
Abstract
Genomic selection based on the single-step genomic best linear unbiased prediction (ssGBLUP) approach is becoming an important tool in forest tree breeding. The quality of the variance components and the predictive ability of the estimated breeding values (GEBV) depends on how well marker-based genomic relationships describe the actual genetic relationships at unobserved causal loci. We investigated the performance of GEBV obtained when fitting models with genomic covariance matrices based on two identity-by-descent (IBD) and two identity-by-state (IBS) relationship measures. Multiple-trait multiple-site ssGBLUP models were fitted to diameter and stem straightness in five open-pollinated progeny trials of Eucalyptus dunnii, genotyped using the EUChip60K. We also fitted the conventional ABLUP model with a pedigree-based covariance matrix. Estimated relationships from the IBD estimators displayed consistently lower standard deviations than those from the IBS approaches. Although ssGBLUP based in IBS estimators resulted in higher trait-site heritabilities, the gain in accuracy of the relationships using IBD estimators has resulted in higher predictive ability and lower bias of GEBV, especially for low-heritability trait-site. ssGBLUP based on IBS and IBD approaches performed considerably better than the traditional ABLUP. In summary, our results advocate the use of the ssGBLUP approach jointly with the IBD relationship matrix in open-pollinated forest tree evaluation.
Similar content being viewed by others
Introduction
Eucalyptus L’Hér. (Myrtaceae) is the most valuable and globally planted forest tree genus. Fast growth, adaptability to a broad diversity of tropical and subtropical regions, combined with versatile wood properties for energy, solid products, pulp, and paper have warranted their outstanding position in current world forestry (de Lima et al. 2019). Eucalyptus dunnii Maiden (hereafter E. dunnii) has become increasingly used in commercial afforestation due to its combined good performance for growth, stem straightness, and frost tolerance, together with suitable wood density and pulp yield.
In a broad sense, genomic selection (GS) is a family of statistical methods developed for predicting the breeding values of non-phenotyped individuals with the assistance of a large number of molecular markers widespread distributed throughout the genome (Meuwissen et al. 2001). These methods exploit co-segregation between markers and quantitative trait loci (QTL) in linkage disequilibrium (LD). In forest trees, GS is of particular benefit due to the extended breeding cycles caused by delayed reproductive maturity and the need for early selection of traits that express late in life (Mphahlele et al. 2020). In this context, GS has a potentially substantial impact on the rate of genetic gain by increasing the intensity and accuracy of selection and, particularly, by shortening the generational interval (Grattapaglia et al. 2018).
The genomic best linear unbiased prediction (GBLUP) is one of the most commonly GS methods. It is basically a variant of the standard BLUP method (hereafter ABLUP, cf. Henderson 1984), where the pedigree-based numerator relationship matrix (A-matrix) is replaced by a genomic relationship matrix (G-matrix, e.g., Habier et al. 2013). Many empirical studies with forest tree species have shown that GBLUP is a very promising approach for tree breeding (e.g., Mphahlele et al. 2020; Resende et al. 2017; Lenz et al. 2019). However, to our knowledge, only two of them have directly investigated the efficiency of genomic prediction using only genotyped trees in E. dunnii through GBLUP (Naidoo et al. 2018; Jones et al. 2019).
Importantly, standard GBLUP can only be implemented when all trees are genotyped, although there are different approaches to produce genomic-enabled breeding value predictions for non-genotyped individuals. One of these is the so-called single-step GBLUP (ssGBLUP), which produces predictions for both genotyped and non-genotyped trees simultaneously in a single evaluation (Misztal et al. 2009; Legarra et al. 2009; Aguilar et al. 2010; Christensen and Lund 2010). The ssGBLUP is nowadays a routinely employed method for genomic evaluation in animal breeding, where it has been shown that it produces more accurate and less biased breeding values prediction than pedigree-based ABLUP (e.g., Legarra et al. 2014) and GBLUP (e.g., Christensen et al. 2012; Croué and Ducrocq 2017). However, the use of ssGBLUP for genomic prediction in forest trees is more recent and scarce (Klápště et al. 2018; Klápště et al. 2020a; Cappa et al. 2019; Ukrainetz and Mansfield 2020; Thavamanikumar et al. 2020). For instance, using data from a Eucalyptus hybrid population, Cappa et al. (2019) showed that ssGBLUP provided the higher predictive ability and lower prediction bias for non-phenotyped but genotyped trees when compared to GBLUP, and concluded that the ssGBLUP model is a promising breeding tool for genomic evaluation in Eucalyptus.
In ssGBLUP, single nucleotide polymorphisms (SNPs) markers are used to infer genetic relationships among the subset of genotyped individuals. The estimated relationships are subsequently used to set up the breeding values covariance matrix. Therefore, precise estimation of variance parameters and accurate prediction of breeding values will depend on how well marker-derived genomic relationships describe genetic relationships realized at unobserved causal loci (de los Campos et al. 2013; Forneris et al. 2016). That is, on how well estimates of genomic relationships capture the signals from the true identity-by-descent (IBD) process in the genome continuum, which in turn is affected by LD, incomplete pedigree information, and inbreeding (Forneris et al. 2016).
Several methods for estimating genomic relationships have been developed. One group of estimators of realized relationships make use of SNP marker information exclusively. Of these, the estimator proposed by VanRaden (2008) (Method 1) is the most widely employed in forestry GS studies (e.g., Bartholomé et al. 2016; Durán et al. 2017; Rambolarimanana et al. 2018). VanRaden’s estimates are computed by means of cross-products of marker genotypes deviated from mean allele frequencies and divided by the total heterozygosity at the locus (VanRaden 2008). Notice that this estimator reflects the actual proportion of marker alleles shared by two individuals, as a deviation from the expected proportion of alleles shared in the population, and thus it is an identity-by-state (IBS) measure (Vela-Avitúa et al. 2015). Also, this estimator gives extra weight to SNPs with high heterozygosity (Meuwissen et al. 2011). To account for this latter fact, Yang et al. (2010) proposed a modified version, where a different weight is used to avoid down-weighting the information from SNPs with low minor allele frequency (MAF).
Although these IBS-based genomic relationship measures ultimately estimate the realized proportion of genome shared by two individuals, they do not take into account either the pedigree or the segmental nature of DNA inheritance (Forneris et al. 2016). Therefore, relatedness estimators that introduce additional information to account for the IBD process, such as marker order and position within the genome or LD pattern, could improve the accuracy of the relationship estimation (Wang et al. 2017). Using a probabilistic hidden Markov model (HMM), Han and Abney (2011) developed a method for estimating relationships that trace markers through the pedigree by linkage analysis while accounting for population LD. A variant of this method, presented some years later by the same authors (Han and Abney 2013), allows estimating relationships when the pedigree is not known but a large amount of SNPs is available.
Even though estimates of realized relationships may play a critical role in the performance of GBLUP and ssGBLUP methods of GS, this has scarcely been investigated in plant and animal breeding. Using both simulated and real data of pigs, Forneris et al. (2016) showed that the IBD estimates of genetic relationships, combining pedigree and marker data, were more precise and this resulted later in higher accuracies in genomic breeding value predictions of candidates to selection when implementing ssGBLUP. Vela-Avitúa et al. (2015), using a GBLUP approach, run a stochastic simulation of a typical aquaculture breeding scheme and showed the superiority of the IBD estimates on the accuracy of genomic predictions compared to IBS estimates, but only at lower marker densities (≤100 SNPs/Mb).
In this study, we obtained and compared the precision of different IBD and IBS measures of genomic realized relationships in five open-pollinated (OP) first-generation progeny trials of E. dunnii. The two IBD estimators proposed by Han and Abney (Han and Abney 2011; Han and Abney 2013) and the IBS estimators of VanRaden (2008) and Yang et al. (2010) were compared. Next, we used these estimates to build up the corresponding genomic covariance matrices and implemented ssGBLUP for the breeding value prediction of diameter at breast height and tree stem straightness in these populations. As a control, we also fitted the conventional pedigree-based model (ABLUP). We compared the performance of the different models in terms of variance components (and functions of them), and the accuracy and bias of the predicted breeding values.
