For three replicates of this experiment performed using three separate plantings of seedlings on three different media (Table 2b), seedlings grown in the absence of ancymidol (control) produced a mean transformation frequency of 103 ± 6%, while seedlings grown on either 2 mg l−1 or 4 mg l−1 ancymidol resulted in mean transformation frequencies of 299 ± 50% and 246 ± 9%, respectively. The two ancymidol treatments did not significantly differ from each other (P = 0.32) but were substantially higher than the control (P < 0.02). All three treatments (with 0, 2 and 4 mg l−1 ancymidol pretreatments) produced T0 plants in which a similar proportion contained a single copy of the integrated T-DNA.
As previously observed for immature embryos32,33, temperature treatment could positively impact downstream transformation results. On the basis of this idea, a subset of PH1V69 seedlings grown on media supplemented with 2 mg l−1 ancymidol at 28 °C were incubated at 45 °C and 70% relative humidity (RH) for three hours, followed by leaf tissue collection and mechanical fragmentation in the presence of Agrobacterium suspension. As shown in Table 2c, the control treatment resulted in a mean transformation frequency of 260 ± 101%. Pretreating seedlings at 45 °C for three hours resulted in a significantly higher transformation frequency of 560 ± 85% (P = 0.002).
Public SS inbred line B104 and Pioneer non-SS maize inbreds respond positively to leaf transformation
To assess whether this method works in the commonly used maize inbred B104, seedlings grown on 2 mg l−1 ancymidol with the three-hour 45 °C/70% RH pretreatment were used as the tissue source, and transformation was carried out as described for PH1V69. For two replicates starting with seven seedlings for each experiment, the numbers of T0 plants produced were 12 and 13 for transformation frequencies of 171% and 186%, respectively (Supplementary Table 5). Of these total 12 and 13 T0 plants, 8 and 7 had single-copy T-DNA insertions, respectively, and further analysis showed that four of eight and five of seven events had no extraneous plasmid backbone sequence (Supplementary Table 5).
The leaf transformation method developed for our SS PH1V69 was also tested in four Pioneer non-SS (NSS) maize inbreds using PHP97334 (containing Nos::Wus2 + 3xEnh–Ubi::Bbm), starting with 21–24 seedlings per genotype with no replicates. Inbreds PH4257 and PNSS01 produced fewer somatic embryos within the first 14 days after Agrobacterium infection, resulting in a lower transformation response ranking compared with the SS inbred PH1V69 (Supplementary Table 6). As a result, PH4257 produced no transgenic plants, and only three T0 plants were regenerated for PNSS01 (13% relative to the number of starting seedlings).
In contrast, the other two NSS inbreds (1PYWK36 and 1PEEA63) demonstrated strong early somatic embryo responses similar to inbred PH1V69. Nevertheless, for these two inbreds, the conversion of these somatic embryos into T0 plants was considerably less efficient, producing only ten and eight T0 plants (48% and 38% transformation frequency, respectively). However, when a different construct (PHP96277) containing Actin-regulated Wus2 was used, the number of regenerated T0 plants increased to 20 and 40 (95% and 190%) for inbreds 1PYWK36 and 1PEEA63, respectively. While the results for these four inbreds were not replicated and are thus preliminary, taken together, we demonstrated that the leaf transformation method worked for six of the seven maize inbreds tested.
Sorghum is amenable to Agrobacterium-mediated seedling-derived leaf tissue transformation
To test the maize leaf transformation method in another crop, we used the same Agrobacterium strain, constructs, growth of seedlings, preparation of leaf material, infection, co-culture, resting culture, selection, Wus2/Bbm excision, somatic embryo maturation and rooting media for sorghum leaf transformation. Even though PHP96037 contained the Hra gene, no selection was used in this set of experiments because no background regeneration was observed from leaf tissue in initial observations in the absence of Wus2/Bbm. In addition, in this set of experiments we compared two resting media (RM): 13266P medium plus 50 mg l−1 meropenem and RM + CB (100 μM cupric sulfate and 0.5 mg l−1 benzylaminopurine).
The results from four sorghum (Tx430 variety) experiments are shown in Table 2d. Sorghum leaf transformation using PHP96037 vector resulted in high regeneration frequencies, calculated on the basis of the number of transgenic T0 plants recovered per starting seedling, with a mean frequency of 166 ± 96% and 297 ± 126% for RM and RM + CB, respectively, with no significant difference between the two media (P = 0.15). The control treatment with no Wus2/Bbm in the T-DNA (PHP86491) did not produce regenerated plants. The mean frequency of high-quality (single-copy T-DNA/backbone-free) T0 sorghum plants when transformed with PHP96037 was between 35% and 37%, with no significant difference between the two media (P = 0.87).
Twenty-three single-copy T0 sorghum plants produced through rapid leaf transformation were grown to maturity in the greenhouse. These plants were healthy, displaying an overall phenotype similar to that of wild-type Tx430 plants. SbS analysis demonstrated that of 23 T0 plants, 17 contained a single copy of the integrated T-DNA (Supplementary Table 4). Twenty-one T0 plants (91%) were fertile, with seed sets ranging from 667 to 3,255 seeds per head and a mean of 2,057 ± 900 seeds per head (Supplementary Table 3).
Leaf transformation facilitates gene dropout in maize
To assess the efficiency of gene dropout in seedling-derived leaf tissue of maize inbred PH1V69, we used a T-DNA vector (PHP98784, Fig. 1b) that contained the previously described components complemented with Cas9 and two guide RNA (gRNA) expression cassettes targeting two sites flanking the endogenous Wx1 gene as previously described by Gao et al.34. For these experiments, 2 mg l−1 ancymidol was used in the germination medium for inbred PH1V69, and a three-hour seedling heat pretreatment was applied prior to Agrobacterium infection.
The results of three Wx1 dropout experiments are summarized in Table 3. High transformation frequencies of 636%, 544% and 363% for a mean of 514 ± 139% were observed. Quantitative PCR analysis performed on T0 plants demonstrated dropout frequencies of 4.4%, 8.1% and 8.0% for these three experiments, respectively, for a mean of 6.8 ± 1.7%.
HDR-mediated targeted gene insertion is efficiently facilitated in maize using leaf transformation
To test HDR-mediated targeted gene insertion in maize leaf cells, two nearly identical T-DNA vectors were constructed (PHP99721 and PHP103735). Both vectors contained Wus2, Bbm, Cre, Cas9, gRNA, a donor template containing a selectable marker gene (NptII) flanked with homology arms and sequences identical to the genomic target site (at 53.66 cM on Chr1 (ref. 35)), a second selectable marker gene (Hra) and ZsGreen1 (PHP99721; Fig. 1c(i)). The two vectors differed in the homology arm sequences specific for the two genotypes used (PHP99721 for PH1V69 and PHP103735 for the Pioneer NSS inbred PHR03). As previously reported36, the presence of flanking target sites leads to the release of the donor template from T-DNA upon expression of gRNA and Cas9 (Fig. 1c(ii)). The use of two different selectable marker genes, NptII and Hra, present inside and outside the donor template, respectively, allowed the comparison of targeted insertion frequencies depending on the selectable marker gene position (Fig. 1c(i)).
