
Abstract
Advances in sequencing technologies have revolutionized wildlife conservation genetics. Analysis of genomic data sets can provide high-resolution estimates of genetic structure, genetic diversity, gene flow, and evolutionary history. These data can be used to characterize conservation units and to effectively manage the genetic health of species in a broad evolutionary context. Here we utilize thousands of genome-wide single-nucleotide polymorphisms (SNPs) and mitochondrial DNA to provide the first genetic assessment of the Australian red-tailed black-cockatoo (Calyptorhynchus banksii), a widespread bird species comprising populations of varying conservation concern. We identified five evolutionarily significant units, which are estimated to have diverged during the Pleistocene. These units are only partially congruent with the existing morphology-based subspecies taxonomy. Genetic clusters inferred from mitochondrial DNA differed from those based on SNPs and were less resolved. Our study has a range of conservation and taxonomic implications for this species. In particular, we provide advice on the potential genetic rescue of the Endangered and restricted-range subspecies C. b. graptogyne, and propose that the western C. b. samueli population is diagnosable as a separate subspecies. The results of our study highlight the utility of considering the phylogeographic relationships inferred from genome-wide SNPs when characterizing conservation units and management priorities, which is particularly relevant as genomic data sets become increasingly accessible.
Similar content being viewed by others
Introduction
Protection and recognition of biodiversity is largely focused at the species level (CITES 2019; IUCN 2019). There is increasing awareness, however, of the need to protect diversity within species (Pollock et al. 2017) and this is now recognized in policy (Aichi Target 13 of SCBD 2010). Implementation of such policies can be supported through research that apportions within-species genetic variation to appropriate taxonomic and conservation units, enabling conservation managers to identify population genetic diversity and prioritize conservation interventions that support long-term retention of adaptive variation. Accordingly, the maximization of species’ evolutionary potential is a fundamental goal of conservation genetics (Ryder 1986; Reed and Frankham 2003; Allendorf and Luikart 2007). Delineation of appropriate genetic units can form the basis for decisions relating to habitat protection, in situ captive population management and ex situ population augmentation (i.e. genetic rescue) (Frankham et al. 2017).
There has been growing awareness and understanding of the complex population-species continuum in which conservation units usually operate (Coates et al. 2018). However, delineating conservation units using one or relatively few genetic markers can be imprecise and misleading, because the demographic and evolutionary signals at different loci are often discordant (Toews and Brelsford 2012; Morales et al. 2017). Thus, the advent of high-throughput sequencing and population genomics has the potential to enhance management of conservation units (Funk et al. 2012; Barbosa et al. 2018; Langin et al. 2018). An emerging paradigm shift recommends an assessment of the genetic health of conservation units (and other population isolates), and the establishment of strategies to mitigate genetic erosion where necessary (Ralls et al. 2018). Hence, knowledge of the relative genetic diversity, interconnectedness and evolutionary history among populations is important for managing the genetic health of a species in a broad evolutionary context.
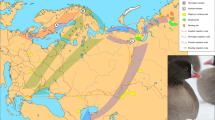
A widespread species whose conservation management is hampered by a lack of genetic data is the red-tailed black-cockatoo (Calyptorhynchus banksii), one of Australia’s most iconic bird species. A large, endemic Australian parrot in the family Cacatuidae (cockatoos), the species is sexually dimorphic for bill colour and the stark red (adult male) or red and orange-yellow (adult female) panels in its tail (Ford 1980). It is widespread throughout Australia from tropical savannahs to desert rangelands and temperate forests and woodlands (Fig. 1). Five subspecies are currently recognized based on body size, bill size and shape, and plumage patterning and colour (Ford 1980; Schodde 1997; see Supplementary Table S1 for subspecies differences in morphology; see Latham 1790; Gould 1843; Mathews 1917 and Schodde 1989 for all original subspecies descriptions).

Circles represent tissue samples (n = 113) and stars represent toe pad samples (n = 29). This distribution map has been adapted from Ford (1980). See Johnstone and Storr (1998) and Johnstone et al. (2013b) for details on distribution in the Western Australian populations. The inset photo of Calyptorhynchus banksii banksii demonstrates the morphological differences between male (right) and female (left) birds. Photo: Patrick Tomkins.
An allozyme study of the systematics of cockatoos (Adams et al. 1984) included single individuals representing four C. banksii subspecies and found relatively low divergence among them. The two southern temperate zone subspecies, C. b. graptogyne Schodde, Saunders & Homberger, 1989, and C. b. naso Gould, 1843 (subspecies epithets used hereafter for brevity where appropriate), occur in south-eastern and south-western Australia, respectively (Fig. 1). Their estimated population sizes are ~1000 and 15,000, respectively, and they are listed as Endangered (graptogyne) and Vulnerable (naso) under Australian Federal and State legislation (Barrett et al. 2003; Department of the Environment 2018a, b). The three tropical northern and arid zone central Australian subspecies that are often recognized, C. b. banksii (Latham, 1790), C. b. macrorhynchus Gould 1843, and C. b. samueli Mathews, 1917, are more abundant, although less well studied. Data on hybridization among subspecies are scarce and anecdotal, and there is uncertainty over whether these divisions accurately reflect the species’ history and long-term barriers to gene flow. Further, samueli as currently construed consists of five populations considered to be allopatric with respect to each other and all other populations (Fig. 1). The south-eastern samueli population is listed as Vulnerable. Whether gene flow, current or historical, has been involved in the evolution of the samueli isolates is unknown.
Despite the threatened status of some C. banskii subspecies and long-standing taxonomic uncertainties, there has been no comprehensive genetic analysis of this species. An updated assessment of how the species should be subdivided would allow more informed conservation priorities to be made, particularly for birds from the poorly studied tropical northern and arid zone central subspecies. Of particular interest is whether gene flow occurs among supposedly allopatric populations, and the extent of cryptic diversity and population structure within the species. Aside from taxonomic recognition of subspecies, the species could potentially be subdivided into conservation units, specifically evolutionarily significant units (ESUs; independently evolving units of genetic variation; Ryder 1986; Moritz 1994; Fraser and Bernatchez 2001) and/or management units (MUs; demographically independent, but not necessarily evolutionarily independent; Moritz 1994; Palsbøll et al. 2007). ESUs and subspecies are implicitly linked and are usually characterized using similar methods. Diagnosis of taxonomic subspecies, however, generally requires multiple lines of evidence and such units do not necessarily represent targets for conservation management and action (Mace 2004; Patten 2015).
Here we use genome-wide single-nucleotide polymorphism (SNP) and mitochondrial DNA (mtDNA) data to conduct a comprehensive population genomic and phylogeographic assessment of C. banksii across its distribution. We substantially expand and apply our earlier methodological work on the use of cryo-frozen tissue and traditional museum samples (Ewart et al. 2019). Our two main objectives are: (1) to assess whether evolutionarily and demographically independent units occur within C. banksii and (2) to evaluate the genetic diversity and estimate the evolutionary history of the species to underpin effective conservation strategies. We also investigate the benefits of analysing genome-wide SNP data in conjunction with mtDNA and morphological differences to expand the original concept of ESUs. Critically, we consider the conservation implications of our findings and determine whether levels of divergence between populations could represent separate taxonomic units (i.e. subspecies or species). We hope that this work will serve as a case study to inform policy on how to characterize and protect biodiversity more broadly, especially in light of Australia’s current extinction crisis.