Materials and methods
Progeny trial data
Data for this study was obtained from five progeny trials of Eucalyptus dunnii Maiden established by the National Institute of Agricultural Technology (Instituto Nacional de Tecnología Agropecuaria, INTA), during 1991 and 1992, in different sites of Argentina; ranging from latitude 26° to 35° S, from longitude 43° to 58° W, and from elevation 35 m to 283 m. These sites were identified as Del Valle, Concepción, Ubajay, Istueta, and Cerro. The field experimental design was the same for all locations: a randomized complete block design with single-tree plots. Each trial contained from 16 to 20 replicates. The spacing was 3 × 3 m. Diameter at breast height (1.3 m, DBH) was measured (in cm) as an indicator trait for growth. In turn, stem straightness (STR) was assessed using a four-point subjective score, and the analyses were performed considering one the most crooked trees and four the straightest trees. This categorical STR trait was transformed into normal scores (Gianola and Norton 1981) and renamed NSTR. Details of the five trials and traits measured are summarized in Table 1. The population in this study consisted of 4860 trees, obtained from 75 OP seed lots: 63 OP families from 6 native stands in Australia and 12 OP families from a landrace originated in Argentina. The 12 Argentinean OP families were derived from trees that were phenotypically selected for growth and stem straightness in a commercial plantation of E. dunnii located near Oliveros, Santa Fe, Argentina. Seeds for these plantations were originally introduced from the Australian provenance known as Moleton, NSW (30°10′ S, 152°10′ E) (Marcó and White 2002). A detailed description of the genetic origin of the trees used in this study can be found in Table S1.
Molecular markers
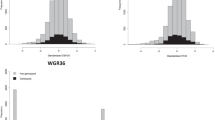
A sample of 642 trees originated from 74 (out of 75) families was genotyped with a range of 2–20 trees per family from three (Del Valle, Concepción, and Ubajay) out of the five sites. The total number of phenotyped trees, with at least one genotyped half-sib, was 2851 (out of 4860) (i.e., 58.66%) (see Table 2 for a summary). The distribution of DBH and STR traits for genotyped and non-genotyped trees are presented in Fig. 1. Generally, genotyped trees follow the same distribution as non-genotyped trees for both traits.

Frequency histograms of diameter at breast height (DBH) and stem straightness (STR, based on the observed original scale 1–4) for the entire Eucalyptus dunnii population (gray = all population) and genotyped trees (black = genotyped).
Genomic DNA was isolated from lyophilized young leaves using the CTAB method as described in Marcucci Poltri et al. (2003). The quality and quantity of the extraction were verified by agarose (1%) gel electrophoresis analysis and spectrometry using Nanodrop equipment (Thermo Fisher Scientific, Waltham, MA, USA). A standard concentration of 500 ng of each dried DNA sample was sent for genotyping with the Illumina chip for Eucalyptus (EUChip60K; Silva-Junior et al. 2015) at GeneSeek, Inc. (a Neogene company, Lincoln, NE, USA). Genotyping SNP was called using GenomeStudio Genotyping module v2.0.3 (Illumina, USA). SNP markers were filtered for MAF > 0.05 and missing values <0.10, using the R-package (www.r-project.org) “synbreed” (Wimmer et al. 2012). As a result, a total of 11,284 SNP markers were used for the genetic analyses providing good genome-wide coverage of the 11 Eucalyptus chromosomes as measured by the pairwise LD (Table S2 and Fig. S1). The average number of SNPs per chromosome was 1026, ranging from 700 on chromosome 1 to 1547 on chromosome 8 (Table S2). Figure S2 shows the distribution of MAF after filtering SNPs with extreme frequencies.
Estimation of pedigree and realized relatedness coefficients
Five different measures of pairwise relatedness coefficients among genotyped trees were computed. The measures are based only on pedigree information, combine pedigree and marker information (and so are referred to as IBD-based), or use only marker information, either incorporating (also referred to as IBD-based) or not LD between SNPs (IBS-based). In this sub-section, we present a brief but detailed description of these five studied measures and the methods used to compute them.
The first measure is Wright’s additive relationship (Wright 1922), defined as twice the coancestry between two individuals. As it is standard, additive relationships can be computed recursively from the pedigree information (Henderson 1984). The procedure returns the standard numerator relationship matrix (A) and was implemented by means of the R-package (http://www.r-project.org) “synbreed” (Wimmer et al. 2012). Pedigree errors were corrected using molecular marker information, before building up the A-matrix, as described by Muñoz et al. (2014).
The two IBD-based measures employed a hidden Markov model (Han and Abney 2011; Han and Abney 2013), where the hidden states are the nine possible condensed identity states (Jacquard 1974) on a locus for a given pair of individuals, and the observed states are the marker genotypes. The first of these methods use the pedigree information to assign hidden state probabilities for the first marker in the chromosome and next to estimate the transition probabilities between states, and the probabilities of the observations given the underlying state. These transition probabilities are calculated according to the actual distances between markers, as well as the pedigree connecting the pair under consideration. The “emission” probabilities of any locus, in turn, depend on the observed allele frequencies and the genotypes at the previous loci, accounting thus for LD between markers. As a result, a multi-point estimation of probabilities of each condensed identity state is given, which is next averaged over the full set of markers to obtain an estimate of the genome-wide IBD sharing for each pair of individuals. These estimates are used to build up the genomic relationship matrix GHA. We implemented this method by means of the IBDLD v3.14 software (Han and Abney 2011) using the “LD-RR” option, keeping the default parameters for the number of previous loci (i.e., window size) equal to 10 and the distance for choosing previous loci equal to 2 cM. A sensitivity analysis showed that when the window size was set to 20 and 40 loci, no big differences were observed in the estimates of relationship coefficients (only differences in the third decimal place) and variance components (also only differences in the third decimal place).
The second IBD-based measure is similar to the previous one, but it does not use pedigree information. In turn, it only uses the marker information of each individual and the genetic map of the markers (Han and Abney 2013). We also implemented this method by means of the IBDLD v3.14 software (Han and Abney 2011) but using the “GIBDLD” option. The resulting genomic relationship matrix is termed GHA-G.
Finally, two measures that use only marker information were also computed. The first one of these is an IBS-based method widely used in forest GS studies (Vanaden 2008, Method 1), in which the realized genomic relationships between all pair of individuals are calculated as:
where W is a matrix with entries equal to wij = gij−2pi, in which gij is the gene content at SNP locus i for tree j, and pi is the current (or observed) allele frequency for marker i. Notice that it assumed that the gene content of any locus is independent of the rest, so GVR does not exploit LD (Gianola et al. 2020). Indeed, a random permutation of the SNPs position will result in the same GVR matrix (Forneris et al. 2016). The GVR matrix was calculated using R-package (www.r-project.org) “synbreed” (Wimmer et al. 2012).
The last measure of relatedness computed, also based solely on molecular markers, was the method developed by Yang et al. (2010). According to this procedure, the genomic covariance matrix (GY) is calculated as:
where Z contains standardized genotypes calculated as \(z_{ij} = \left( {g_{ij} - 2p_i} \right)/\sqrt {2p_i\left( {1 - p_i} \right)}\) for which gij and pi are the same as described above, and m is the number of SNPs. This method was described by Meuwissen et al. (2011) as the best estimate of genetic relationships when a high proportion of loci with low MAF are used. We employed the R-package (http://www.r-project.org) “AGHmatrix” (Amadeu et al. 2016) to implement this method.