For these experiments, seedlings were grown on germination medium with ancymidol. A total of five experiments were conducted using two different Pioneer proprietary genotypes. Experiments 1–4 were performed on inbred PH1V69, while Experiment 5 was conducted using inbred PHR03. In Experiment 1, there was no seedling heat pretreatment, while in Experiments 2 and 3, half of the seedlings were exposed to 45 °C at 70% RH for three hours prior to Agrobacterium infection and the other half had no heat pretreatment. In Experiments 4 and 5, all seedlings were subjected to the heat pretreatment. In Experiments 1, 2, 3 and 5, NptII (inside the donor template) was used as the selectable marker gene. In Experiment 4, one set of T0 plants was regenerated using G418 as the selection agent (for NptII), while the second set of plants was regenerated on ethametsulfuron-containing media (for Hra).
The results of all five experiments are summarized in Table 4. In Experiments 1–3 when no heat pretreatment was applied, the transformation frequency (the number of T0 plants per seedling) was 140%, 262% and 230%, respectively, with a mean of 211 ± 63%. In contrast, in Experiments 2–4 when the temperature pretreatment was used, transformation frequencies increased significantly (P = 0.001) to 560%, 570% and 524%, respectively, with a mean of 551 ± 24%. The escape rate (the number of regenerants with no selectable marker gene detected) ranged from 9.0% to 10.9%. T0 plants were first analysed for HDR-mediated insertion events by diagnostic HR1- and HR2-junction qPCR (Fig. 1c(iii)) as described in the Methods. T0 plants that were qPCR positive for either an HR1 or HR2 junction alone ranged between 5% and 12.6%. T0 plants that were positive for both junctions (HR1&HR2 column in Table 4) demonstrated that the targeted insertion frequencies were similar (P = 0.71) across the four experiments; 4.5%, 6.8% and 5.2% with no heat pretreatment and 7.0%, 5.6% and 5.0% with pretreatment.
Using long PCR that spans the entire insertion site (from the genomic sequence outside the HR1 arm across the integrated donor to the sequence outside the HR2 arm; Fig. 1c(iii)), we were able to detect the frequency of putative perfect insertion events. For both the non-heat-pretreated and the heat-pretreated replicates in Experiments 1–4, the frequencies were very similar: 3.2%, 2.8% and 3.0% and 2.7%, 2.5% and 2.6%, respectively. These results clearly demonstrate that although heat pretreatment resulted in a substantial increase in the initial recovery of T0 plants, targeted gene insertion frequencies were very consistent. In addition, the number of long-PCR-positive events relative to the total number of HR1- and HR2-junction-positive T0 plants provides an indication of the attrition associated with incomplete and/or complex insertions that occurred through a combination of HDR and NHEJ pathways. These frequencies in Experiments 1–3 were 72%, 41% and 58% for non-heat-pretreated replicates, while for both heat-pretreated replicates the values were 40% and 45%.
Targeted insertion frequencies were also compared using two different selectable marker genes located either inside the donor template (NptII), therefore integrating into the target site, or outside the donor template (Hra), in which case the selectable marker gene is left in the randomly integrated T-DNA (Experiment 4). Comparing these two treatments, the initial transformation frequency was similar: 524% (with a 9.8% escape frequency) when selected on medium with G418 and 579% (with an escape frequency of 12.6%) when selected on medium with ethametsulfuron. For the treatment with the selectable marker gene inside the donor template, the frequency of HR1&HR2-qPCR-positive events was 5.0%, similar to the frequencies observed in the first three experiments. In comparison, when the selectable marker gene was outside the donor template, the frequency of targeted gene insertion events was lower at 2.9%. The frequencies of long-PCR-positive events for the two groups were 2.6% and 1.5%, respectively.
Experiment 5 was conducted to evaluate targeted gene insertion frequency in the NSS inbred PHR03 (Table 4). Using the same T-DNA design and NptII as the selectable marker gene (located inside the donor template), the transformation frequency was 205% (with a 9.6% escape frequency), while the combined HR1&HR2 rate was 2.2% and the final long-PCR frequency was 1.2%.
To evaluate transmission and segregation patterns of the HDR-based targeted integrations, we analysed 32 T1 plants of three T0 insertion events from Experiment 1 (Table 4) using qPCR for the presence of gene insertion and T-DNA components (Cas9, gRNA, Bbm, Wus2, Hra and NptII). The results of this analysis are summarized in Supplementary Table 7. All three T0 plants showed transmission of the insertion to the T1 generation with a segregation ratio of approximately 1:1 (40%, 44% and 56%), consistent with stable Mendelian inheritance of a mono-allelic locus. As expected, approximately 50% of the T1 plants with insertion were transgene-free.
T1 insertion-positive plants were further analysed by Sanger sequencing to verify the quality and integrity of the insertion. Long PCR products from ten T1 selected plants (three or four plants for each of the three T0 events) were sequenced and assembled into contigs. Comparison of all ten contigs showed no sequence variation, confirming that every plant had a precise, HDR-mediated gene insertion.
Leaf transformation across several selected species within the Poaceae
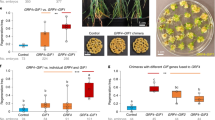
Encouraged by our results in maize and sorghum, we performed a preliminary evaluation of leaf transformation using nine species across the Poaceae, including Eragrostis tef, Panicum virgatum (switchgrass), Cenchrus americanus (syn. Pennisetum glaucum, pearl millet), Setaria italica (foxtail millet), Triticum aestivum (Pioneer Spring wheat), Secale cereale (rye), Hordeum vulgare (barley), Saccharum officinarum (sugarcane) and Oryza sativa (rice) (Fig. 4a). The method optimized for maize, but without the ancymidol or heat pretreatments of seedlings, was used for this assessment. Leaf tissue was harvested from the basal portion of the seedling and manually sliced into 1.5 to 3 mm fragments (Extended Data Fig. 2). The leaf pieces were then infected with Agrobacterium containing PHP97334 (Fig. 1a). Between six and 120 seedlings were used for different species/varieties (small numbers in some species were due to the limited availability of starting material). Using fluorescence microscopy to evaluate ZsGreen1 expression four to five days after infection, we observed good T-DNA delivery in all species tested (Fig. 4b, left micrographs). Further culture of the leaf tissue for four to five weeks resulted in fluorescent embryogenic calli (Fig. 4b, centre micrographs). After Wus2/Bbm excision, the tissue was cultured for three weeks on embryo maturation medium and then for two weeks on rooting medium, at which point regenerating plantlets were photographed (Fig. 4b, right photographs).

a, The Poaceae family tree indicating the species used in the experiment and the corresponding subfamilies. Species that were successfully transformed and regenerated, producing T0 plants, are shown in black; species that were transformed and produced callus tissue but did not regenerate are shown in red. b, Results for all successfully transformed grass species. The images on the left show transient expression of ZsGreen1 three to four days after Agrobacterium infection (scale bars, 500 µm), demonstrating the relative T-DNA delivery in various crops; the centre images show examples of green-fluorescent embryogenic callus formation three to four weeks after infection (scale bars, 1 mm); and the images on the right show recovered plantlets with shoots and roots ready for transfer to soil five to eight weeks after infection. The images are representative of three independent experiments.