Methods
Single-nucleotide polymorphisms
We used SNP data previously obtained from 131 individuals of C. banksii (excluding replicate samples) from across its Australian distribution (Ewart et al. 2019; Fig. 1; Supplementary Table S2). These data were generated using the DArTseq™ platform (Sansaloni et al. 2011; Kilian et al. 2012; Cruz et al. 2013). Briefly, genome complexity was reduced using restriction enzyme digestion and fragment size selection. DNA was digested using a combination of two restriction enzymes (PstI and SphI), then processed following Kilian et al. (2012), using two different adaptors. The library was then subjected to a competitive PCR and sequenced using an Illumina Hiseq2500. The library preparation and sequencing was performed by Diversity Arrays Technology (DArT) in Canberra, Australia. Additional library preparation details can be found in Ewart et al. (2019).
In addition to the SNPs generated by Ewart et al. (2019), we included SNPs genotyped from tissue samples of three glossy black-cockatoos (Calyptorhynchus lathami), the sister species of C. banksii (White et al. 2011), to provide an outgroup for phylogenetic analyses. Genomic DNA from samples of C. lathami was extracted following the manufacturer’s protocols for the Bioline Isolate II Genomic DNA kit (Bioline, Australia). DNA concentration was measured using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Australia) and 15 µL of DNA was sent to DArT for further processing (Kilian et al. 2012; Cruz et al. 2013).
To test the effects of using different SNP-calling methods, we ran analyses using SNPs called with DArTsoft14 (a proprietary DArT SNP-calling pipeline) and with STACKS (Catchen et al. 2011, 2013; Shafer et al. 2016). The resultant SNP data sets are referred to as the ‘DArTsoft14 SNP’ and ‘STACKS SNP’ data sets, respectively. Variation in the number of samples genotyped in populations can lead to ascertainment bias, because allele frequency distributions are biased towards more common alleles when using a smaller sample size (Lachance and Tishkoff 2013). Hence, we also called SNPs based on rarefaction (four samples per putative population), referred to as the ‘rarefied SNP’ data set, to investigate the effects of ascertainment bias and uneven sampling. We only included contemporary tissue samples (all collected after 1977) in this rarefaction sampling to account for potential temporal biases arising from the inclusion of older samples (some >100 years old). Further, when including the three outgroup samples, a separate SNP-calling analysis was performed (using DArTsoft14). The resultant SNP data set was used for the phylogenetic analysis requiring an outgroup (i.e. the maximum-likelihood analysis below). Details of different sampling schemes and SNP-calling methods are provided in the Supplementary Material (Appendix I).
We filtered the SNPs for quality and missing data, based on reproducibility, minor allele frequency, log likelihood of SNP calls and call rate. Additional filters were applied for analyses with different requirements and assumptions, such as linkage equilibrium and Hardy–Weinberg equilibrium. Filtering was performed using the R packages dartR 1.0.5 (Gruber and Georges 2018) and adegenet 2.1.0 (Jombart 2008). Details of SNP-filtering strategies for the different analyses are provided in the Supplementary Material (Appendix II).
Mitochondrial DNA sequences
Mitochondrial genomic data were generated via low-coverage whole genomic sequencing and assembled using Geneious 10.2.4 (Kearse et al. 2012) and NOVOPlasty (Dierckxsens et al. 2017) for six samples (indicated in Supplementary Table S2). We subsequently performed PCRs on 34 samples (indicated in Supplementary Table S2) using primers designed to target the mitochondrial ND1, ND2 and ND5 genes. Amplicons were Sanger sequenced at the Australian Genome Research Facility (Australia) and Apical Scientific Sdn Bhd (Malaysia). Further information on whole-genome sequencing, mitochondrial genome assemblies, primers and PCR conditions is provided in the Supplementary Material (Appendix III).
Genetic structure
We used four methods to investigate population structure and identify population units based on SNPs. These included discriminant analysis of principal components (DAPC; Jombart 2008), STRUCTURE (version 2.3; Pritchard et al. 2000), pairwise FST (Weir and Cockerham 1984) and pairwise population kinship coefficients (Loiselle et al. 1995).
DAPC was carried out using the R package adegenet. DAPC differs from principal components analysis by minimizing variation within groups and maximizing variation between groups, making it an effective approach to investigate population clustering. We assessed multiple values of K to investigate whether the identification of genetic clusters was precluded by high gene flow (Younger et al. 2017). We reran the DAPC within some groups to investigate intrapopulation structure, and to further examine genetic structure inferred from the mtDNA analysis (see mtDNA results in Results section).
STRUCTURE analyses were run for 500,000 steps with a burn-in of 100,000 steps. We modelled up to 10 ancestral populations (i.e. K = 1–10), replicating each model 10 times and assuming admixture and independent allele frequencies. We ran these analyses without the use of location information to establish priors. We used a hierarchical approach: first we analysed all individuals to detect the uppermost layer of structure, then repeated the analysis for distinct genetic clusters that were identified. To determine the optimal value of K, we used six different estimators in StructureSelector (Li and Liu 2018). Results of replicate runs were merged and plots were produced using CLUMPAK (Kopelman et al. 2015), which we ran through StructureSelector.
To calculate pairwise FST values (Weir and Cockerham 1984), we used the R package hierfstat 0.4.22 (Goudet 2005). We performed 1000 bootstraps to test for significance and to construct 95% confidence intervals. Kinship coefficients based on Loiselle et al. (1995) were estimated for each pair of individuals using GENODIVE (Meirmans and Van Tienderen 2004); we subsequently calculated the average of the pairwise estimates between groups.
To investigate population structure based on mtDNA data, we constructed a haplotype network using 40 concatenated sequences of ND1, ND2 and ND5 (a total of 1998 bp) via the statistical parsimony TCS method (Clement et al. 2000) in PopART (Leigh and Bryant 2015).
Genetic diversity
We measured the genetic diversity of each group by computing four metrics from the SNP data. First, we computed the average expected and observed heterozygosities using GenAlEx 6.503 (Peakall and Smouse 2006, 2012). We then calculated allelic richness using the R package diveRsity 1.9.9 (Keenan et al. 2013) with 1000 bootstrap replicates to account for uneven sample sizes. Finally, we calculated intrapopulation kinship coefficients using GENODIVE, as described above for the measurements of pairwise population kinship coefficients (i.e. the average of the pairwise estimates within groups).
For the mtDNA data, we calculated the genetic diversity (i.e. number of haplotypes and nucleotide diversity) of each group using the R package pegas 0.1 (Paradis 2010).
SNP phylogenetic analyses
We performed phylogenetic analyses to identify whether genetic clusters exhibited reciprocal monophyly, and to investigate the evolutionary history of the species. We conducted a maximum-likelihood analysis using RAxML (Stamatakis 2014) based on concatenated SNPs, using the GTR substitution model with Γ-distributed rates among sites. This is the most complex time-reversible model that does not allow a proportion of invariant sites. We used the Lewis-type ascertainment bias correction to account for our exclusion of invariant sites. Node support was estimated using 1000 bootstrap replicates. We removed SNPs that did not exhibit both homozygote genotypes at least once, because the Lewis correction requires all three SNP genotypes at every marker to be present (details of this filtering strategy are in Supplementary Material, Appendix II). We performed this RAxML analysis using the same samples for which we had obtained mtDNA data (n = 40) to allow a direct comparison. We performed these analyses using SNP data sets called with and without an outgroup. The trees were rooted using the outgroup method (implemented in Figtree 1.4.2; Rambaut 2009), midpoint method (implemented in Figtree) and minimal ancestor deviation method (using R scripts from Tria et al. 2017). Three rooting methods were used to check for consistency in clustering and topology.