Statistical analysis
Due to spatial heterogeneity within trials, as well as for reasons of computational efficiency, the statistical analyses were conducted in two stages. In the first stage, a pedigree-based single-trait single-site classical a priori block design model (Block) and a posteriori spatial model with a first-order autoregressive error (co)variance structure (AR1 × AR1) (Cappa et al. 2019), were fitted to data from each combination of trait and site. The most appropriate model (i.e., either Block or AR1 × AR1) was then chosen based on the spatial distribution (heatmap) and semivariogram of residuals (Gilmour et al. 1997), and the Akaike information criterion (Table S3). Fig. S3 illustrates an example of the spatial patterns (i.e., spatial effects) found from the Block and AR1 × AR1 models for DBH and NSTR at the site Del Valle. The goal of this stage was to generate spatially adjusted phenotypes for each tree accounting for environmental variation in each of the five sites.
More explicitly, the single-trait single-site a priori block design model (Block) was as follows:
where y* is the vector of original individual-tree observations, β is the vector of fixed effects for genetic groups formed according to provenance; b is the vector of random block effects distributed as \({{b}}\sim {{N}}\left( {0,{{I}}\sigma _b^2} \right)\), where I is the identity matrix and \(\sigma _b^2\) is the block variance; a is a vector of random effects that represents the genetic effects (or breeding values) distributed as \({{a}}\sim {{N}}\left( {0,{{A}}\sigma _a^2} \right)\) where A is the numerator relationship matrix derived from the pedigree (Henderson 1984) and \(\sigma _a^2\) is the additive variance. Finally, e is the vector of random residuals distributed as \({{e}}\sim {{N}}\left( {0,{{I}}\sigma _e^2} \right)\) where \(\sigma _e^2\) is the residual variance. In turn, for the spatial model (AR1 × AR1), the vector e was partitioned into spatially dependent (ξ) and spatially independent (η) residuals. Therefore, the residual (co)variance matrix can be expressed as \(\sigma_{\upxi}^2\left[ {{{AR}}1\left( {\rho _{col}} \right) \otimes {{AR}}1\left( {\rho _{row}} \right)} \right] + \sigma _{\upeta}^2I\), where \(\sigma _\xi ^2\) is the spatially dependent variance, \(\sigma _\eta ^2\) is the spatially independent variance, AR1(ρ) is the first-order autoregressive correlation process, ρcol and ρrow are autocorrelations parameters for columns and rows, respectively, and ⊗ denotes the Kronecker product. The X, Zb, and Za are all incidence matrices for their respective effects.
In the second stage, the spatially adjusted phenotypes were obtained for each tree and trait and at each site by subtracting the estimated block (NSTR for all sites except at the site Del Valle, see Table S3) and the autoregressive residual effects (ξ, model [1]) (DBH for all sites and NSTR at the site Del Valle, see Table S3) from the original phenotypes. After adjusting phenotypes, a pedigree-based (ABLUP) multiple-trait multiple-site mixed model was fitted as:
or, more compactly as:
where \({{y}} = \left[ {{{y}}_{11}^\prime , \ldots ,{{y}}_{ij}^\prime , \ldots ,{{y}}_{25}^\prime } \right]\) is the vector of spatially adjusted phenotypes for each i trait (I = DBH and NSTR) and j site (j = Del Valle, Concepción, Ubajay, Istueta, and Cerro); \({{\beta }} = \left[ {{{\beta }}_{11}^\prime , \ldots ,{{\beta }}_{ij}^\prime , \ldots ,{{\beta }}_{25}^\prime } \right]\) is the vector of fixed effects for provenance for each trait-site combination; the random vector of genetic effects in \({{a}} = \left[ {{{a}}_{11}^\prime , \ldots ,{{a}}_{ij}^\prime , \ldots ,{{a}}_{25}^\prime } \right]\) is distributed as \({{a}}\sim {{N}}\left( {0,{{{\Sigma}}}_0 \otimes {{A}}} \right)\), where Σ0 is the (co)variance matrix of genetic effects for each combination of the two traits (DBH and NSTR) and the five sites (Del Valle, Concepción, Ubajay, Istueta, and Cerro) with dimension 10 × 10, and A is the numerator relationship matrix (Henderson 1984) containing the additive relationships among all trees: 75 mothers without records plus 4860 offspring with data in y. Finally, \({{e}} = \left[ {{{e}}_{11}^\prime , \ldots ,{{e}}_{ij}^\prime , \ldots ,{{e}}_{25}^\prime } \right]\) is the vector of random residual distributed as \({{e}}\sim {{N}}\left( {0,{{R}}_0 \otimes {{I}}} \right)\) where R0 is the residual (co)variance matrix for each combination for the two traits and five sites with dimension 10 × 10. We assumed an unstructured (co)variance matrix for the genetic effects (Σ0). However, for the residual (co)variance matrix (R0), we set the covariances between traits across sites to zero, given that the sites were assessed separately. The matrices Xij and Zij relate the spatially adjusted phenotypes to the means of the provenances \(\left[ {{{\beta }}_{ij}^\prime } \right]\), and the genetic effects in \(\left[ {{{a}}_{ij}^\prime } \right]\). The symbol ′ indicates the transpose operation. For the prediction of the genetic effects, the A-matrix based on the pedigree is used to solve the following mixed model equations (MME, Henderson 1984):
In order to fit the ssGBLUP models, the A-matrix of model [2] and MME [3] was replaced by the H-matrix, of the same dimension as the A-matrix. This matrix is a function of the genomic relationship matrices described above. Actually, only the inverse of H is needed to fit the ssGBLUP models. The inverse of the H-matrix (H−1) was obtained as follows (Misztal et al. 2009; Legarra et al. 2009; Aguilar et al. 2010; Christensen and Lund 2010):
where λ scales the differences between genomic and pedigree-based information, G−1 is the inverse of the corresponding genomic relationship matrix (either, as described above, GHA, GHA-G, GVR, or GY), and \({{A}}_{22}^{ - 1}\) is the inverse of the pedigree-based relationship matrix for the genotyped individuals. In all our analyses, the weight on the pedigree was λ = 0.95. Genomic relationship matrices calculated using only marker information (GHA-G, GVR, and GY) were scaled to have the same diagonal and off-diagonal averages as the corresponding A-matrix, as previously described Christensen et al. (2012) (Eq. (4)).
The narrow-sense heritability \(\widehat h^2\) for each trait-site combination was estimated as:
where \(\widehat \sigma _{a_{ij}}^2\) is the estimated genetic variance for the i trait and j site, and \(\widehat \sigma _{e_{ij}}^2\) the estimated residual variance for the i trait and j site from the multiple-trait multiple-site model [2]. The genetic correlations, \(\widehat r\), were estimated as:
where \(\widehat \sigma _{a_{ii}}^2\) and \(\widehat \sigma _{a_{jj}}^2\) are the genetic variances for the traits or sites i and j, respectively, and \(\widehat \sigma _{a_{ij}}\) is the estimated covariance between traits or sites i and j from the multiple-trait multiple-site model [2].
The ABLUP and the four ssGBLUP (ssGBLUPHA, ssGBLUPHA-G, ssGBLUPVR, and ssGBLUPY) evaluation models described above were fitted in R (www.r-project.org), with the function remlf90 from the package “breedR” (Muñoz and Sánchez 2014), using the expectation-maximization (EM) algorithm followed by one round of an average information (AI) algorithm to compute the standard deviations of the variance components, heritabilities, and genetic correlations (Chateigner et al. 2020). The remlf90 function in the R-package “breedR” is based on the REMLF90 (for the EM algorithm) and AIREMLF90 (for the AI algorithm) of the BLUPF90 family (Misztal et al. 2018). The program preGSf90, also from the BLUPF90 family (Misztal et al. 2018), was used to build up the inverse of the H-matrices (\({{H}}_{{\mathrm{HA}}}^{ - 1},\,{{H}}_{{\mathrm{HA}} - {\mathrm{G}}}^{ - 1},\,{{H}}_{{\mathrm{VR}}}^{ - 1},\,{\mathrm{and}}\,{{H}}_{\mathrm{Y}}^{ - 1}\)).