Embryogenic callus was produced in all species tested (the individual experiments for each species are summarized in Supplementary Table 8), and T0 plantlets with healthy shoots and roots were regenerated in seven of nine species (Fig. 4b). Two species, wheat and sugarcane, failed to produce T0 plants (Extended Data Fig. 3). In the case of Pioneer wheat variety PH456D, although somatic embryos were produced within the first 10–14 days (based on the early growth phenotype), embryogenic potential deteriorated and the callus became non-regenerable over the next 2–3 weeks. In the case of sugarcane, somatic embryogenic callus was produced and maintained, but embryogenic morphology was quickly lost after exposure to the 45 °C heat treatment used to stimulate Cre-mediated excision. The experiment with sugarcane will be repeated in the future, replacing the heat-inducible promoter with an abscisic acid/desiccation-inducible Rab17 promoter driving Cre to facilitate excision as described in Lowe et al.10.
Discussion
The use of morphogenic genes to improve Agrobacterium-mediated transformation and/or Cas9-mediated gene editing in grass species continues to gain momentum. The combination of Wus2 and Bbm to enhance immature embryo transformation in maize was first reported for several recalcitrant maize inbreds10. The application of these genes was further extended to sorghum transformation10,12, sorghum genome editing37, maize public inbred transformation14,15,38 and maize genome editing15,35,36,39. Recently, Wox5 has also been demonstrated to improve transformation in numerous wheat varieties, with potential application in other cereals such as Triticum monococcum, triticale, rye, barley and maize19. Similarly, Grf5 has been shown to improve transformation in maize inbred line A18840, and the Grf4/Gif1 gene combination has been shown to stimulate growth and regeneration of transgenic events in wheat, rice and triticale18. However, the above progress continues to rely on immature embryo transformation, which has been a constraint for many grass family crops, motivating researchers to explore alternative explants for tissue culture and transformation.
The leaf base has been an attractive tissue culture explant for almost four decades, with callus initiation and regeneration being demonstrated in numerous Poaceae species including wheat41,42,43,44, oat45,46, barley47,48, rye49, maize23, rice50 and various apomictic grasses51. Accordingly, researchers have used leaf bases to generate embryogenic callus as the target explant for Agrobacterium infection and the regeneration of transgenic plants—for example, in maize23, indica rice24,52, Ma bamboo25 and teosinte26. However, except for a report in orchard grass (Dactylis glomerata)53 where transgenic plants were produced, studies where leaf-base tissue has been used as the direct transformation target were limited to demonstrating only transient transgene expression in rice54, wheat55 and maize56.
Lowe et al.10 have shown that the use of morphogenic genes Wus2 and Bbm facilitated direct Agrobacterium-mediated transformation of basal leaf tissue and the recovery of maize plants with random transgene integration. Recently, this method has been extended to random transformation in Panicum virgatum27 and Cas9-mediated mutagenesis in Eragrostis tef28, demonstrating that Wus2/Bbm can facilitate direct leaf-base transformation in other grass species. However, to support the true potential of Cas9-mediated genome editing, substantially higher transformation rates are still required to produce the large numbers of plants necessary to recover complex genome modifications, such as targeted HDR-mediated gene insertion and large-scale chromosomal rearrangements9.
To address this need, we show that Agrobacterium-mediated delivery of Wus2 and Bbm regulated by new promoters can significantly elevate leaf-base transformation in maize and sorghum. Previously, two promoter combinations have been used for Wus2/Bbm-mediated transformation of immature embryos. In the first one, the use of the Nos and Ubi promoters driving the expression of Wus2 and Bbm, respectively, stimulated the development of embryogenic callus but required weeks of culture followed by morphogenic gene excision prior to plant regeneration10,12. The second promoter combination, Axig1 for Wus2 and Pltp for Bbm, stimulated rapid somatic embryo formation and did not require later excision11,14,15,57. Lowe et al.10 demonstrated that maize leaf transformation using Nos::Wus2 and Ubi::Bbm initiates weak embryogenic callus that barely develops within the first two weeks after infection (Table 1, PHP81858), and the use of Axig1::Wus2 and Pltp::Bbm, although leading to strong transient expression in leaf explants, did not result in subsequent somatic embryo formation (Table 1, PHP83652). This led us to test many different promoter combinations, some of which produced somatic embryos at slow (++) or faster (+++) rates (Table 1). Although these promoter combinations are not optimal for maize, they may prove useful in other grass species with further experimentation. The best results were obtained for a group of constructs when Nos, Actin or Ubi promoters controlled Wus2 expression and Bbm was regulated by a FMV::PCSV::MMV::Ubi promoter (three viral enhancers upstream of the Ubi promoter). For maize and sorghum, we focused on Nos::Wus2 and 3xEnh–Ubi::Bbm for further method development.
Transformation frequencies were further enhanced by growing seedlings on ancymidol and providing heat pretreatment before leaf explant harvesting and Agrobacterium infection. The benefit of heat exposure immediately before infection is similar to that seen for immature embryo transformation in rice58, maize44,58 and sorghum32, where it has been suggested that the accumulation of heat shock proteins may mitigate against the stress of Agrobacterium infection. A link between ancymidol exposure and stress mitigation is less clear. Instead, one possible explanation for the enhanced embryogenic response may be related to the observation that ancymidol-treated leaf bases accumulate noticeable amounts of starch in the developing mesophyll cells (Extended Data Fig. 4). A correlation between starch accumulation and improved somatic embryogenesis has been noted in other systems59,60,61, with the suggestion that starch is an easily metabolizable source of energy to support somatic embryo development. As a result, for PH1V69 we achieved average transformation frequencies (the number of T0 regenerants per starting seedling) of 260% with ancymidol and 560% with a combination of ancymidol and heat pretreatment (Table 2c). The protocol developed for PH1V69 was also successfully used to transform Pioneer NSS inbred PHR03 and public line B104 with frequencies around 200% and 180%, respectively; other tested NSS lines had transformation frequencies between 13 and 190%. Currently, we do not have a common metric to compare the efficiency of immature embryo transformation with that of seedling-derived leaf transformation simply because the explants used for Agrobacterium infection are so different. Also, when using the mini-Cuisinart for mechanized leaf fragmentation, the population of individual leaf pieces produced is too variable in number and size to provide a reproducible starting point to calculate efficiency. We have thus resorted to using the number of starting seedlings as our common denominator when comparing leaf transformation frequencies between experiments, between inbreds and between grass species. On the basis of this measure, we can unequivocally say that maize inbred PH1V69 and sorghum variety Tx430 produced high numbers of T0 transgenic events per starting seedling and that the other inbreds tested (such as B104) produced lower results. For B104 and other inbreds, leaf transformation frequencies will probably continue to improve as more laboratories work with this tissue for Agrobacterium infection and as new improvements (such as a modified ternary plasmid system for Agrobacterium-mediated immature embryo transformation developed in B104 (ref. 62) are brought to bear on leaf tissue.