We also performed model comparison in SNAPP (Bryant et al. 2012), implemented in BEAST 2.4 (Bouckaert et al. 2014), to test a number of ‘species’ hypotheses (subspecies or ESUs in this case). SNAPP is a Bayesian coalescent-based method that can estimate species trees using independent biallelic SNPs. We tested four ‘species’ hypotheses to assess the conflicting scenarios supported by our population genetic analyses and by morphology (Table 2). For this analysis, to improve computational tractability, we used four individuals from each putative genetic cluster identified in the population genetic analyses, and 1000 randomly selected SNPs after filtering (filtering details are provided in the Supplementary Material, Appendix II). SNAPP samples the position of the root and does not require an outgroup. We ran the analysis for four million Markov chain Monte Carlo (MCMC) steps, sampling every 1000 steps after a burn-in of 400,000 steps. We used allele frequencies for the forward and backward mutation rates, and the default settings for priors. Next, we compared models using the AICM (Akaike information criterion through MCMC) method, which we chose for its computational tractability. The best model was considered to be the one with the lowest AICM score. We ran three replicates of the best-supported scenario. After checking for convergence in Tracer 1.6 (Rambaut et al. 2014), we combined samples from the three runs. The tree was visualized using Figtree and DensiTree 2 (Bouckaert and Heled 2010).
Mitochondrial DNA phylogenetic analysis
We conducted phylogenetic analyses of mtDNA to provide a comparison with the results of our analyses of nuclear DNA SNPs, because mitochondrial phylogenies frequently form the basis of conservation decisions. We analysed mtDNA (1998 bp of concatenated ND1, ND2 and ND5) using Bayesian inference in MrBayes 3.2 (Ronquist et al. 2012) and maximum likelihood in RAxML. For both the maximum-likelihood and Bayesian analyses, we used the HKY + G substitution model, following model selection in MEGA 6.06 (Tamura et al. 2013), and partitioned the sequence data by codon position. MrBayes was performed using four independent Markov chains, each run for 100 million steps and sampled every 1000 steps. Convergence diagnostics were calculated every 1000 steps; we regarded values of ≤0.01 for the average standard deviation of split frequencies as indicative of convergence. For the RAxML analysis, we ran 1000 bootstrap replicates. As above, trees were rooted using the outgroup, midpoint or minimal ancestor deviation methods.
Molecular dating analysis
The divergence times among C. banksii and other cockatoo taxa were estimated using a Bayesian phylogenetic analysis in BEAST 1.10 (Suchard et al. 2018). We analysed mtDNA haplotypes from C. banksii along with homologous sequences from C. lathami, C. baudinii, C. latirostris, Cacatua moluccensis, Cacatua pastinator and outgroup taxon Melopsittacus undulatus from White et al. (2011) (Supplementary Table S2). We ran the analysis using the GTR + G substitution model and using a strict clock with the highest (0.0119 substitutions/site/Myr) and lowest (0.0032 substitutions/site/Myr) estimates of mitochondrial substitution rates (based on C. lathami, the closest relative of C. banksii) estimated by Nabholz et al. (2016). We enforced monophyly for the Cacatuidae, Calyptorhynchus and C. banksii groups. To check the sensitivity of the results to the choice of tree prior (Ritchie et al. 2017), we performed the analysis using tree priors based on the constant-size coalescent and birth–death speciation model. Posterior distributions were estimated from samples drawn every 5000 steps over 20 million MCMC steps. After checking for convergence and sufficient sampling in Tracer, we removed 1% of the samples as burn-in and generated a maximum-clade-credibility tree using TreeAnnotator v.10 (part of the BEAST package).
Results
Population structure inferred from SNPs
Genetic clusters identified using DAPC when K = 5 included a northern group (macrorhynchus and banksii), a western samueli group (north- and south-western samueli isolates), a central/eastern samueli group (central, north-eastern and south-eastern samueli isolates; Fig. 1), a naso group and a graptogyne group (Fig. 2a). However, the optimal number of clusters identified with DAPC was K = 4 (based on the Bayesian information criterion for successive k-means), whereby the northern (i.e. banksii and macrorhynchus) and the western samueli groups clustered together (Fig. 2b). Other analyses (described below) indicated that five groups are likely to be present in the data set. Clustering was consistent when using different SNP data sets (Fig. 2a, Supplementary Fig. S1). We found that a range of principal components was considered optimal, and therefore ran each analysis multiple times with different numbers of principal components. Each of these analyses produced qualitatively similar results (not shown). One outlier sample was evident in the DAPC analysis (an individual naso) (Fig. 2a, b). This is likely to have been due to high levels of missing data (this sample had 76.29% missing data in the full DArTsoft14 SNP data set; missing data were replaced by the mean allele frequency).

Discriminant analysis of principal components for a K = 5, based on 60 principal components, 131 C. banksii samples and 11,918 SNPs from the DArTsoft14 SNP data set; and b K = 4 with the same settings and data as described for K = 5. Inset barplots represent the DAPC eigenvalues of the analysis (top right of each panel) and the PCA eigenvalues that were retained for the analysis (top left of each panel). STRUCTURE plots under various sampling schemes, based on SNPs from the DArTsoft14 SNP data set, for c K=4, based on 130 individuals across all C. banksii populations and 7219 SNPs; d K = 2, based on 63 individuals (C. b. banksii and C. b. macrorhynchus) and 7013 SNPs; and e K = 2, based on 15 individuals from central/eastern C. b. samueli and 3727 SNPs. Each individual is represented by a vertical bar showing the percentage ancestry attributable to each of the pertinent genetic groups identified by different colours.
To investigate whether there was any genetic structure within the northern group that aligned with its current recognition as two subspecies (i.e. macrorhynchus and banksii, based on morphology), we isolated samples from the northern group (and re-filtered the SNP data set) and ran a separate DAPC using K = 2. The two clusters identified did not coincide with the current morphological subspecies boundary (Supplementary Fig. S2). However, since we forced the analysis to detect two clusters, this division does not necessarily represent true genetic structure. Similarly, to investigate whether the central/eastern samueli group could be further subdivided as suggested by our mtDNA results (see below), we isolated the central/eastern samueli samples and ran a separate DAPC using K = 2–4. Individuals from the south-eastern samueli isolate consistently clustered separately from the other samueli isolates, while individuals from north-eastern samueli separated into two clusters when K = 2 and K = 3 (Supplementary Fig. S3; see Fig. 1 for the distribution of samueli). Individuals from the central samueli isolate either clustered with north-eastern samueli (K = 3) or formed a separate cluster (K = 4).
The optimal number of ancestral populations for the STRUCTURE analysis varied across different estimators, although most supported K = 4. We present the major modes generated by CLUMPAK for K = 3, K = 4 and K = 5 (Fig. 2c–e, Supplementary Fig. S4). The outlier individual identified in the DAPC analysis, with a high proportion of missing data, was removed from the STRUCTURE results. Clear genetic differentiation was apparent among the five groups in all analyses, regardless of the K value or SNP data set used. However, resolution was reduced when we analysed the rarefied SNP data set (based on 20 individuals) (Supplementary Fig. S4d).
We identified significant differentiation among the five groups identified in the clustering analyses based on the pairwise FST analysis. Values ranged from 0.130 (between the northern and western samueli groups) to 0.435 (between naso and graptogyne) (Table 1). Pairwise FST results were mostly consistent between SNP data sets (Supplementary Table S3). FST estimates were considerably higher than the Hudson FST estimates from Ewart et al. (2019). This discrepancy is likely to be due to the use of a different FST estimator and differing sampling regimes (Willing et al. 2012; Bhatia et al. 2013). Most of the pairwise population kinship coefficients were negative (Table 1), indicating a lack of recent gene flow. There were some slight inconsistencies in the patterns of pairwise population kinship coefficients between the DArTsoft14 and rarefied SNP data sets (Supplementary Table S3).
Population structure based on mtDNA
We identified 20 mtDNA haplotypes across the distribution of C. banksii. With one exception, the haplotype network showed the same but less resolved patterns of population structure as those inferred from nuclear SNPs (Fig. 3a). Some central/eastern samueli mtDNA haplotypes clustered within the northern group and were relatively distant from the other central/eastern samueli haplotypes, in contrast with population clustering by nuclear SNPs.