Relatedness coefficients and model performance
The mean and variance of the five measures of pairwise relatedness coefficients across either half-sibs or unrelated trees were calculated and compared. Self-relationships were also considered. Additionally, to study the population (and family) structure of the E. dunnii dataset, we generated a network visualization of these five types of relatedness coefficients. Following the work by Rincent et al. (2012), we represented each genotyped or non-genotyped tree in a network in which two individuals are either linked when their estimated relationship coefficient is larger than 0.05, or unlinked. We used the R-package (http://www.r-project.org) “network” (Butts 2008) to generate and plot this representation.
In turn, ABLUP and the four ssGBLUP models were compared in terms of their predictive ability and prediction bias. Tenfold cross-validation with ten replications was carried out, in which one random subsample was used as the validation set and the remaining nine samples as the training set. All trees with phenotypic data were in the training population at least once in each replication. The variance components were fixed to the respective variance components calculated with all the available trees with phenotypic data in the cross-validation analysis. The predictive ability was determined as the Pearson correlation between the estimated breeding values from the ABLUP, ssGBLUPHA, ssGBLUPHA-G, ssGBLUPVR, and ssGBLUPY model obtained by fitting the full data set (i.e., using all the available phenotyped trees, 4860 trees) and those of the validation set predicted from the respective model, multiplied by the square root of the narrow-sense heritability of each trait-site combination calculated using ABLUP for all available trees (Legarra et al. 2008). Standard errors (SE) were computed using the following expression: \({\mathrm{SE}} = \sigma /\sqrt n\), where σ is the standard deviation across replicates, and n is the number of replicates (10).
The prediction bias was calculated by regressing the observed breeding values of the trees estimated using the full data set, on those predicted with either the ABLUP, ssGBLUPHA, ssGBLUPHA-G, ssGBLUPVR, or ssGBLUPY model. A slope equal to one is consistent with no bias, while an estimate greater or smaller than one indicates deflated or inflated predictions, respectively.
In this study, only the predictive ability and prediction bias of the genotyped trees from the validation set of the three genotyped sites (Del Valle, Concepción, and Ubajay) are presented for all site-trait combinations examined.
An analysis of variance (ANOVA) on the predicted ability and the prediction bias measures was performed to test differences in performance across the pedigree (ABLUP) and the four ssGBLUP models. A Tukey’s multiple comparison test was employed at a significance level α = 0.05.
Programs from the BLUPF90 family (Misztal et al. 2018) were used for cross-validation analyses. A customized R-script was written to automate the cross-validation analyses.
Results
Relatedness and genomic relationship matrices
Statistics of the distribution of pairwise relatedness coefficients for both half-sibs and unrelated genotyped trees, as well as for self-relationship coefficients, are presented in Table 3 and Fig. 2. For the 642 genotyped trees, a total of 205,761 pairwise relationships were estimated, 98.38% (202,426) of which involved estimates for unrelated individuals (according to the pedigree) and only 1.62% (3335) represented half-sibs. This latter proportion varied across sites with genotyped trees: 3.00% for Del Valle, 1.82% for Concepción, and 1.30% for Ubajay.

The dotted line represents Wright’s additive relationship for each group.
As expected, the estimated pairwise coefficients were consistently larger for related trees than for unrelated ones (the average across different marker-based measures was 0.278 against 0.000, respectively). However, these averages exhibited differences across the four IBD- and IBS-based methods. For related individuals, Table 3 and Fig. 2 show that the IBS-based methods yielded, on average, the largest estimated pairwise coefficients (GVR = 0.291 and GY = 0.290), followed by the IBD-based methods GHA-G (0.270) and GHA (0.261). The same trend was observed for the standard deviations (SD), where the IBS-based values were consistently larger (GVR = GY = 0.092) than the IBD-based ones (GHA = 0.055 and GHA-G = 0.086); on average, SD of IBS-based relationships was 30.50% larger than IBD-based relationships. For unrelated individuals, the actual relatedness estimates obtained were, on average, close to the expected value of zero for all methods. However, the SD followed the same trend as for related individuals, i.e., the IBS-based approaches showed larger SD (GVR = GY = 0.036) than the IBD-based approaches (GHA = 0.000 and GHA-G = 0.022); on average, SD of IBS-based relationships was 227.27% larger than IBD-based relationships.
On average, the self-relationships coefficients were equal to the expected value of one for all genomic matrices. In fact, this is by design for the case of matrices calculated using only marker information. Recall that these matrices were scaled so that the means of diagonals (and off-diagonals) are the same as in the pedigree relationship matrix (Christensen et al. 2012). Instead, the SD of these diagonal entries were more close to zero for IBD-based estimators (GHA = 0.003 and GHA-G = 0.004) than for IBS-based ones (GVR = 0.048 and GY = 0.046).
Pedigree and combined pedigree-genomic relationship matrices
In ssGBLUP, the relationship matrix H is obtained by combining the marker-based relationships between genotyped trees with the additive relationships between non-genotyped trees. For each genomic relationship matrix, a corresponding H-matrix was built up. Network visualization of these H-matrices is presented in Fig. 3. In the figure, each node represents either a genotyped (black) or a non-genotyped (white) tree, and pairs of individuals with a relationship coefficient larger than 0.05 are linked by an edge (black) (Rincent et al. 2012). At a first glance, a clear pattern arises, clustering tightly the trees belonging to each of the 75 half-sib families. For the pedigree-based (A) and HHA matrices, these clusters appear unlinked with the other clusters, meaning that a model with this covariance structure assumes no correlations between breeding values beyond the family level. Instead, all other H-matrices show connections among families. The color palette helps relating these connections to the seven different provenances of the families. Interestingly, the HHA-G matrix showed lower connectivity among trees from families of the same provenances, and lower clustering of families of the same provenance, than matrices HVR and HY. Moreover, in the HHA-G matrix, the provenance Unumgar State Forest (in green) has not connected with the other provenances (i.e., the relationship coefficient is lower than 0.05). In the HHA-G, HVR, and HY matrices, is also clear one isolated cluster from one half-sib family of the Dead Horse provenance (in violet), with all non-genotyped trees (i.e., all-white node).

Each H-matrix was built up based on a different relatedness measure between genotyped trees (see text for references). The colored circles represent the 75 half-sibs Eucalyptus dunnii families, whereas larger clusters and edges represent connections among these families. The network connects trees that have a kinship > 0.05. White and black smallest dots represent trees with pedigree and with genomic information, respectively. The colors represent the seven provenances: Acacia Creek (yellow), Boomi Creek (blue), Dead Horse Track (violet), Oaky Creek (brown), South Yabra State Forest (orange), Unumgar State Forest (green), and Argentinean landrace (Oliveros, light blue).
(Co)variance components estimation
Table 4 displays the estimates of genetic parameters for diameter at breast height (DBH) and stem straightness normal score (NSTR) within each progeny trial for each of the five models fitted. Overall, there are clear and important differences in the estimates between the models that fitted IBD-relatedness measures (ABLUP, ssGBLUPHA, and ssGBLUPHA-G) and the models that fitted an IBS-based genomic matrix (ssGBLUPVR and ssGBLUPY). The latter, in general, produced the largest heritabilities, with some variation across sites (remarkably, Del Valle site against the others). The ssGBLUP model estimates showed lower SE than the ABLUP estimates, except for those from the ssGBLUPHA-G model that was similar to the ABLUP one.