Direct leaf transformation provides other advantages allowing automation and improved handling of the tissue culture material. Specifically, the tedious embryo isolation procedure is replaced with simple seedling-derived leaf tissue preparation, fragmentation is automated using a blender (potentially reducing variability in explant preparation) and Agrobacterium infection happens directly in the blending vessel. Moreover, while immature embryos are subcultured by transferring each embryo onto new medium individually, the transformed leaf fragments are placed on filter paper, which can be moved easily from one medium to the next, substantially reducing time and labour. Combined with eliminating the need for year-round embryo production in addition to seasonal variation that has plagued immature embryo methods for years15, these advantages make transformation easier, more practical, reproducible, ergonomically friendly and widely applicable to a broad variety of important grass species.
From the earliest days of immature embryo culture and transformation in grass species, it was always abundantly clear that somatic embryo formation occurred on the scutellar epithelium63,64. However, for leaf-base culture, the origin of the culture response remained unresolved. Despite many reports on leaf-base tissue responding to in vitro culture manipulations and callus production across a variety of Poaceae species24,25,43,47,50, the specific cell types involved were not clear. Our histological analysis along with observations of fluorescing multicellular clusters five to seven days after Agrobacterium infection (Extended Data Fig. 1) confirm earlier observations in Lowe et al.10 that somatic embryos predominantly appear to form from interior leaf cells, an observation that is consistent with embryo growth from leaf tissue in orchard grass65 and with observations in rice leaves constitutively expressing the OsBbm1 gene66. On the basis of the abundant early formation of fluorescent multicellular clusters (Extended Data Fig. 1a–c) and our current frequency of recovering transgenic plants from only a fraction of such early growth responses, there is probably a potential for further improvement of this method by simply reducing the rate of attrition during the culture process.
We also demonstrated that the leaf-base transformation level achieved in Pioneer proprietary maize SS inbred PH1V69 supports the recovery of plants with various Cas9-mediated genome modifications (NHEJ-based mutagenesis, gene dropouts and HDR-facilitated targeted gene insertions) at frequencies that approach ones obtained for immature embryos35,36,39. Another important aspect to consider for genome editing is the contribution of growth stimulation by morphogenic genes, concomitantly accelerating cell cycle progression and cell division, which contribute to the overall efficiency of Cas9-mediated modifications. Accordingly, the advantage of using Wus2/Bbm in immature embryos has been demonstrated in maize to facilitate the recovery of gene dropouts35,34 and HDR-mediated integration events36,39. Furthermore, the frequency of recovered gene dropouts in T0 sorghum plants is enhanced by Wus2 beyond the level that can be accounted for by simply producing more T0 plants17. As speculated by Peterson et al.36, an added advantage of using morphogenic genes in genome modification experiments is the stimulation of cell division, providing a cellular environment conducive to more efficient complex forms of genome editing, including gene dropouts and HDR-mediated insertions. Our results with maize leaf bases are consistent with these interpretations.
In summary, our results clearly demonstrate the potential advantages of using leaf bases as the starting explant for transformation and genome editing in maize and sorghum. For maize, we have shown the potential of the method in terms of high efficiencies for both transgenic T0 plant production and genome editing. Moreover, successful leaf transformation and plantlet regeneration were demonstrated for multiple maize inbreds and a representative group of grass species. A single method can now be used across a wide spectrum of grasses, opening up the possibility of expanding transformation and genome editing methods across the entire Poaceae family.
Methods
Plant material
Seeds of Pioneer proprietary Zea mays lines PH1V69, PHR03, PH4257, PNSS01, 1PYWK36 and 1PEEA63 were used in the transformation experiments, in addition to the B104 public line. For Sorghum bicolor, seeds of line Tx430 were used. Transformation was carried out on additional Poaceae species: Eragrostis tef (teff variety DZ-01-354), Panicum virgatum (switchgrass variety Performer), Pennisetum glaucum (syn. Cenchrus americanus, pearl millet variety ICMB93333), Setaria italica (foxtail millet), Triticum aestivum (Pioneer Spring wheat variety PH456D), Secale cereale (rye, winter varieties Emerald and Pierre), Hordeum vulgare (barley, varieties Golden promise and Morex SPDL2), Saccharum officinarum (sugarcane variety CPCL02) and Oryza sativa (both indica variety IRV95 and japonica variety Kitaake).
In vitro seed germination and preparation of leaf tissue explants
Mature seeds were surface sterilized in an 80% ethanol solution for three min, followed by incubation in 30% Clorox bleach solution containing 0.1% Tween-20 for 20 min and finally rinsing three times for five min each in autoclaved sterile water. The sterilized seeds were transferred onto solid 90O medium to germinate (Supplementary Table 9). In vitro germination and seedling growth were carried out under 80 μmol m−2 s−1 light intensity at 28 °C with a 16 hour light/eight hour dark photoperiod.
Seedling pretreatments prior to harvesting leaf tissue for transformation
Ancymidol pretreatment
Surface-sterilized seeds were sown onto germination medium containing either no ancymidol (control medium 90O containing 0 mg l−1 ancymidol) or 90O medium plus 2 or 4 mg l−1 ancymidol (Sigma Aldrich, product number A9431). The germination and growth of seedlings occurred under 80 μmol m−2 s−1 light intensity using a 16-hour photoperiod at 28 °C, and the seedlings were harvested for transformation at 14 days for all replicate experiments and treatments.
Heat pretreatment
Surface-sterilized seeds were sown onto germination medium 90O plus 2 mg l−1 ancymidol. The germination and growth of seedlings occurred as described above. Before explant harvest, 12- to 18-day-old seedlings were placed into a 45 °C/70% RH incubator for three hours.
Preparation of Agrobacterium suspension
In prior studies, the combination of helper plasmid pVIR9 (PHP71539) in Agrobacterium strain LBA4404 THY- has been highly efficacious for use in maize transformation29 and has been an integral component in optimizing Wus2/Bbm methods for immature embryos10,11,16 and now leaf tissue. Agrobacterium tumefaciens strain LBA4404 TD THY- harbouring helper plasmid PHP71539 and a binary donor vector was streaked out from a −80 °C frozen aliquot onto solid 12R medium (Supplementary Table 10a) and cultured at 28 °C in the dark for two days to make a master plate. A working plate was prepared by streaking four or five colonies from the 12R-grown master plate across fresh 810K medium (Supplementary Table 10b) and incubating overnight in the dark at 28 °C prior to using it for Agrobacterium infection. Immediately prior to seedling leaf tissue preparation, Agrobacterium was suspended and dispersed in 700F infection medium (Supplementary Table 11b).
If the seedlings were manually dissected (see below), a small volume of infection solution (700F) was prepared by mixing 10 ml of 700J medium (Supplementary Table 11a), 20 µl of acetosyringone (100 mM stock) and 0.02% (v/v) surfactant (BREAK-THRU S 233, Evonik Industries GmbH) in a 50 ml conical tube. Agrobacterium was collected from the working plate, transferred to the infection medium in the 50 ml tube and then vortexed until uniformly suspended. The optical density (OD) of the suspension was measured at 550 nm and adjusted to an OD of 0.6. The final Agrobacterium suspension was aliquoted into Corning six-well plates containing 0.4 µm permeable culture inserts (Falcon, Part Numbers 353046 and 353090, respectively) with each well containing about 8 ml of the suspension.
For mechanized leaf fragmentation in a Cuisinart Mini-Prep Food-Processor (see below), the infection mixture (700F) was prepared by combining 100 ml of 700J medium, 200 µl of acetosyringone and 0.02% (v/v) BREAK-THRU S 223. Approximately one half to a full working plate of Agrobacterium was transferred to the 100 ml mixture and adjusted to an OD of 0.6 at 550 nm.