a TCS-based haplotype network based on 1998bp of concatenated sequences of mitochondrial ND1, ND2 and ND5. Dashes on haplotype network branches indicate mutations between haplotypes, and the sizes of circles are proportional to the number of samples for each haplotype. b Phylogeny and divergence times of C. banksii based on 1998 bp concatenated sequences of mitochondrial ND1, ND2 and ND5 (isolated from Supplementary Fig. S11a). Blue bars on the tree correspond to the 95% credibility intervals of the estimated node ages.
Genetic diversity
Estimates of genetic diversity varied with the SNP data set being analysed, but their patterns remained relatively consistent (Supplementary Table S4). For all diversity metrics, graptogyne had the lowest genetic diversity, while the northern group had the highest. Calyptorhynchus banksii graptogyne also had the highest mean population kinship coefficient (Table 1; Supplementary Table S3), corroborating the other diversity estimates. The ranking of mtDNA-based estimates of diversity differed somewhat from those obtained using nuclear SNP data (Supplementary Table S4). Interpretation of the mtDNA diversity measures is not straightforward because the sampling is uneven and the mtDNA markers have low variability. Patterns of genetic diversity were relatively consistent when we accounted for ascertainment bias and uneven sampling (geographically and temporally).
Phylogeny inferred from SNPs
Phylogenetic analyses of nuclear SNPs (i.e. the maximum-likelihood analysis and Bayesian model selection) identified the same five distinct groups that we identified in the population genomic analyses. One exception was the northern group (containing banksii and macrorhynchus), which did not form a monophyletic group in the maximum-likelihood tree. Instead, rooting the tree with C. lathami as the outgroup rendered this northern clade polyphyletic (Supplementary Fig. S5). The relatively large distance between the outgroup and the ingroup might have caused an unreliable rooting of the ingroup clade in this case (Whitfield and Lockhart 2007). When excluding the outgroup and rooting the tree at the midpoint or using minimal ancestor deviation, each of the five clusters in Fig. 2 was found to be monophyletic (however the monophyly of the northern group received low support; Fig. 4). The effects that ascertainment bias, uneven sampling and SNP-calling pipeline had on phylogenetic inference are described in Supplementary Material (Appendix IV).

The tree was rooted using the midpoint method. Bootstrap support (%) is given above important branches.
Bayesian model selection gave the highest support to the same five groups (Table 2). In the tree associated with the best-supported scenario (i.e. five populations), two clades were well substantiated: one comprising naso and western samueli, and one comprising central/eastern samueli and the northern group (Supplementary Fig. S8). However, the placement of graptogyne was unresolved.
Phylogeny and divergence times inferred from mtDNA
In our phylogenetic analyses of mtDNA, as with the results from our analyses of nuclear SNPs, use of an outgroup caused the ingroup to be rooted at an unexpected position (Supplementary Fig. S9). The use of different Calyptorhynchus outgroup species (C. baudinii and C. latirostris) did not improve the inferred placement of the root (not shown). When excluding the outgroup and rooting the trees using the midpoint method or minimal ancestor deviation, three of the five groups were monophyletic: naso, western samueli and graptogyne (Supplementary Fig. S10). Similar to the haplotype network (Fig. 3a), central/eastern samueli individuals did not group together, with some clustering instead with individuals from the northern group (i.e. banksii and macrorhynchus). Individuals from the northern group were scattered throughout the tree.
The posterior mean of age of the most recent common ancestor (MRCA) of C. banksii ranged from 0.28 to 1.28 million years (Myr), depending on which tree prior and substitution rate was used (Fig. 3b, Supplementary Fig. S11). When using the higher substitution rate estimate from Nabholz et al. (2016) in this analysis (i.e. 0.0119 substitutions/site/Myr), the age of the Calyptorhynchus MRCA falls within the 95% credibility intervals reported by White et al. (2011). However, when using the lower substitution rate estimate (i.e. 0.0032 substitutions/site/Myr), the age of the MRCA of C. banksii increased to 1.28 Myr, and the Calyptorhynchus MRCA was older than the upper limits of the 95% credibility intervals reported by White et al. (2011).
Discussion
We have performed the first species-wide genomic assessment of an Australian parrot, the red-tailed black-cockatoo C. banksii, which has a continent-wide but fragmented distribution. Our analysis of mtDNA sequences and nuclear SNP data enabled high-resolution delineation and genetic characterization of novel ESUs within this morphologically and ecologically diverse species, while finding insufficient support for further differentiation of MUs. These data have provided insights into the interconnectedness and evolutionary history of this species, which will be critical for establishing effective conservation strategies that maximize its evolutionary potential and chances of persistence in the wild. Our population genomic and phylogeographic inferences have also allowed us to reassess the species’ taxonomy, which is currently based on morphology.
Conservation units within C. banksii
Our analyses reveal five divergent groups within C. banksii: a northern group (comprising nominal subspecies banksii and macrorhynchus), a central/eastern inland group (comprising the central and eastern populations of samueli), a western inland group (comprising the western populations of samueli), a south-western group (naso), and a south-eastern group (graptogyne). Phylogenetic analyses of nuclear SNPs supported the monophyly of each of these five groups. Although monophyly of the northern group was poorly supported in some cases, Bayesian model selection provided the highest support for a scenario in which macrorhynchus and banksii were combined into a single group (Table 2). Our analysis of mtDNA markers generally produced the same genetic clusters, with the exception that some individuals of central/eastern samueli are placed within the northern group. This discrepancy may be due to introgression between the central/eastern samueli and the northern groups (discussed below). However, it might also be due to incomplete lineage sorting; in two sister lineages, the alleles at any individual locus can take hundreds of thousands to millions of years to achieve reciprocal monophyly (Fraser and Bernatchez 2001). Numerous other studies have demonstrated that nuclear SNPs are effective markers for detecting population genetic structure that is not detected using mtDNA markers (e.g. Leslie and Morin 2016; Rodríguez‐Ezpeleta et al. 2016; Younger et al. 2017).
The five identified groups appear to have diverged in allopatry. They display significant divergence in nuclear allele frequencies and evolutionary distinctiveness (based on phylogenetic analyses); hence, we propose that each be considered an ESU (Moritz 1994). The main difference of this result from the current understanding of C. banksii taxonomy is the finding that western samueli is distinct from central and eastern samueli. Utilizing thousands of genome-wide SNPs allowed us to consider the phylogenetic patterns of the nuclear genome, and to reduce the period of evolutionary time required to permit detection of ESUs relative to that based on mtDNA (Fraser et al. 2016). ESUs are typically determined by assessing whether putative conservation units are reciprocally monophyletic in mitochondrial trees (Moritz 1994). By analysing SNPs in a phylogenetic framework, we were able to retain the ESU criterion of reciprocal monophyly, instead of relying on arbitrary levels of differentiation (e.g. FST), which are more sensitive to sampling design and marker selection. Furthermore, we were able to compare different ESU hypotheses using formal Bayesian model comparison (based on AICM), rather than relying on interpretation of phylogenetic node support values. These results demonstrate the value of genomic data for establishing ESUs (Funk et al. 2012; Peters et al. 2016; Coates et al. 2018; Langin et al. 2018).