We also estimated genetic correlations between DBH and NSTR traits within and across sites. Overall, our results showed that the genetic correlations within sites estimated using the ABLUP and ssGBLUP models were moderate to high and positive at the sites Del Valle, Istueta, and Cerro, ranging from 0.533 to 0.849 (average of 0.668, Table S4). However, at the Concepción and Ubajay sites, the genetic correlations between these traits were low and ranging from −0.024 to 0.166 (average of 0.093) for the ABLUP and the models that fitted the IBD-based genomic matrix (ssGBLUPHA and ssGBLUPHA-G), and large and positive (from 0.645 to 0.736; an average of 0.696) for the models that fitted an IBS-based genomic matrix (ssGBLUPVR and ssGBLUPY) (Table S4). Estimates of genetic correlations between sites from the ABLUP and ssGBLUP were high for DBH (average of 0.761, Table S5) with values ranging from moderate to high (from 0.432 to 0.969), with the lowest values between the sites Istueta and Ubajay (average of 0.573) and Istueta and Cerro (average of 0.520). Slightly higher genetic correlations were obtained for NSTR (average of 0.841, from 0.445 to 0.999) (Table S5). As we observed for the estimates of heritability and genetic correlation between traits, higher estimates of the correlation between sites were founded for models that fitted the IBS-based genomic matrix (average across sites 0.717) compared to the IBD-based genomic matrix (average across sites 0.400).
Accuracy and bias of predicted breeding values
Predictive abilities and prediction bias of each of the five models fitted to DBH and NSTR data are presented in Fig. 4. Our empirical study in E. dunnii showed that the ssGBLUP models provided larger predictive abilities (average correlation across sites = 0.648 and 0.632, for DBH and NSTR, respectively) than the pedigree-based ABLUP model (average correlation across sites = 0.406 and 0.400, for DBH and NSTR, respectively), with a single exception for NSTR at the site Concepción. As expected, this superiority of the genomic predictions over the traditional pedigree predictions was more pronounced when heritabilities were lower. For instance, the ssGBLUPHA breeding value prediction for DBH was 104.85% more accurate (i.e., the model showed larger predictive ability) than the pedigree-based one when heritability was 0.246 (site Concepción), while with a heritability of 0.808 (site Del Valle), the corresponding difference was only 9.27%. Similar results were obtained for the IBS-based model ssGBLUPVR, which showed 90.69 and 13.18% greater accuracies than the pedigree-based method for heritabilities of 0.246 and 0.808, respectively.

Vertical bars represent standard error and different letters above bars indicate significant differences (p-value < 0.05) between values of predictive accuracy or prediction bias for the five evaluation models as assessed by Tukey’s multiple comparison tests.
Among the ssGBLUP models, the ones that fitted an IBD-based covariance structure showed clear superiority over the ones that fitted an IBS-based one (Fig. 4). There was only one exception to this, in Del Valle site, where the ssGBLUPY predictions for DBH and ssGBLUPVR predictions for NSTR outperformed the ssGBLUPHA predictions. The differences, however, were small compared to the other cases; the large heritability in this site leveled out all differences in performance among models. Within each group of models (i.e., IBS- and IBD-based), differences were smaller, although statistically significant for IBD-based ones, favoring ssGBLUPHA-G, and not significant for the IBS-based ones, with a few exceptions.
Regarding prediction bias, the least-square estimates of the regression coefficients for the two studied traits and the three sites with genotyped trees ranged from 0.726 to 0.915 for the ABLUP model, from 0.720 to 0.943 for the IBD models (ssGBLUPHA and ssGBLUPHA-G), and from 0.480 to 0.879 for the IBS models (ssGBLUPVR and ssGBLUPY) (Fig. 4). Averaged across sites, the IBD-based ssGBLUPHA-G model showed the lowest bias. Within the IBS-based ssGBLUP models, ssGBLUPVR predictions showed no differences in bias with respect to predictions from ssGBLUPY, except for NSTR at Concepción and Ubajay sites.
Discussion
GS based on combining phenotype, pedigree, and genomic information by means of the ssGBLUP is becoming an important tool in forest tree breeding. Predictive ability using ssGBLUP depends on the choice of genomic relationship matrix (Speed and Balding 2015). Here, using data from an E. dunnii breeding population we compared the accuracy and bias of predicted genomic breeding values of four multiple-trait multiple-site ssGBLUP models, each with a different genomic relationship matrix derived from two IBD- and two IBS-based relatedness measures. As a reference, we also fitted a standard pedigree-based model without the use of marker information. To the best of our knowledge, neither multiple-trait and multiple-site models nor IBD-based genomic relationship matrices have been previously employed in GS in forestry.
Relatedness and genomic relationship matrices
Accurate genomic prediction of breeding values from GS models depends entirely on how well marker-derived genomic relationships describe actual genetic relationships realized at unobserved causal loci (de los Campos et al. 2013). This is, to the extent to which marker-based relationships properly describe the unobserved genetic relationships at trait loci (Hill 2014). In connection to this, we presented descriptive statistics of the different relatedness measures we calculated across trees with the same pedigree relationship. IBD-based measures of relatedness have consistently lower standard deviations than those from IBS-based approaches (see Table 3 and Fig. 2). These findings are in agreement with Forneris et al. 2016, who reported for a pig dataset that standard deviations of IBD-based relationship coefficients (GHA, as used here) were smaller than their IBS-based counterparts (e.g., GVR as used here) for different types of relatives. In related work, García-Baccino et al. (2017) found that the actual relationship of half-sib pigs, estimated with relatedness measures that do not use pedigree information (e.g., GVR as used here), had larger standard deviations than the ones estimated with genomic and pedigree data (e.g., GHA as used here). As discussed by Wang et al. (2017), these results are expected, given that IBD-based estimators make use of additional sources of information through linkage analysis and LD between markers and, thus, follow the true inheritance of DNA that is segmental in nature. Actual LD patterns are informative of the joint inheritance across markers.
Heritability estimates
Estimates of heritability from ssGBLUP models with IBS genomic relationship matrices (GVR and GY) were larger than those estimated from both IBD ones (GHA and GHA-G) and pedigree (A) (Table 4). Kumar et al. (2016) demonstrated that in structured populations, like the one analyzed here, BLUP analyses with genomic relationship matrices based solely on marker information produced biased heritability estimates with unreliable SE. According to these authors, the bias arises because, in structured populations, thousands of eigenvalues from IBS G-matrices are closely packed (near 0) and have large sampling errors associated with their values. This bias is proportional to the skewness of the genomic relationship matrix eigenvalues distribution. Network representation revealed some population structure in the E. dunnii population used in this study (Fig. 3). Skewness values founded for the eigenvalues of the IBS G-matrices were twice as large (GVR = 7.29 and GY = 7.29) as those from IBD G-matrices (GHA = 3.47 and GHA-G = 4.32). The same patterns were observed when we analyzed the eigenvalues of the corresponding H-matrices.
Several authors have also studied from a theoretical perspective the problem of biases in the heritability estimates when fitting genomic models. de los Campos et al. (2015) warned about problems arising when GBLUP approaches are used for variance components inferences in complex traits, especially in populations with distantly related individuals as the E. dunnii one studied here. In these populations, when large numbers of markers are in linkage equilibrium with the underlying QTLs, G-matrices build up using IBS-based measures can lead to an incorrect specification of genomic relationships and this can result in potential inconsistencies of estimates of genomic heritability. Wang and Thompson (2019) investigated the properties of the heritability estimates when the genetic correlation matrix differs from the truth. They show that heritability estimates can be biased if the genetic correlation matrix is misspecified, especially, when the population sample contains many remotely related individuals. Cuyabano et al. (2018) also concluded that if the covariance structure specified by the genomic model provides a poor description of that specified by the true inheritance process, then the likelihood misspecification may lead to the biased inference of the variance parameters.
All together, our results suggest that heritability estimates obtained for this E. dunnii population using IBS models could have been biased. However, this assertion should be taken with caution, because the results were not uniform across all the traits and sites studied; e.g., one exception was observed for the NSTR trait at the Del Valle site (Table 4). As stated by de los Campos et al. (2015), further research is needed to understand under what circumstances GS approaches based on IBS-based methods can be used to correctly estimate genetic parameters of interest.