Seedling leaf preparation and Agrobacterium infection
The seedlings were grown as described above. The leaf-base segment (an approximately 2.5–3.0 cm section above the mesocotyl) was harvested from each 12- to 18-day-old in-vitro-germinated seedling with sterilized scissors, and the outer leaf was peeled away and discarded. The remaining leaf-base cylinders were bisected longitudinally into two lengthwise halves using a sterile no. 10 scalpel blade. The bisected leaf whorl halves were then either further dissected manually or processed mechanically in a blender.
Manual leaf explant preparation and Agrobacterium infection
The bisected leaf whorl segments were cross-sectioned into smaller leaf explants (~3 mm), which were transferred into a permeable culture insert sitting in a six-well culture plate containing 8 ml of premade Agrobacterium suspension (medium 700F, Supplementary Table 11b) and infected for 20 min at room temperature (Extended Data Fig. 2). After infection, the culture insert containing the Agrobacterium-infected leaf segments was removed from the six-well plate and placed on an autoclaved dry filter paper to wick up and remove any residual Agrobacterium solution. The infected leaf segments were then transferred onto a fresh filter paper (VWR 7.5 cm diameter) resting on 710N solid co-cultivation medium (Supplementary Table 12). Forceps were used to evenly disperse the leaf segments on the filter paper and to ensure that they had enough room to grow. The infected leaf tissues were incubated at 21 °C in the dark for two days.
Mechanized leaf chopping and Agrobacterium infection in the Cuisinart Mini-Prep Food-Processor
The bisected leaf whorls were directly dropped into the bowl of a Cuisinart Mini-Prep Food-Processor (MODEL DLC-1SSTG, pre-sterilized using 70% ethanol) containing 100 ml of premade Agrobacterium infection medium (700F). For each experimental replicate, the total number of leaf whorls (corresponding to the total number of seedlings used to produce each bisected leaf whorl) varied; the number listed in the results table is the number of starting seedlings in each replicate. The leaf segments were chopped at the low-speed setting with ten quick pulses. The chopped leaf tissues were left in the container with the Agrobacterium suspension for 20 min and gently agitated two or three times during this infection period. After infection, all chopped leaf tissues in the suspension were poured into an autoclaved stainless-steel mesh sieve (Target.com item no. 53142252) to drain out the Agrobacterium suspension. The infected leaf tissues were transferred onto two layers of autoclaved filter papers in an empty petri dish to blot-dry the residual Agrobacterium suspension. The infected leaf tissues from roughly one or two seedings were then transferred onto an autoclaved filter paper sitting on the surface of solid co-cultivation medium 710N and dispersed evenly. The infected leaf tissues were incubated at 21 °C in the dark for two days.
Tissue culture, selection and regeneration
After two days of co-cultivation, the filter papers supporting the leaf segments were transferred from 710N to 13266P RM (Supplementary Table 13) and cultured for one week in the dark at 28 °C without selection. The filter papers subtending the tissue fragments were then transferred onto selection medium (13266P plus 150 mg l−1 G418 for NptII selection or 0.1 mg l−1 ethametsulfuron for Hra selection; see Supplementary Table 14a,b, respectively). After three weeks of culture on selection medium, the plates were placed into a controlled-temperature/humidity incubator (45 °C/70% RH) for two hours to stimulate the Hsp17 promoter and induce Cre-mediated excision of Wus2, Bbm and Cre recombinase cassettes. The plates were removed from the incubator and kept at room temperature for one to two hours until the plates had cooled down.
After the heat treatment and temperature equilibration at room temperature, leaf segments with newly developed somatic embryos were transferred onto maturation medium 404 (Supplementary Table 15) without filter papers, cultured in the dark at 28 °C for two weeks and then moved into a 26 °C light room for an additional week. Leaf segments that now supported small shoots were transferred onto rooting medium 272M (Supplementary Table 16) for an additional two to three weeks until well-formed roots had developed, at which point the plantlets were ready for transfer to soil in the greenhouse. To ensure that each T0 plant was an independent event, only the most vigorously growing plantlet from each leaf piece (which often developed multiple somatic embryos) was transferred to rooting medium and finally the greenhouse.
Molecular analysis of T0 plants
Molecular analyses of transformed leaf pieces for transcript levels of Wus2 and Bbm were carried out as described previously67 with some modifications. Briefly, total RNA was isolated by grinding samples in RB Lysis Buffer (Omega Bio-tek), followed by adding an equal volume of 70% ethanol and transferring them to a Nunc Glass binding purification plate (Thermo Fisher Scientific) atop a 2 ml deep-well plate. The plates were covered with a Qiagen AirPore sheet (Qiagen), centrifuged and washed sequentially with RNA wash buffer I and II (Omega Bio-tek). RNA was eluted with nuclease-free water heated to 95 °C after a two min incubation in the Nunc Glass binding plate followed by centrifugation to collect the RNA in a 96-well plate. DNA was removed from the RNA samples using Roche Recombinant RNase-free DNAse I (Roche Diagnostics Corp.), and complementary DNA was synthesized using Applied Biosystems High Capacity cDNA Reverse Transcription kits (Thermo Fisher Scientific) according to the manufacturer’s instructions. Quantitative PCR was done using hydrolysis probes. The primers and probes were designed using Applied Biosystems Primer Express software. Hydrolysis-probe-based PCR was performed using Bioline Sensi-fast mix (Bioline) on an Applied Biosystem Viia7 Real-Time PCR instrument using the manufacturer’s conditions. Relative gene expression was calculated by normalizing against maize Eukaryotic Initiation Factor 4-Gamma gene (GenBank accession no. EU967723). A list of primers and probes used for the transcript analysis is given in Supplementary Table 17.
Molecular analyses of dropout and HDR-mediated gene insertion events were carried out as referenced in Peterson et al.36. Quantitative PCR was performed with QuantiTect Multiplex PCR Master Mix (Qiagen) according to the manufacturer’s directions (except for HR1/HR2 detection) on a ViiA 7 Real-Time PCR System (Thermo Fisher Scientific). Supplementary Table 18 lists the primers and probes used to detect Wx1 dropout products, HR1/HR2 junctions, T-DNA components and HDR target site mutations. To detect Wx1 dropout products (110 base pairs), endogenous Wx1 target sites (3.8 kilobases) were flanked by primers/probes. The primers and probes were used to characterize T-DNA expression cassette components (Wus2, BBM, Cas9, gRNA and Hra) for T1 segregation copy number as well as HDR-mediated insertion product components (SiUbi promoter, NptII and SiUbi terminator). T1 qPCR characterization provides the best understanding of event integrity when coupled with long insert spanning PCR using LongAmp Hot Start Taq 2X Master Mix (New England Biolabs) followed by agarose gel electrophoresis. Genomic primers (forward primer 5′-GCGTGCGTGCTTACATGATG-3′ and reverse primer 5′-GTGCGACATTAAACAGTGTTAGTTGTAGCC-3′) that flank the HDR target site were designed to amplify both the intact insert product (~5.0 kilobases) and the allele without insertion (1.0 kilobases). Various insertions or deletions indicated by varied band sizes exist and may represent double-stranded break repair via the NHEJ pathway36. The detection of HR junctions was also completed with fluorogenic probe-based detection. Although the assays (Supplementary Table 18) spanned from the genomic region into the insertion products (HR1 (600 base pairs) and HR2 (586 base pairs)), modifications to the standard qPCR cycling protocol allowed for the digital detection of HR products. The following cycling parameters in a three-step cycling approach were used: 95 °C denaturation for 15 mins followed by 40 cycles of 95 °C/20 s denaturation, 62 °C/60 s annealing and 68 °C/30 s extension. The qPCR reaction mix contained 50 ng of template DNA, 900 nM primers and 100 nM probe. Although the HR1 product contained additional fluorescent background, the use of a proprietary normalizer (VIC) of similar size provided qPCR-based copy number prediction for both junctions. The background in the HR1 reaction was subtracted out on the basis of higher quantitation values. For T1 long-PCR-positive segregation events, the full insert products were Sanger sequenced with the primers noted in Supplementary Table 18. SbS analysis was performed as described by Zastrow-Hayes et al.30.