We investigated additional genetic structure within ESUs. Our analyses of mtDNA recovered some genetic differentiation between the central/eastern samueli isolates. These isolates are tightly associated with different rivers and drainage systems and are known to be geographically disjunct from one another (Fig. 1; Ford 1980; Schodde 1997). However, there is minimal genetic structure among these isolates based on SNPs. When we constrained the DAPC analysis to detect two to four populations within central/eastern samueli using SNPs (i.e. K = 2–4), the clustering differed from that inferred from analysis of mtDNA. These inconsistencies and the lack of genetic structure inferred from the SNP analyses suggest that the three samueli isolates are linked by gene flow. Therefore, patterns of mtDNA genetic structure might be due to secondary contact between the north-eastern samueli isolate and the northern group. Given these inferences, there is insufficient evidence to treat these central/eastern samueli isolates as separate MUs.
In the large continuous northern group (macrorhynchus and banksii), we found no divisions across the sparsely wooded plains south of the Gulf of Carpentaria, which is currently recognized as the boundary between these subspecies. This suggests that that region, known as the Carpentarian Barrier, is not a significant biogeographic barrier for this species as it is in many bird species (Jennings and Edwards 2005; Lee and Edwards 2008; Toon et al. 2010; Baldassarre et al. 2014; but see Kearns et al. 2010; Dorrington et al. 2020). Therefore, they should be treated as a single ESU and MU.
Among the ESUs identified, the endangered south-eastern group (graptogyne) had the lowest genetic diversity, and is also known to have the smallest population size (Department of the Environment 2018a) (Supplementary Table S4). Low genetic diversity can decrease the average fitness of a population due to inbreeding and the accumulation of deleterious alleles, while Allee effects due to small population size can exacerbate these problems (Frankham et al. 2010; Crates et al. 2017). The high degree of relatedness between graptogyne individuals based on the mean population kinship coefficient is also indicative of inbreeding and warrants further monitoring to inform conservation management. The Vulnerable south-western group (naso) had the second-lowest level of genetic diversity according to most metrics, although not markedly lower than the other populations. Both graptogyne and naso are threatened by habitat loss and climate change (Joseph 1982; Maron 2005; Johnstone et al. 2013b; Garnett and Franklin 2014), which is particularly concerning given that low genetic diversity is associated with poor adaptive potential in changing environments (Frankham et al. 2010). It is unsurprising that the northern group is the most genetically diverse according to most metrics, given that it is the most abundant and widespread population.
Evolution and biogeography of C. banksii
Calyptorhynchus banksii is likely to have diversified within the past ~1.28 Myr, in the Pleistocene, based on our molecular dating analyses. The Pleistocene is characterized by oscillations of arid and mesic climates in Australia (Byrne et al. 2018). The strong genetic differentiation within C. banksii is consistent with the proposal that Pleistocene populations of some species were isolated in localized refugia from which they have since expanded (Byrne et al. 2008). Calyptorhynchus banksii subspecies/populations appear to have evolved via vicariance, as morphological and neutral genetic differentiation mostly coincide (Joseph and Wilke 2006).
The multiple genetically distinct groups found within C. banksii contrast with other widespread Australian bird species that diversified during the Pleistocene. These other species, including the singing honeyeater (Gavicalis virescens), spiny-cheeked honeyeater (Acanthagenys rufogularis), black-faced woodswallow (Artamus cinereus), and pied butcherbird (Cracticus nigrogularis) (Joseph and Wilke 2007; Kearns et al. 2010), typically exhibit limited phylogeographic divergence across their range. This difference may be due to the reliance of C. banksii on specific habitats for nesting; that is, inland rivers lined with tall trees having deep hollows (Joseph et al. 1991; Kurucz 2000; Johnstone et al. 2013b). Being constrained to these habitats, such as large river systems, the birds may have experienced restricted historical gene flow among populations. These patterns are more consistent with those of the galah (Eolophus roseicapilla), another widespread cockatoo species, which exhibits broad genetic differentiation between the northern, western, and eastern portions of its range (Engelhard et al. 2015).
Unfortunately, the order and timing of divergence of the C. banksii subspecies/populations could not be determined with confidence, because the root of the tree could not be resolved. Calyptorhynchus banksii diverged from its closest relative (glossy black-cockatoo, C. lathami) ~7 Myr ago (White et al. 2011). This ancient divergence is likely to have contributed to the uncertain placement of the root when using C. lathami as an outgroup (Whitfield and Lockhart 2007). Further, it is notoriously difficult to resolve phylogenetic relationships for taxa that have undergone recent or rapid evolutionary radiations (Whitfield and Lockhart 2007; Giarla and Esselstyn 2015).
The mito-nuclear discordance that we have identified raises the possibility of introgression from the northern group to the central/eastern samueli group, particularly since the placement of the two central/eastern samueli clades is consistent among the different mtDNA analyses (Leavitt et al. 2017). Uncovering potential past hybridization event(s) might lead to a clearer phylogenetic resolution for this species. However, discordance might have also been caused by a range of other factors, including sex-biased dispersal, marker-specific selection, and/or incomplete lineage sorting (Toews and Brelsford 2012).
The phylogeography of C. banksii supports the occurrence of species-specific responses to biogeographic barriers (Ford and Blair 2005; Byrne et al. 2008). As well as its idiosyncratic response to the Carpentarian Barrier (i.e. no apparent genetic structure), C. banksii exbibits no genetic differentiation at the Pilbara region (i.e. the north-western samueli isolate; Fig. 1) or Melville Island (part of the range of macrorhynchus), contrasting with other species such as the pied butcherbird and blue-winged kookaburra (Kearns et al. 2010; Dorrington et al. 2020). Calyptorhynchus banksii is a highly mobile species, hence likely capable of dispersing over these well-recognized biogeographical barriers (Higgins 1999).
Implications for taxonomy
Various subspecies delineations have been proposed for C. banksii over the past 50 years (cf. Condon 1975; Ford 1980; Schodde 1989, 1997), but the ESUs we have characterized are largely consistent with the morphologically diagnosed subspecies (sensu Schodde 1997) that have prevailed for over 20 years. Two notable exceptions, however, lead us to make two proposals.
Our first proposal is based on the lack of genetic evidence for the recognition of two subspecies across northern Australia or a divergence at the Carpentarian Barrier. The two subspecies were originally diagnosed as distinct by Gould (1843) on differences in tail colouration. Our findings are consistent with the lack of morphological divergence acknowledged by Ford (1980) and only tentative recognition of the subspecies by Schodde (1997). Hence, we recommend synonymizing C. b. macrorhynchus and Calyptorhynchus banksii banksii as Calyptorhynchus banksii banksii. Any phenotypic differences might have appeared through variable rates of divergent evolution in one or few genomic regions (Baldassarre et al. 2014; Lamichhaney et al. 2015; Stryjewski and Sorenson 2017).
Second, we infer that the western population of samueli as currently construed forms a separate subspecies. Western samueli and central/eastern samueli are more genetically divergent from each other than other pairs of C. banksii subspecies/populations (e.g. the central/eastern samueli group and the northern group). Further, individuals from western samueli form a clade that is more closely related to naso than to central/eastern samueli based on phylogenetic analyses of nuclear SNPs and mtDNA. The holotype of nominate samueli is a central Australian specimen and there are no synonyms (see Schodde 1997). Therefore, we propose that this western population of Calyptorhynchus banksii samueli be named:
Calyptorhynchus banksii escondidus Ewart, Joseph & Schodde, subsp. nov.