Now, comparing the models with IBS genomic relationship matrices, both resulted in similar heritabilities estimates (Table 4). This result shows that the different scaling that these two genomic relationship matrices performed didn’t have a large impact on variance component estimation for the two studied traits (DBH and NSTR) in the E. dunnii population. Given that this is the first GS forestry study that contrasts these two IBS-matrices, a comparison with other studies is not feasible. However, in animal breeding, using a GBLUP model, Choi et al. (2017) also observed no differences in the variance components and heritability for intramuscular fat trait assessed in Korean cattle using the G-matrices proposed by VanRaden (2008) and Yang et al. (2010). These two IBS approaches were compared in a Merino sheep dataset by Clark et al. (2013) and they neither observed differences in the estimates of variance components and heritability.
Predictive ability and bias
In this study, the ssGBLUP models showed larger predictive ability than the standard ABLUP model for all the trait-site combinations studied (Fig. 4). Under the pedigree-based ABLUP model, and in the absence of inbreeding and of assortative mating, the accuracy of prediction of breeding values is limited given that the Mendelian segregation term can explain the 50% of the individual’s breeding value variance (Daetwyler et al. 2013). However, when genomic information from a large number of markers is available, the Mendelian sampling term can be estimated with great accuracy early in its life even when an individual’s own record or records from progeny is not yet available. Cantet and Vitezica (2014) demonstrated that this gain in accuracy of predicted breeding values is explained by markers accounting for a greater proportion of the individual’s breeding value variance and a concomitant reduction in the variance of the Mendelian residuals.
As expected, the superiority of the ssGBLUP GS models over the ABLUP model was more important for those trait-site combinations with lower heritability. Previous studies on marker-assisted selection have shown that selection response rates are relatively larger for traits with lower heritability estimates (Lande and Thompson 1990; Meuwissen and Goddard 1996). However, as discussed by Su et al. (2010), these calculations were conditional on the fact that QTL had been identified, which is much more difficult for low-heritability traits because of the low statistical power of detection. In GS approaches, however, information from a large number of SNP markers is used to estimate breeding values without having a precise knowledge of where the QTLs are located, and this is the reason that accurate breeding values can be obtained even for low-heritability traits. As in our work, several studies in forest tree species using both simulated and empirical data have shown that GS is especially effective for low-heritability traits over traditional ABLUP selection. For instance, Stejskal et al. (2018) showed the advantage of GS (GBLUP) over the conventional phenotypic selection (ABLUP) using stochastic simulations. Similar findings were reported for Eucalyptus grandis and using ssGBLUP (Cappa et al. 2019) and in Pinus radiata and Eucalyptus nitens using GBLUP GS approaches (Klápště et al. 2020b).
The application of GS models for growth, stem straightness, and wood quality traits in E. dunnii has been understudied. For DBH, the predictive ability reported here is consistent with recent results in E. dunnii by Jones et al. (2019) and larger than those reported by Naidoo et al. (2018). Though GBLUP and ssGBLUP GS approaches has been scarcely studied in E. dunnii, there is extensive literature on tree species in general (e.g., Isik et al. 2016; Li et al. 2019; Calleja-Rodriguez et al. 2020) and Eucalyptus in particular (e.g., Resende et al. 2012; Klápště et al. 2018; Thavamanikumar et al. 2020). For instance, a similar predictive ability of GS was reported for stem straightness in Pinus radiata D. Don (0.55; Li et al. 2019), Eucalyptus nitens (0.65; Klápště et al. 2018), and for stem sweep (a measure of tree stem straightness) in Pinus pinaster Ait. (0.49; Isik et al. 2016). Predictive abilities reported for growth traits were also quite similar to those found in our work (Klápště et al. (2018) in Eucalyptus nitens, 0.63; Resende et al. (2012) in two Eucalyptus hybrid populations, 0.51–0.54; Isik et al. (2016) in Pinus radiata D. Don, 0.43–0.47; Thavamanikumar et al. (2020) in Eucalyptus pellita, 0.60–0.82; Calleja-Rodriguez et al. (2020) in Pinus sylvestris L., 0.66–0.75).
Across the two traits and three sites with genotyped trees, greater prediction ability and lower bias were associated with ssGBLUP models that used IBD-based G-matrices (Table 3 and Fig. 4). Other researchers have also compared predictive performance between IBD and IBS models. Our results agree with previous findings in the simulation study carried out by Forneris et al. (2016). On the other hand, in the simulation study carried out by Vela-Avitúa et al. (2015), where a strong family structure (full-sib families) was simulated, the genomic prediction based on IBS relatedness measures was slightly superior to the one based on IBD information when dense marker panels were used. At lower densities (≤100 SNPs/Mbp) the IBD model was more accurate. The IBD model used in the study by Vela-Avitúa et al. (2015) didn’t include information on LD and this could explain the different results they obtained compared to our own work. Fitting a GBLUP model to data on scanned trait eye muscle depth in Merino sheep, Clark et al. (2013) found that there was no difference in accuracy between the IBS and the IBD methods. Luan et al. (2012) obtained small favorable differences in accuracies of predicted breeding values of dairy bulls when using an IBD relatedness estimator.
We hypothesize that the increase in the predictive ability of the ssGBLUP models based on IBD relatedness measures was due to a better account for the realized relationship between related (and unrelated) trees, and consequently to the increased similarity between the IBD-based covariance matrix and the true genetic relationships at unobserved causal loci (de los Campos et al. 2013) compared with the IBS-based matrix used in ssGBLUP. As we stated above, IBD-based estimators make use of additional sources of information through pedigree and LD between markers. Further evidence in support to our hypothesis was attained when we calculated the predictive ability in terms of correlations between predicted breeding values and adjusted phenotypes (see r2 in Klápště et al. 2020a) (results not shown). Lower predictive ability than the one obtained in terms of correlations between breeding values was observed. In addition, predictive ability from the ssGBLUP models with IBD-based genomic matrices was also higher than the one estimated from IBS-based matrices for the two studied traits. These findings suggest a limited short-range LD between markers and putative QTLs underlying the trait in this E. dunnii population and support our previous interpretation: predictive performance is mainly driven by the ability of markers to capture realized relatedness. Therefore, and as other authors have also observed (Forneris et al. 2016; de los Campos et al. 2013), our results point towards the idea that the better ability in capturing the true patterns of realized genetic relationships at unobserved causal loci of the IBD-based relationship matrices should be seen as the key factor for the higher predictive ability obtained from the IBD-based ssGBLUP models in this population.
Comparing the two IBD ssGBLUP models, the one that fitted a genomic covariance matrix without pedigree aid (GHA-G) resulted, in general, in larger predictive abilities than the one that used pedigree information (GHA) (Fig. 4). The latter infers relationships by tracing the transmission of markers throughout the pedigree (linkage analysis) while accounting for population LD or background sharing beyond the pedigree (Forneris et al. 2016). In our case, given that families were unrelated, it only captured information from known half-sibs, which typically share long chromosomal segments IBD (Ødegård and Meuwissen 2014). In contrast, the former, which also considers LD but whose estimates are not conditional to the pedigree, tends to pick up short-range hidden relationships (e.g., among founding families). Arguably, the additional information from hidden relationships between individuals helped to improve the accuracy of the predicted breeding values. Therefore, this larger accuracy could be explained by a deeper exploration of the background IBD sharing (i.e., beyond the pedigree) while also accounting for the population-wide LD.
Heritability vs predictive performance
As we have already pointed out, IBS models produced larger heritabilities but lower predictive performance than IBD models. Using simulations and real human data from related and unrelated individuals, de los Campos et al. (2013) also reported that the estimates of genomic heritability did not follow the same patterns as those of predictive ability. As these authors explained, the entries of the derived genomic relationships matrices represent, in different ways, the patterns of realized genetic relationships at causal loci: some of these elements (off-diagonal elements of related individuals and diagonal elements) represent very well the true covariance function while others (off-diagonal elements of distant relatives and pairs of unrelated individuals) do not. In addition, as they pointed out, variance components estimators are functions of diagonal and off-diagonal elements of G-matrix whereas the predictive ability is mainly determined by the off-diagonal entries of G-matrix and, therefore, these two tasks (inference vs. prediction) are driven, in part, by different forces (de los Campos et al. 2013). Given that the E. dunnii breeding population we studied here showed a larger proportion of pairs of unrelated trees compared to related ones plus self-relationships (Table 3), we believe our contrasting results on heritability estimates and predictive ability could be explained by these considerations. These should be taken into account in further GS forestry studies.