Statistics and reproducibility
Comparison of means for all pairs was done by one-sided Tukey–Kramer honestly significant difference tests using JMP version 16.0.0 (SAS Institute Inc.). The numbers of replicates used in each experiment are either described in the legends of the respective figures and tables or presented as individual data points in the tables.
Histology of leaf tissue
Non-transformed leaf tissue was collected after the tissue was cut into small fragments and fixed for histological examination (see below). Transformed leaf tissue pieces at five, ten and 15 days after infection were collected and placed into freshly prepared fixative, 2.5% glutaraldehyde (Electron Microscopy Sciences) in phosphate buffer, pH 7.0, and fixed overnight at room temperature. The tissue was washed in three changes of phosphate buffer (three to five min per change), and the tissue pieces were dehydrated in a graduated ethanol series to 100% ethanol. The material was infiltrated with activated Technovit 7100 (Heraeus Kulzer GmbH), starting at a 1:1 mixture of 100% ethanol:Technovit 7100 for one hour, followed by two changes of 100% Technovit 7100, the first for one hour and the second overnight. The samples were polymerized in polytetrafluoroethylene moulds after the addition of 1 ml Technovit 7100 hardener to 15 ml of activated Technovit 7100. Polymerization was allowed to proceed overnight under vacuum in a vacuum desiccator.
Sections were cut at 2.5 µm with a sapphire knife on a microtome (Leica 2050, Leica Biosystems). The sections were floated on drops of distilled water on glass slides (Fisherbrand SuperFrost Plus) and dried onto the slides on a hot plate at 50 °C. The sections were stained with periodic acid Schiff reagent for polysaccharides68 followed by a five min counterstain for proteins with 0.1% aniline blue black (in 7.0% acetic acid)69. Images were captured on a Nikon E800 microscope equipped with a Nikon DS-Ri1 camera (Nikon).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that all data supporting the findings of this study are available within the Article and its Supplementary Information files and Extended Data. Corteva Agriscience will provide plasmids to academic investigators for non-commercial research under an applicable material transfer agreement subject to proof of permission from any third-party owners of all or parts of the material and to governmental regulation considerations. Completion of a stewardship plan is also required. The Pioneer maize inbred lines PH1V69, PH4257, PNSS01, 1PEEA63 and 1PYWK36 described in this research are proprietary.
References
Horsch, R. B. et al. A simple and general method for transferring genes into plants. Science 227, 1229–1231 (1985).
Potrykus, I. Gene transfer to plants: assessment of published approaches and results. Annu. Rev. Plant Physiol. Plant Mol. Biol. 42, 205–225 (1991).
Nasti, R. A. & Voytas, D. F. Attaining the promise of plant gene editing at scale. Proc. Natl Acad. Sci. USA 118, e2004846117 (2021).
Manghwar, H. et al. CRISPR/Cas systems in genome editing: methodologies and tools for sgRNA design, off-target evaluation, and strategies to mitigate off-target effects. Adv. Sci. 7, 1902312 (2020).
Wada, N., Ueta, R., Osakabe, Y. & Osakabe, K. Precision genome editing in plants: state-of-the-art in CRISPR/Cas9-based genome engineering. BMC Plant Biol. 20, 234 (2020).
Zess, E. & Begemann, M. CRISPR–Cas9 and beyond: what’s next in plant genome engineering. In Vitro Cell. Dev. Biol. Plant 57, 584–594 (2021).
Jaganathan, D., Ramasamy, K., Sellamuthu, G., Jayabalan, S. & Venkataraman, G. CRISPR for crop improvement: an update review. Front. Plant Sci. 9, 985 (2018).
Yassitepe, J. E. d. C. T. et al. Maize transformation: from plant material to the release of genetically modified and edited varieties. Front. Plant Sci. 12, 766702 (2021).
Gordon-Kamm, B. et al. Strategies for CRISPR/Cas9-mediated genome editing: from delivery to production of modified plants. In Willmann, M.R. (ed) Genome editing for precision crop breeding. 1-36 (Burleigh Dodds Science Publishing, Cambridge, 2021).
Lowe, K. et al. Morphogenic regulators Baby boom and Wuschel improve monocot transformation. Plant Cell 28, 1998–2015 (2016).
Lowe, K. et al. Rapid genotype “independent” Zea mays L. (maize) transformation via direct somatic embryogenesis. In Vitro Cell. Dev. Biol. Plant 54, 240–252 (2018).
Mookkan, M., Nelson-Vasilchik, K., Hague, J., Zhang, Z. J. & Kausch, A. P. Selectable marker independent transformation of recalcitrant maize inbred B73 and sorghum P898012 mediated by morphogenic regulators BABY BOOM and WUSCHEL2. Plant Cell Rep. 36, 1477–1491 (2017).
Nelson-Vasilchik, K., Hague, J., Mookkan, M., Zhang, Z. J. & Kausch, A. Transformation of recalcitrant sorghum varieties facilitated by Baby Boom and Wuschel2. Curr. Protoc. Plant Biol. 3, e20076 (2018).
Masters, A. et al. Agrobacterium-mediated immature embryo transformation of recalcitrant maize inbred lines using morphogenic genes. J. Vis. Exp. 156, e60782 (2020).
Aesaert, S. et al. Optimized transformation and gene editing of the B104 public maize inbred by improved tissue culture and use of morphogenic regulators. Front. Plant Sci. 13, 883847 (2022).
Hoerster, G. et al. Use of non-integrating Zm-Wus2 vectors to enhance maize transformation. In Vitro Cell. Dev. Biol. Plant 56, 265–279 (2020).
Che, P. et al. Wuschel2 enables highly efficient CRISPR/Cas-targeted genome editing during rapid de novo shoot regeneration in sorghum. Commun. Biol. 5, 344 (2022).
Debernardi, J. M. et al. A GRF–GIF chimeric protein improves the regeneration efficiency of transgenic plants. Nat. Biotechnol. 38, 1274–1279 (2020).
Wang, K. et al. The gene TaWOX5 overcomes genotype dependency in wheat genetic transformation. Nat. Plants 8, 110–117 (2022).