Description and diagnosis
Morphologically distinguishing characteristics, evident in adult females but not males, are spotting on head and shoulders moderately dense and bright cream-yellow, averaging 3–4 mm wide on shoulders; bars on ventrum rather bright and clearly defined, moderately clear cream-yellow, the bars cf. 2 mm wide, ranging from bright yellowish-orange to deep orange on under-tail coverts; reddish wash in tail panels extensive, deep orange-red, washing over distal two-thirds or more of inner vane on under-surface of outer 4–5 rectrices; spotting over under-wing coverts dense and coarse, clear mid cream-yellow. Bill small, with culmen from cere to tip cf. 43–47 mm, the maxilla notched under tip and, in females, leaden-bone; notch at tip of mandible ultra-shallow, <2.0 mm deep (Supplementary Table S5). Calyptorhynchus banksii escondidus differs from its genetically closest subspecies, C. b. naso, by its smaller, less robust bill and duller ventral barring and wing covert spotting in females (Supplementary Table S1), and from other small-bodied and small-billed ground-feeding populations in central and eastern Australia (C. b. samueli) in its more brightly marked females with orange barring in under-tail coverts, leaden tone to the maxilla in females and more shallowly notched tip to the mandible (Supplementary Table S5).
Holotype
Australian National Wildlife Collection (ANWC) B37847, adult female, from 13 miles NE of Morawa, Western Australia, 11 July 1978, Latitude −29.05, Longitude 116.1.
Paratypes
WAM A15957, adult female, ~48 km north-east of Wubin; WAM A15962, adult female, ~92 km north of Northampton; WAM A15960, juvenile male, ~92 km north of Northampton; ANWC B37843, adult male, ~20 km north-east of Morawa; ANWC B37153, immature male, ~10 km west of Mingenew.
Geographical range
The arid and semi-arid zones of Western Australia south of the Kimberley, here tentatively (e.g. Ford 1987) including the presumed isolated population inhabiting the north-eastern Pilbara in the upper drainages of the Oakover and de Grey Rivers (Ford 1980; Johnstone et al. 2013a), and definitely including the Wheatbelt. The range of this form specifically excludes the Kimberley region of far northern Western Australia inhabited by C. b. banksii (or by C. b. macrorhynchus if that taxon is recognized) and the more humid, forested southwest of Western Australia inhabited by naso (Fig. 1). For more details, see Ford (1980, 1987), Johnstone and Storr (1998), Schodde (1997) and Johnstone et al. (2013a, b).
Etymology
The subspecific epithet escondidus is the masculine gender of the Spanish and Portuguese adjective meaning ‘hidden’ and refers to this subspecies having been ‘hidden in plain sight’.
Other notes
All populations that have been assigned to samueli are predominantly ground feeders, whereas other C. banksii populations are mostly arboreal feeders (Forshaw and Cooper 2002; Cameron 2007). Morphological similarities between the form described here as escondidus and central/eastern samueli are thus likely to be convergent due to similar feeding ecologies (Homberger 1990, 2003).
Implications for conservation
Calyptorhynchus banksii contains two subspecies, graptogyne and naso, that are considered as Threatened under Australian Federal legislation, and/or State legislation. Recovery plans have been established for both (Department of the Environment 2018a, b). Identification of the five ESUs in this study, and the estimates of genetic diversity within these units, provide valuable information for establishing conservation priorities. Conservation strategies should aim to conserve the evolutionary potential of ESUs and to mitigate genetic erosion where necessary. We have identified some groups for which management could be improved and/or altered to take into account the results of our study, namely, the western samueli group and the south-eastern graptogyne group.
The effective genetic isolation of what we here recognize as C. b. escondidus has significant conservation implications, because protecting its evolutionarily distinct genetic diversity must now be considered. This newly recognized entity needs to be assessed to investigate whether it is threatened. Any natural or anthropogenic factors that may affect the genetic health and persistence of escondidus need to be evaluated. For example, escondidus feeds primarily on double-gee (Emex australis), a noxious weed found commonly in crop and pasture paddocks throughout south-west Western Australia (Saunders 1991). Although escondidus continues to expand its range into the Wheatbelt of Western Australia, significant control of double-gee by the agricultural industry could potentially have negative impacts (Saunders 1991; Scott et al. 1996). Therefore, additional research and forward planning are vital to ensure the persistence of this subspecies amidst control of their major and recently available food sources.
Management of genetic diversity in isolated populations is one of the major issues in conservation biology (Frankham 2010a, b). Calyptorhynchus banksii graptogyne has the lowest genetic diversity, is the most divergent from the other groups, and has the highest mean population kinship coefficient, indicating that the subspecies will be likely to suffer from the negative effects of inbreeding. Further, its low abundance (~1000 individuals; Department of the Environment 2018a) is of particular concern, since the rate of adverse genetic change is inversely related to population size (Frankham 1996). Genetic erosion is clearly a serious threat for graptyogyne, making it a prime candidate for genetic rescue: the re-establishment of gene flow between populations to alleviate the effects of genetic erosion in small isolated populations (Tallmon et al. 2004; Frankham 2015; Whiteley et al. 2015). To maximize the genetic diversity benefits, a source population for genetic rescue is chosen based on the lowest mean pairwise population kinship to the target inbred population (Frankham et al. 2017). However, genetic rescue usually involves establishing gene flow between populations that have been recently fragmented (within the last 500 years). Because graptogyne diverged from the other populations much earlier (up to ~1.28 Myr ago; Fig. 3b), the most sensible approach might involve utilizing the central/eastern samueli group, which is the most closely related group based on the highest mean pairwise population kinship coefficient. If the mean kinship of graptogyne is successfully reduced (without significant adverse effects), inbreeding is likely to decrease, hence genetic diversity will be maximized in subsequent generations.
Unfortunately, management authorities often do not have adequate data to mitigate threats caused by reductions in genetic diversity, or do not take them seriously (Frankham 2010b). Fears of outbreeding depression and the uncertainty in predicting the outcomes inhibit uptake of genetic rescue strategies (Frankham et al. 2017; Ralls et al. 2018). This is germane to graptogyne, since it is considerably divergent from central/eastern samueli (Table 2). However, studies have demonstrated that crossing highly divergent populations in a genetic rescue can be beneficial (Osborne et al. 2000; Garnett et al. 2011; Kronenberger et al. 2017; Weeks et al. 2017; Kelly and Phillips 2019), and further research can assist in predicting the outcomes of a genetic rescue. Relatively simple cytogenetic experiments can be conducted to determine whether the chromosomes of different populations are compatible, and aviary-based experiments investigate the effects of local adaptation of hybrid birds (e.g. through observing the feeding behaviour of a hybrid) (Frankham et al. 2017). Increased genome-level knowledge will assist investigations into the genetic basis of adaptive characteristics and ecologically important traits, such as bill shape and size, and might also assist in the predictions and management of crosses (Zhang et al. 2014; Lamichhaney et al. 2015; Johnson et al. 2018).
Conclusion
We have combined an extensive SNP data set with comprehensive population genomic and phylogenetic analyses to shed light on the evolutionary history of C. banksii. The result is an effective approach to investigate evolutionary processes that have occurred on relatively short evolutionary timescales, and to characterize conservation units and genetic diversity in a widespread species. We have successfully used genome-wide SNP data to establish ESUs within C. banksii, where mtDNA and morphological data have been unable to provide adequate resolution, and have provided a taxonomic reassessment of the group. This study represents the first comprehensive genetic analysis of C. banksii, one of Australia’s most iconic bird species. We hope that it will inform specific conservation management strategies and associated policy for this species in a broad evolutionary context.
Data archiving
All SNP data and the raw sequence reads used to call SNPs are available for download on the Dryad Digital Repository (https://doi.org/10.5061/dryad.rb15131) and short read archive (SRA accession: PRJNA557608), respectively. All mitochondrial data generated in the paper are available in GenBank (accession numbers MT308416–MT308535).