Conclusion
This is the first study that investigated the potential benefit of using IBD measures of relatedness in the context of ssGBLUP methods for the prediction of breeding values in forestry data. Our study, that fitted data on an E. dunnii population, showed that marker-based IBD genomic relationship matrices produced consistently larger predictive abilities than standard IBS genomic matrices, over a range of two different traits and five different experimental sites. The benefit of using ssGBLUP methods, compared to standard pedigree-based approaches, was more relevant for trait-site combinations with lower heritability.
Data availability
Information of the Eucalyptus dunnii trials including phenotypic, pedigree, and genomic data are available in the Zenodo repository, https://doi.org/10.5281/zenodo.4906814.
References
Aguilar I, Misztal I, Johnson DL, Legarra A, Tsuruta S, Lawlor TJ (2010) Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J Dairy Sci 93:743–752
Amadeu RR, Cellon C, Olmstead JW, Garcia AAF, Resende MFR, Muñoz PR (2016) AGHmatrix: R package to construct relationship matrices for autotetraploid and diploid species: a blueberry example. Plant Genome 9.
Bartholomé J, Van Heerwaarden J, Isik F, Boury C, Vidal M, Plomion C et al. (2016) Performance of genomic prediction within and across generations in maritime pine. BMC Genom 17:1–14
Butts CT (2008) network: a package for managing relational data in R. J Stat Softw 24:1–36
Calleja-Rodriguez A, Pan J, Funda T, Chen Z, Baison J, Isik F et al. (2020) Evaluation of the efficiency of genomic versus pedigree predictions for growth and wood quality traits in Scots pine. BMC Genom 21:1–17
Cantet RJC, Vitezica ZG (2014) Properties of Mendelian residuals when regressing breeding values using a genomic covariance matrix. WCGALP 10:17–22
Cappa EP, de Lima BM, da Silva-Junior OB, Garcia CC, Mansfield SD, Grattapaglia D (2019) Improving genomic prediction of growth and wood traits in Eucalyptus using phenotypes from non-genotyped trees by single-step GBLUP. Plant Sci 284:9–15
Chateigner A, Lesage-Descauses MC, Rogier O, Jorge V, Leplé JC, Brunaud V et al. (2020) Gene expression predictions and networks in natural populations supports the omnigenic theory. BMC Genom 21:1–16
Choi T, Lim D, Park B, Sharma A, Kim JJ, Kim S et al. (2017) Accuracy of genomic breeding value prediction for intramuscular fat using different genomic relationship matrices in Hanwoo (Korean cattle). Asian-Australas J Anim Sci 30:907
Christensen OF, Lund MS (2010) Genomic prediction when some animals are not genotyped. Genet Sel Evol 42:2
Christensen OF, Madsen P, Nielsen B, Ostersen T, Su G (2012) Single-step methods for genomic evaluation in pigs. Animal 6:1565–1571
Clark SA, Kinghorn BP, Van Der Werf JH (2013) Comparisons of identical by state and identical by descent relationship matrices derived from SNP markers in genomic evaluation. Proc Assoc Advmt Anim Breed Gene 20:261–265
Croué I, Ducrocq V (2017) Genomic and single-step evaluations of carcass traits of young bulls in dual-purpose cattle. J Anim Breed Genet 134:300–307
Cuyabano BCD, Sørensen AC, Sørensen P (2018) Understanding the potential bias of variance components estimators when using genomic models. Genet Sel Evol 50:41
Daetwyler HD, Calus MP, Pong-Wong R, de Los Campos G, Hickey JM (2013) Genomic prediction in animals and plants: simulation of data, validation, reporting, and benchmarking. Genetics 193:347–365
de Lima BM, Cappa EP, Silva-Junior OB, García C, Mansfield SD, Grattapaglia D (2019) Quantitative genetic parameters for growth and wood properties in Eucalyptus “urograndis” hybrid using near-infrared phenotyping and genome-wide SNP-based relationships. PLoS ONE 14:1–24
de los Campos G, Vazquez AI, Fernando RL, Klimentidis YC, Sorensen D (2013) Prediction of complex human traits using the genomic best linear unbiased predictor. PLoS Genet 9.
de los Campos G, Sorensen D, Gianola D (2015) Genomic heritability: what is it? PLoS Genet 11:e1005048
Durán R, Isik F, Zapata-Valenzuela J, Balocchi C, Valenzuela S (2017) Genomic predictions of breeding values in a cloned Eucalyptus globulus population in Chile. Tree Genet Genomes 13:1–12
Forneris NS, Steibel JP, Legarra A, Vitezica ZG, Bates RO, Ernst CW et al. (2016) A comparison of methods to estimate genomic relationships using pedigree and markers in livestock populations. J Anim Breed Genet 133:452–462
García-Baccino CA, Munilla S, Legarra A, Vitezica ZG, Forneris NS, Bates RO et al. (2017) Estimates of the actual relationship between half-sibs in a pig population. J Anim Breed Genet 134:109–118
Gianola D, Norton HW (1981) Scaling threshold characters. Genetics 99:357–364
Gianola D, Fernando RL, Schön CC (2020) Inferring trait-specific similarity among individuals from molecular markers and phenotypes with Bayesian regression. Theor Popul Biol 132:47–59
Gilmour AR, Cullis BR, Verbyla AP (1997) Accounting for natural and extraneous variation in the analysis of field experiments. J Agric Biol Environ Stat 2:269–293
Grattapaglia D, Silva-Junior OB, Resende RT, Cappa EP, Müller BS, Tan B et al. (2018) Quantitative genetics and genomics converge to accelerate forest tree breeding. Front Plant Sci 9:1693
Habier D, Fernando RL, Garrick DJ (2013) Genomic BLUP decoded: a look into the black box of genomic prediction. Genetics 194:597–607
Han L, Abney M (2011) Identity by descent estimation with dense genome-wide genotype data. Genet Epidemiol 35:557–567
Han L, Abney M (2013) Using identity by descent estimation with dense genotype data to detect positive selection. Eur J Hum Genet 21:205–211
Henderson CR (1984) Applications of linear models in animal breeding. University of Guelph, Guelph
Hill WG (2014) Applications of population genetics to animal breeding, from Wright, Fisher and Lush to genomic prediction. Genetics 196:1–16
Isik F, Bartholomé J, Farjat A, Chancerel E, Raffin A, Sanchez L et al. (2016) Genomic selection in maritime pine. Plant Sci 242:108–119
Jacquard A (1974) The genetic structure of populations. Springer, New York, NY
Jones NB, Naidoo S, Kanzler A, Myburg A (2019) Genomic prediction by combining data across Eucalyptus dunnii populations. IUFRO tree biotechnology conference, Raleigh, 23–28.