Toki, S. et al. Early infection of scutellum tissue with Agrobacterium allows high-speed transformation of rice. Plant J. 47, 969–976 (2006).
Sahoo, R. K. & Tuteja, N. Development of Agrobacterium-mediated transformation technology for mature seed-derived callus tissues of indica rice cultivar IR64. GM Crops Food 3, 123–128 (2012).
Thakur, T. et al. Efficient genetic transformation of rice for CRISPR/Cas9 mediated genome-editing and stable overexpression studies: a case study on rice lipase 1 and galactinol synthase encoding genes. Agronomy 12, 179 (2022).
Ahmadabadi, M., Ruf, S. & Bock, R. A leaf-based regeneration and transformation system for maize (Zea mays L.). Transgenic Res. 16, 437–448 (2007).
Tran, T. N. & Sanan-Mishra, N. Effect of antibiotics on callus regeneration during transformation of IR 64 rice. Biotechnol. Rep. (Amst.) 7, 143–149 (2015).
Ye, S. et al. An efficient plant regeneration and transformation system of ma bamboo (Dendrocalamus latiflorus Munro) started from young shoot as explant. Front. Plant Sci. 8, 1298 (2017).
Zobrist, J. D. et al. Transformation of teosinte (Zea mays ssp. parviglumis) via biolistic bombardment of seedling-derived callus tissues. Front. Plant Sci. 12, 773419 (2021).
Xu, N., Kang, M., Zobrist, J. D., Wang, K. & Fei, S. Z. Genetic transformation of recalcitrant upland switchgrass using morphogenic genes. Front. Plant Sci. 12, 781565 (2021).
Beyene, G. et al. CRISPR/Cas9-mediated tetra-allelic mutation of the `Green Revolution' SEMIDWARF-1 (SD-1) gene confers lodging resistance in tef (Eragrostis tef). Plant Biotechnol. J. 20, 1716–1729 (2022).
Anand, A. et al. An improved ternary vector system for Agrobacterium-mediated rapid maize transformation. Plant Mol. Biol. 97, 187–200 (2018).
Zastrow-Hayes, G. M. et al. Southern-by-sequencing: a robust screening approach for molecular characterization of genetically modified crops. Plant Genome 8, eplantgenome2014.2008.0037 (2015).
Leopold, A. C. Antagonism of some gibberellin actions by a substituted pyrimidine. Plant Physiol. 48, 537–540 (1971).
Gurel, S. et al. Efficient, reproducible Agrobacterium-mediated transformation of sorghum using heat treatment of immature embryos. Plant Cell Rep. 28, 429–444 (2009).
Hiei, Y., Ishida, Y. & Komari, T. Progress of cereal transformation technology mediated by Agrobacterium tumefaciens. Front. Plant Sci. 5, 628 (2014).
Gao, H. et al. Superior field performance of waxy corn engineered using CRISPR–Cas9. Nat. Biotechnol. 38, 579–581 (2020).
Gao, H. et al. Complex trait loci in maize enabled by CRISPR–Cas9 mediated gene insertion. Front. Plant Sci. 11, 535 (2020).
Peterson, D. et al. Advances in Agrobacterium transformation and vector design result in high-frequency targeted gene insertion in maize. Plant Biotechnol. J. 19, 2000–2010 (2021).
Aregawi, K. et al. Morphogene-assisted transformation of Sorghum bicolor allows more efficient genome editing. Plant Biotechnol. J. 20, 748–760 (2022).
Zhang, Q. et al. A novel ternary vector system united with morphogenic genes enhances CRISPR/Cas delivery in maize. Plant Physiol. 181, 1441–1448 (2019).
Barone, P. et al. Efficient gene targeting in maize using inducible CRISPR–Cas9 and marker-free donor template. Mol. Plant 13, 1219–1227 (2020).
Kong, J. et al. Overexpression of the transcription factor GROWTH-REGULATING FACTOR5 improves transformation of dicot and monocot species. Front. Plant Sci. 11, 572319 (2020).
Wernicke, W. & Milkovits, L. Developmental gradients in wheat leaves—response of leaf segments in different genotypes cultured in vitro. J. Plant Physiol. 115, 49–58 (1984).
Rajyalakshmi, K., Grover, A., Maheshwari, N., Tyagi, A. K. & Maheshwari, S. C. High frequency regeneration of plantlets from the leaf-bases via somatic embryogenesis and comparison of polypeptide profiles from morphogenic and non-morphogenic calli in wheat (Triticum aestivum). Physiol. Plant. 82, 617–623 (1991).
Haliloglu, K. Efficient regeneration system from wheat leaf base segments. Biol. Plant. 50, 326–330 (2006).
Yu, G.-r et al. Optimization of Agrobacterium tumefaciens-mediated immature embryo transformation system and transformation of glyphosate-resistant gene 2mG2-EPSPS in maize (Zea mays L.). J. Integr. Agric. 12, 2134–2142 (2013).
Chen, Z., Zhuge, Q. & Sundqvist, C. Oat leaf base: tissue with an efficient regeneration capacity. Plant Cell Rep. 14, 354–358 (1995).
Gless, C., Lörz, H. & Jähne-Gärtner, A. Establishment of a highly efficient regeneration system from leaf base segments of oat (Avena sativa L.). Plant Cell Rep. 17, 441–445 (1998).
Pasternak, T. P., Rudas, V. A., Lörz, H. & Kumlehn, J. Embryogenic callus formation and plant regeneration from leaf base segments of barley (Hordeum vulgare L.). J. Plant Physiol. 155, 371–375 (1999).
Becher, T., Haberland, G. & Koop, H. U. Callus formation and plant regeneration in standard and microexplants from seedlings of barley (Hordeum vulgare L.). Plant Cell Rep. 11, 39–43 (1992).
Haliloglu, K. & Aydin, M. Efficient regeneration system from rye leaf base segments. Springerplus 5, 2005 (2016).
Ramesh, M., Murugiah, V. & Gupta, A. K. Efficient in vitro plant regeneration via leaf base segments of indica rice (Oryza sativa L.). Indian J. Exp. Biol. 47, 68–74 (2009).
Yaguinuma, D. H., Takamori, L. M., Mendonça de Oliveira, A., Vieira, L. G. E. & Ribas, A. F. In vitro regeneration from leaf-base segments in three genotypes of Urochloa spp. Crop Pasture Sci. 69, 527–534 (2018).
Karthikeyan, A., Shilpha, J., Karutha Pandian, S. & Ramesh, M. Agrobacterium-mediated transformation of indica rice cv. ADT 43. Plant Cell Tissue Organ Cult. 109, 153–165 (2012).
Denchev, P. D., Songstad, D. D., McDaniel, J. K. & Conger, B. V. Transgenic orchardgrass (Dactylis glomerata) plants by direct embryogenesis from microprojecticle bombarded leaf cells. Plant Cell Rep. 16, 813–819 (1997).
Dekeyser, R. A. et al. Transient gene expression in intact and organized rice tissues. Plant Cell 2, 591–602 (1990).
Chugh, A. & Khurana, P. Regeneration via somatic embryogenesis from leaf basal segments and genetic transformation of bread and emmer wheat by particle bombardment. Plant Cell Tissue Organ Cult. 74, 151–161 (2003).