Change history
11 February 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41437-020-0343-7
References
Adams M, Baverstock PR, Saunders DR, Schodde R, Smith GT (1984) Biochemical systematics of the Australian cockatoos (Psittaciformes: Cacatuinae). Aust J Zool 32:363–377
Allendorf FW, Luikart G (2007) Conservation and the genetics of populations. Blackwell, Malden
Bhatia G, Patterson NJ, Sankararaman S, Price AL (2013) Estimating and interpreting FST: the impact of rare variants. Genome Res 23:1514–1521
Baldassarre DT, White TA, Karubian J, Webster MS (2014) Genomic and morphological analysis of a semipermeable avian hybrid zone suggests asymmetrical introgression of a sexual signal. Evolution 68:2644–2657
Barbosa S, Mestre F, White TA, Paupério J, Alves PC, Searle JB (2018) Integrative approaches to guide conservation decisions: using genomics to define conservation units and functional corridors. Mol Ecol 27:3452–3465
Barrett G, Silcocks A, Barry S, Cunningham R, Poulter R (2003) The new atlas of Australian birds. Royal Australasian Ornithologists Union, Melbourne
Bouckaert RR (2010) DensiTree: making sense of sets of phylogenetic trees Bioinformatics 26:1372–1373
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D et al. (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537
Bryant D, Bouckaert R, Felsenstein J, Rosenberg NA, RoyChoudhury A (2012) Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol Biol Evol 29:1917–1932
Byrne M, Joseph L, Yeates DK, Roberts JD, Edwards D (2018) Evolutionary history. In: Lambers H (ed) On the ecology of Australia’s arid zone. Springer, Cham, pp 45–75
Byrne M, Yeates DK, Joseph L, Kearney M, Bowler J, Williams MAJ et al. (2008) Birth of a biome: insights into the assembly and maintenance of the Australian arid zone biota. Mol Ecol 17:4398–4417
Cameron M (2007) Cockatoos. CSIRO Publishing, Melbourne
Catchen J, Amores A, Hohenlohe P, Cresko W, Postlethwait J (2011) Stacks: building and genotyping loci de novo from short‐read sequences. G3-Genes Genom Genet 1:171–182
Catchen J, Hohenlohe P, Bassham S, Amores A, Cresko W (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies Mol Ecol 9:1657–1659
Coates DJ, Byrne M, Moritz C (2018) Genetic diversity and conservation units: dealing with the species-population continuum in the age of genomics. Front Ecol Evol 6:1–13
Condon HT (1975) Checklist of birds of Australia: non-passerines. Royal Australasian Ornithologists Union, Melbourne
Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES) (2019). http://www.cites.org.
Crates R, Rayner L, Stojanovic D, Webb M, Heinsohn R (2017) Undetected Allee effects in Australia’s threatened birds: implications for conservation. Emu 117:207–221
Cruz VM, Kilian A, Dierig DA (2013) Development of DArT marker platforms and genetic diversity assessment of the U.S. collection of the new oilseed crop lesquerella and related species. PLoS ONE 8:e64062
Department of the Environment (2018a) Calyptorhynchus banksii graptogyne in species profile and threats database. Department of the Environment, Canberra. http://www.environment.gov.au/sprat
Department of the Environment (2018b) Calyptorhynchus banksii naso in species profile and threats database. Department of the Environment, Canberra. http://www.environment.gov.au/sprat
Dierckxsens N, Mardulyn P, Smits G (2017) NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res 45:e18
Dorrington A, Joseph L, Hallgren W, Mason I, Drew A, Hughes JM et al. (2020) Phylogeography of the blue-winged kookaburra Dacelo leachii across tropical northern Australia and New Guinea. Emu 120:33–45
Engelhard D, Joseph L, Toon A, Pedler L, Wilke T (2015) Rise (and demise?) of subspecies in the Galah (Eolophus roseicapilla), a widespread and abundant Australian cockatoo. Emu 115:289–301
Ewart KM, Johnson RN, Ogden R, Joseph L, Frankham GJ, Lo N (2019) Museum specimens provide reliable SNP data for population genomic analysis of a widely distributed but threatened cockatoo species. Mol Ecol Resour 19:1578–1592
Ford J (1980) Morphological and ecological divergence and convergence in isolated populations of the Red-tailed Black-Cockatoo. Emu 80:103–120
Ford JR (1987) Notes on Kimberley birds. Western Australian. Naturalist 16:181–184
Ford F, Blair D (2005) Neat patterns with a messy history: savannah refuges in northern Australia. Mamm Study 30:S45–S50
Forshaw JM, Cooper WT (2002) Australian parrots, 3rd edn. Alexander Editions, Robina
Frankham R (2010a) Challenges and opportunities of genetic approaches to biological conservation Biol Conserv 143:1919–1927
Frankham R (2010b) Where are we in conservation genetics and where do we need to go? Conserv Genet 11:661–663
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508
Frankham R (2015) Genetic rescue of small inbred populations: meta-analysis reveals large and consistent benefits of gene flow. Mol Ecol 24:2610–2618
Frankham R, Ballou JD, Briscoe DA (2010) Introduction to conservation genetics, 2nd edn. Cambridge University Press, Cambridge
Frankham R, Ballou JD, Ralls K, Eldridge MDB, Dudash MR, Fenster CB et al. (2017) Genetic management of fragmented animal and plant populations. Oxford University Press, Oxford
Fraser CI, McGaughran A, Chuah A, Waters JM (2016) The importance of replicating genomic analyses to verify phylogenetic signal for recently evolved lineages. Mol Ecol 25:3683–3695
Fraser DJ, Bernatchez L (2001) Adaptive evolutionary conservation: towards a unified concept for defining conservation units. Mol Ecol 10:2741–2752
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol 27:489–496
Garnett S, Franklin D (2014) Climate change adaptation plan for Australian birds. CSIRO Publishing, Melbourne
Garnett ST, Olsen P, Butchart SH, Hoffmann AA (2011) Did hybridization save the Norfolk Island boobook owl Ninox novaeseelandiae undulata? Oryx 45:500–504
Giarla TC, Esselstyn JA (2015) The challenges of resolving a rapid, recent radiation: empirical and simulated phylogenomics of Philippine shrews. Syst Biol 64:727–740
Goudet J (2005) Hierfstat, a package for R to compute and test hierarchical F-statistics Mol Ecol Notes 5:184–186
Gould J (1843) Descriptions of thirty new species of birds from Australia. Proc Zool Soc Lond 1842:131–140
Gruber B, Unmack PJ, Berry OF, Georges A (2018) DARTR: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol Ecol Resour 18:691–699
Higgins PJ (1999) Handbook of Australian, New Zealand & Antarctic Birds. vol 4, Parrots to Dollarbird. Oxford University Press, Melbourne
Homberger DG (1990) Filing ridges and transversal step of the maxillary rhamphotheca in Australian cockatoos (Psittaciformes: Cacatuidae): a homoplastic structural character evolved in adaptation to seed shelling. In: van den Elzen R, Schuchmann K-L, Schmidt-Koenig K (eds) Proceedings of the International 100th meeting of the German Ornithologists Society: current topics in avian biology. Bonn, Germany, pp 43–48
Homberger DG (2003) The comparative biomechanics of a prey–predator relationship: the adaptive morphologies of the feeding apparatus of Australian Black-Cockatoosand their foods as a basis for the reconstruction of the evolutionary history of the Psittaciformes. In: Bels VL, Gasc J-P, Casinos A (eds) Vertebrate Biomechanics and Evolution. BIOS Scientific Publishers, Scientific Publishers, Oxford, Chapter 13, pp 203–228
IUCN (2019) The IUCN red list of threatened species. Version 2019-1. http://www.iucnredlist.org.