Klápště J, Suontama M, Dungey HS, Telfer EJ, Graham NJ, Low CB et al. (2018) Effect of hidden relatedness on single-step genetic evaluation in an advanced open-pollinated breeding program. J Hered 109:802–810
Klápště J, Dungey HS, Graham NJ, Telfer EJ (2020a) Effect of trait’s expression level on single-step genomic evaluation of resistance to Dothistroma needle blight. BMC Plant Biol 20:1–13
Klápště J, Dungey HS, Telfer EJ, Suontama M, Graham NJ, Li Y et al. (2020b) Marker selection in multivariate genomic prediction improves accuracy of low heritability traits. Front Genet 11:499094
Kumar SK, Feldman MW, Rehkopf DH, Tuljapurkar S (2016) Limitations of GCTA as a solution to the missing heritability problem. Proc Natl Acad Sci USA 113:E61–E70
Lande R, Thompson R (1990) Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics 124:743–756
Legarra A, Robert-Granié C, Manfredi E, Elsen JM (2008) Performance of genomic selection in mice. Genetics 180:611–618
Legarra A, Aguilar I, Misztal I (2009) A relationship matrix including full pedigree and genomic information. J Dairy Sci 92:4656–4663
Legarra A, Christensen OF, Aguilar I, Misztal I (2014) Single step, a general approach for genomic selection. Livest Sci 166:54–65
Lenz PR, Nadeau S, Mottet MJ, Perron M, Isabel N, Beaulieu J et al. (2019) Multi-trait genomic selection for weevil resistance, growth, and wood quality in Norway spruce. Evol Appl 13:76–94
Li Y, Klápště J, Telfer E, Wilcox P, Graham N, Macdonald L et al. (2019) Genomic selection for non-key traits in radiata pine when the documented pedigree is corrected using DNA marker information. BMC Genom 20:1–10
Luan T, Woolliams JA, Ødegård J, Dolezal M, Roman-Ponce SI, Bagnato A et al. (2012) The importance of identity-by-state information for the accuracy of genomic selection. Genet Sel Evol 44:1–7
Marcó M, White TL (2002) Genetic parameter estimates and genetic gains for Eucalyptus grandis and E. dunnii in Argentina. Genet 9:205–215
Marcucci Poltri S, Zelener N, Traverso JR, Gelid P, Hopp HE (2003) Selection of a seed orchard of Eucalyptus dunnii based on genetic diversity criteria calculated using molecular markers. Tree Physiol 23:625–632
Meuwissen THE, Goddard ME (1996) The use of marker haplotypes in animal breeding schemes. Genet Sel Evol 28:161–176
Meuwissen TH, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Meuwissen TH, Luan T, Woolliams JA (2011) The unified approach to the use of genomic and pedigree information in genomic evaluations revisited. J Anim Breed Genet 128:429–439
Misztal I, Legarra A, Aguilar I (2009) Computing procedures for genetic evaluation including phenotypic, full pedigree, and genomic information. J Dairy Sci 92:4648–4655
Misztal I, Tsuruta S, Lourenco DAL, Masuda Y, Aguilar I, Legarra A et al. (2018) Manual for BLUPF90 family programs. University of Georgia, Athens, USA
Mphahlele MM, Isik F, Mostert-O’Neill MM, Reynolds SM, Hodge GR, Myburg AA (2020) Expected benefits of genomic selection for growth and wood quality traits in Eucalyptus grandis. Tree Genet Genomes 16:1–12
Muñoz PR, Resende Jr MF, Huber DA, Quesada T, Resende MD, Neale DB et al. (2014) Genomic relationship matrix for correcting pedigree errors in breeding populations: impact on genetic parameters and genomic selection accuracy. Crop Sci 54:1115–1123
Muñoz F, Sánchez L (2014) breedR: statistical methods for forest genetic resources analysts, R package.
Naidoo R, Jones N, Kanzler A, Myburg A (2018) Genomic selection modelling of growth and wood properties in Eucalyptus dunnii. In: Cirad (ed.) Eucalyptus 2018: managing eucalyptus plantation under global changes, IUFRO: Montpellier, p 100.
Ødegård J, Meuwissen TH (2014) Identity-by-descent genomic selection using selective and sparse genotyping. Genet Sel Evol 46:1–8
Rambolarimanana T, Ramamonjisoa L, Verhaegen D, Tsy JMLP, Jacquin L, Cao-Hamadou TV et al. (2018) Performance of multi-trait genomic selection for Eucalyptus robusta breeding program. Tree Genet Genomes 14:1–13
Resende MDV, Resende Jr MFR, Sansaloni CP, Petroli CD, Missiaggia AA, Aguiar AM et al. (2012) Genomic selection for growth and wood quality in Eucalyptus: capturing the missing heritability and accelerating breeding for complex traits in forest trees. New Phytol 194:116–128
Resende RT, Resende MDV, Silva FF, Azevedo CF, Takahashi EK, Silva-Junior OB et al. (2017) Assessing the expected response to genomic selection of individuals and families in Eucalyptus breeding with an additive-dominant model. Heredity 119:245–255
Rincent R, Laloë D, Nicolas S, Altmann T, Brunel D, Revilla P et al. (2012) Maximizing the reliability of genomic selection by optimizing the calibration set of reference individuals: comparison of methods in two diverse groups of maize inbreds (Zea mays L.). Genetics 192:715–728
Silva-Junior OB, Faria DA, Grattapaglia D (2015) A flexible multi-species genome wide 60K SNP chip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytol 206:1527–1540
Speed D, Balding DJ (2015) Relatedness in the post-genomic era: is it still useful? Nat Rev Genet 16:33–44
Stejskal J, Lstibůrek M, Klápště J, Čepl J, El-Kassaby YA (2018) Effect of genomic prediction on response to selection in forest tree breeding. Tree Genet Genomes 14:1–9
Su G, Guldbrandtsen B, Gregersen VR, Lund MS (2010) Preliminary investigation on reliability of genomic estimated breeding values in the Danish Holstein population. J Dairy Sci 93:1175–1183
Thavamanikumar S, Arnold RJ, Luo J, Thumma BR (2020) Genomic studies reveal substantial dominant effects and improved genomic predictions in an open-pollinated breeding population of Eucalyptus pellita. G3 (Bethesda) 10:3751–3763
Ukrainetz NK, Mansfield SD (2020) Prediction accuracy of single-step BLUP for growth and wood quality traits in the lodgepole pine breeding program in British Columbia. Tree Genet Genomes 16:64
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
Vela-Avitúa S, Meuwissen TH, Luan T, Ødegård J (2015) Accuracy of genomic selection for a sib-evaluated trait using identity-by-state and identity-by-descent relationships. Genet Sel Evol 47:1–6
Wang B, Thompson E (2019) Realized genome sharing in heritability estimation using random effects models. G3 9:1385–1391
Wang B, Sverdlov S, Thompson E (2017) Efficient estimation of realized kinship from single nucleotide polymorphism genotypes. Genetics 205:1063–1078
Wimmer V, Albrecht T, Auinger HJ, Schön CC (2012) synbreed: a framework for the analysis of genomic prediction data using R. Bioinformatics 28:2086–2087
Wright S (1922) Coefficients of inbreeding and relationship. Am Nat 56:330–338
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR et al. (2010) Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42:565–569
Acknowledgements
The authors sincerely acknowledge Dr. Carolina García-Baccino for her help in implementing the IBDLD v3.14 software. Also, thanks go to Dr. “Fito” Cantet who helped us in the interpretation of the results obtained in this study. Particular thanks to Pedro Ernesto Gelid, Victor Oscar Botto, Santiago Becerro, Mauro Rodrigo Surenciski, Santiago Álvarez Cettour, and Nicolás Alanis for help in establishing, maintaining, and assessing the trials and for assistance with field sampling. They would also like to thank the landowner associated with the field trials involved in this study.
Funding
This research was supported by the BIOTECH II platform (grant number 373-780) and the Instituto Nacional de Tecnología Agropecuaria (PNFOR-1104062 and PNFOR-1104064). EJJ’s was supported by doctoral fellowships from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET). EPC’s research was partially supported by the Agencia Nacional de Promoción Científica y Tecnológica of Argentina PICT-2016 1048.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Jurcic, E.J., Villalba, P.V., Pathauer, P.S. et al. Single-step genomic prediction of Eucalyptus dunnii using different identity-by-descent and identity-by-state relationship matrices. Heredity 127, 176–189 (2021). https://doi.org/10.1038/s41437-021-00450-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-021-00450-9