Kirienko, D. R., Luo, A. & Sylvester, A. W. Reliable transient transformation of intact maize leaf cells for functional genomics and experimental study. Plant Physiol. 159, 1309–1318 (2012).
Gu, Y., Chen, X., Song, R. & Qi, W. Establishment of a bivector genetic transformation system in recalcitrant maize inbred lines. Agriculture 11, 663 (2021).
Hiei, Y., Ishida, Y., Kasaoka, K. & Komari, T. Improved frequency of transformation in rice and maize by treatment of immature embryos with centrifugation and heat prior to infection with Agrobacterium tumefaciens. Plant Cell Tissue Organ Cult. 87, 233–243 (2006).
Feng, M.-Q. et al. miR156 regulates somatic embryogenesis by modulating starch accumulation in citrus. J. Exp. Bot. 73, 6170–6185 (2022).
Wu, G., Wei, X., Wang, X. & Wei, Y. Changes in biochemistry and histochemical characteristics during somatic embryogenesis in Ormosia henryi Prain. Plant Cell Tissue Organ Cult. 144, 505–517 (2021).
Wang, Y., Chen, F., Wang, Y., Li, X. & Liang, H. Efficient somatic embryogenesis and plant regeneration from immature embryos of Tapiscia sinensis Oliv., an endemic and endangered species in China. HortScience 49, 1558–1562 (2014).
Kang, M. et al. An improved Agrobacterium-mediated transformation and genome-editing method for maize inbred B104 using a ternary vector system and immature embryos. Front. Plant Sci. 13, 860971 (2022).
Vasil, V., Lu, C.-Y. & Vasil, I. K. Histology of somatic embryogenesis in cultured immature embryos of maize (Zea mays L.). Protoplasma 127, 1–8 (1985).
McCain, J. W. & Hodges, T. K. Anatomy of somatic embryos from maize embryo cultures. Bot. Gaz. 147, 453–460 (1986).
Conger, B. V., Hanning, G. E., Gray, D. J. & McDaniel, J. K. Direct embryogenesis from mesophyll cells of orchardgrass. Science 221, 850–851 (1983).
Khanday, I., Skinner, D., Yang, B., Mercier, R. & Sundaresan, V. A male-expressed rice embryogenic trigger redirected for asexual propagation through seeds. Nature 565, 91–95 (2019).
Stephenson, E. et al. Over-expression of the photoperiod response regulator ZmCCT10 modifies plant architecture, flowering time and inflorescence morphology in maize. PLoS ONE 14, e0203728 (2019).
O'Brien, T.P. & McCully, M.E. The study of plant structure: principles and selected methods. (Termarcarphi Pty. Ltd., Melbourne, 1981).
Fisher, D.B. Localization of endogenous RNA polymerase activity in frozen sections of plant tissues. J. Cell Biol. 39, 745–749 (1968).
Acknowledgements
Micro-propagated shoots of Saccharum officinarum (sugarcane variety CPCL02) and Panicum virgatum (switchgrass variety Alamo) were provided by the labs of F. Altpeter (University of Florida) and W. Parrott (University of Georgia), respectively, for use in the leaf transformation screening experiment.
Author information
Authors and Affiliations
Contributions
N.W., L.R., N.S., E.W., A.A., K.L., P.C., P.B., S.S., T.J. and W.G.-K. designed the experiments. N.W., L.R., B.L., A.W., D.v.D. and T.J. conducted the experiments. All authors contributed to analysing the results, and N.W., L.R., N.S., E.W., B.L., A.W., D.v.D., P.B., S.S., T.J. and W.G.-K. contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
N.W., L.R., N.S., E.W., B.L., K.L., P.C., A.A., A.W., D.v.D., P.B., S.S., T.J. and W.G.-K. have competing interests due to their employment with Corteva Agriscience at the time this research was conducted. The authors’ employer (Corteva Agriscience) has applied for and has obtained patents covering one or more aspects of this work.
Peer review
Peer review information
Nature Plants thanks Sadiye Hayta, Kirankumar Mysore and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Early growth of somatic embryos.
a, Fluorescence image of transient ZsGreen1 expression four days after Agrobacterium infection with PHP97334. b, Growing, independent fluorescent foci as somatic embryos begin growing nine days post-infection. c, Higher magnification at nine days post-infection showing fluorescent somatic embryos emerging from the surface of the leaf. d-f, Micrographs of transverse sections of leaf tissue stained with PAS and counter-stained with aniline blue black, showing d, small clusters of dividing cells depicting early developing somatic embryos (arrows) in leaf segments five days after infection, e, cluster of dividing cells ready to emerge through the epidermal layer in leaf segments ten days after infection, and f, larger somatic embryos pushing through the surface of the leaf (arrows) in leaf segments 15 days after infection. See end of Supplementary section for histology methods. Images are representative of over ten independent experiments.
Extended Data Fig. 2 Manual explant preparation and infection.
a, Agrobacterium suspension aliquoted into permeable culture inserts sitting in a six-well culture plate. b, Maize seedling depicting the region harvested for manual explant preparation. c, Manually cut leaf tissue explants in Agrobacterium suspension. d,e, Leaf tissues in a permeable culture insert with excess Agrobacterium suspension being blotted off, and leaf tissues cultured on a filter paper sitting on surface of solid medium.
Extended Data Fig. 3 Transformation of Saccharum officinarum and Triticum aestivum leaf explants.
Transient expression of ZsGreen1 three to four days after Agrobacterium infection (panels on left), demonstrating the relative T-DNA delivery. Green-fluorescent embryogenic callus formation three to four weeks after infection (panels on right). Images are representative of three independent experiments.
Extended Data Fig. 4 Sections of maize leaf base tissue from 14 day-old seedlings grown in the absence (a) or presence (b) of 2 mg/l ancymidol.
a,b, Sections of maize leaf base tissue from 14 day-old seedlings grown in the absence (a) or presence (b) of 2 mg/l ancymidol. Sections are stained with periodic acid-Schiff for polysaccharides and aniline blue-black for protein. Arrow indicates magenta-stained amyloplasts. Note extensive presence of amyloplasts in developing mesophyll of ancymidol-treated leaf tissue and general lack of amyloplasts in tissue grown in the absence of ancymidol. Scale bar = 50 µm.
Supplementary information
Supplementary Information
Supplementary Tables 1–18, description of histology methods and references.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, N., Ryan, L., Sardesai, N. et al. Leaf transformation for efficient random integration and targeted genome modification in maize and sorghum. Nat. Plants 9, 255–270 (2023). https://doi.org/10.1038/s41477-022-01338-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41477-022-01338-0
This article is cited by
-
Development of an in vitro regeneration system from immature inflorescences and CRISPR/Cas9-mediated gene editing in sudangrass
Journal of Genetic Engineering and Biotechnology (2023)
-
Leaf transformation in grasses
Nature Plants (2023)
-
Promoting genotype-independent plant transformation by manipulating developmental regulatory genes and/or using nanoparticles
Plant Cell Reports (2023)
-
Antisense RNA (asRNA) technology: the concept and applications in crop improvement and sustainable agriculture
Molecular Biology Reports (2023)
-
Epigenetic modifications and miRNAs determine the transition of somatic cells into somatic embryos
Plant Cell Reports (2023)