Jennings WB, Edwards SV (2005) Speciational history of Australian grass finches (Poephila) inferred from thirty gene trees. Evolution 59:2033–2047
Johnson RN, O’Meally D, Chen Z, Etherington GJ, Ho SYW, Nash WJ et al. (2018) Adaptation and conservation insights from the koala genome. Nat Genet 50:1102
Johnstone RE, Burbidge AH, Darnell JC (2013a) Birds of the Pilbara region, including seas and offshore islands, Western Australia: distribution, status and historical changes Rec West Aust Mus Suppl 78:343–441
Johnstone RE, Kirby T, Sarti K (2013b) The breeding biology of the forest red-tailed black cockatoo Calyptorhynchus banksii naso Gould in south-western Australia. I. Characteristics of nest trees and nest hollows. Pac Conserv Biol 19:121–142.
Johnstone RE, Storr GM (1998) Handbook of Western Australian birds Volume 1. Non-passerines. Western Australian Museum, Perth
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Joseph L (1982) The Red-tailed Black-Cockatoo in south-eastern Australia. Emu 82:42–45
Joseph L, Emison WB, Bren WM (1991) Critical assessment of the conservation status of red-tailed black-cockatoos in south-eastern Australia with special reference to nesting requirements. Emu 91:46–50
Joseph L, Wilke T (2006) Molecular resolution of population history, systematics and historical biogeography of the Australian ringneck parrots Barnardius: are we there yet? Emu 106:49–62
Joseph L, Wilke T (2007) Lack of phylogeographic structure in three widespread Australian birds reinforces emerging challenges in Australian historical biogeography. J Biogeogr 34:612–624
Kearns A, Joseph L, Cook L (2010) The impact of Pleistocene climatic and landscape changes on Australian birds: a test using the pied butcherbird (Cracticus nigrogularis). Emu 110:285–295
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) diveRsity: an R package for the estimation of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788
Kelly E, Phillips B (2019) How many and when? Optimising targeted gene flow for a step change in the environment. Ecol Lett 22:447–457
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H et al. (2012) Diversity Arrays Technology: a generic genome profiling technology on open platforms. In: Pompanon F, Bonin A (eds) Data production and analysis in population genomics. Humana Press, Totowa, pp 67–89
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191
Kronenberger JA, Funk WC, Smith JW, Fitzpatrick SW, Angeloni LM, Broder ED et al. (2017) Testing the demographic effects of divergent immigrants on small populations of Trinidadian guppies. Anim Conserv 20:3–11
Kurucz N (2000) The nesting biology of the Red-tailed Black Cockatoo (Calyptorhynchus banksii macrorhynchus) and its management implications in the Top End of Australia. MSc Thesis, Northern Territory University.
Lachance J, Tishkoff SA (2013) SNP ascertainment bias in population genetic analyses: why it is important, and how to correct it. BioEssays 35:780–786
Lamichhaney S, Berglund J, Almén MS, Maqbool K, Grabherr M, Martinez-Barrio A et al. (2015) Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518:371
Langin KM, Aldridge CL, Fike JA, Cornman RS, Martin K, Wann GT et al. (2018) Characterizing range-wide divergence in an alpine-endemic bird: a comparison of genetic and genomic approaches. Conserv Genet 19:1471–1485
Latham J (1790) Index ornithologicus. Leigh & Sotheby, London
Leavitt DH, Marion AB, Hollingsworth BD, Reeder TW (2017) Multilocus phylogeny of alligator lizards (Elgaria, Anguidae): testing mtDNA introgression as the source of discordant molecular phylogenetic hypotheses. Mol Phylogenet Evol 110:104–121
Lee JY, Edwards SV (2008) Divergence across Australia’s Carpentarian barrier: statistical phylogeography of the red‐backed fairy wren (Malurus melanocephalus). Evolution 62:3117–3134
Leigh JW, Bryant D (2015) popart: full‐feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Leslie MS, Morin PA (2016) Using genome-wide SNPs to detect structure in high-diversity and low-divergence populations of severely impacted eastern tropical Pacific spinner (Stenella longirostris) and pantropical spotted dolphins (S. attenuata). Front Mar Sci 3:253
Li YL, Liu JX (2018) StructureSelector: a web based software to select and visualize the optimal number of clusters using multiple methods. Mol Ecol Resour 18:176–177
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Mace GM (2004) The role of taxonomy in species conservation. Philos Trans R Soc Ser B 359:711–719
Maron M (2005) Agricultural change and paddock tree loss: implications for an endangered subspecies of Red‐tailed Black‐Cockatoo. Ecol Manag Restor 6:207–212.
Mathews GM (1917) The birds of Australia, volume 6. Witherby and Co., London
Meirmans PG, Van Tienderen PH (2004) Genotype and Genodive: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4:792–794
Morales H, Pavlova A, Joseph L, Sunnucks P (2017) Perpendicular axes of incipient speciation generated by mitochondrial introgression. Mol Ecol 26:3241–3255
Moritz C (1994) Defining “evolutionarily significant units” for conservation. Trends Ecol Evol 9:373–375
Nabholz B, Lanfear R, Fuchs J (2016) Body mass‐corrected molecular rate for bird mitochondrial DNA. Mol Ecol 25:4438–4449
Osborne MJ, Norman JA, Christidis L, Murray ND (2000) Genetic distinctness of isolated populations of an endangered marsupial, the mountain pygmy-possum, Burramys parvus. Mol Ecol 9:609–613
Palsbøll PJ, Berube M, Allendorf FW (2007) Identification of management units using population genetic data. Trends Ecol Evol 22:11–16
Patten MA (2015) Subspecies and the philosophy of science. Auk: Ornithol Adv 132:481–485
Paradis E (2010) pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26:419–420
Peakall R, Smouse PE (2006) GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peakall R, Smouse PE (2012) GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Peters JL, Lavretsky P, DaCosta JM, Bielefeld RR, Feddersen JC, Sorenson MD (2016) Population genomic data delineate conservation units in mottled ducks (Anas fulvigula). Biol Conserv 203:272–281
Pollock LJ, Thuiller W, Jetz W (2017) Large conservation gains possible for global biodiversity facets. Nature 546:141–144
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rambaut A (2009) FigTree: tree figure drawing tool version 1.4.2. http://tree.bio.ed.ac.uk/software/figtree.
Rambaut A, Suchard MA, Xie D, Drummond AJ (2014) Tracer v1.6. http://beast.bio.ed.ac.uk/Tracer.
Ralls K, Ballou JD, Dudash MR, Eldridge MD, Fenster CB, Lacy RC et al. (2018) Call for a paradigm shift in the genetic management of fragmented populations. Conserv Lett 11:e12412
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237
Ritchie AM, Lo N, Ho SYW (2017) The impact of the tree prior on molecular dating of data sets containing a mixture of inter- and intraspecies sampling. Syst Biol 66:413–425
Rodríguez‐Ezpeleta N, Bradbury IR, Mendibil I, Álvarez P, Cotano U, Irigoien X (2016) Population structure of Atlantic mackerel inferred from RAD‐seq‐derived SNP markers: effects of sequence clustering parameters and hierarchical SNP selection. Mol Ecol Resour 16:991–1001
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S et al. (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Ryder OA (1986) Species conservation and systematics: the dilemma of subspecies. Trends Ecol Evol 1:9–10
Sansaloni C, Petroli C, Jaccoud D, Carling J, Detering F, Grattapaglia D et al. (2011) Diversity Arrays Technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc 5:P54
Saunders DA (1991) The effect of land clearing on the ecology of Carnabyas Cockatoo and the inland Red-tailed Black-Cockatoo in the wheatbelt of Western Australia Proc Int Ornithol Congr 20:658–665
SCBD (2010) Secretariat of the Convention On Biological Diversity. COP-10 Decision X/2 Target 13.
Schodde R (1989) New subspecies of Australian birds. Canberra Bird Notes 13:119–22
Schodde R (1997) Cacatuidae. In: Houston W, Wells A (eds) Aves (Columbidae to Coraciidae), zoological catalogue of Australia, vol 2. CSIRO Publishing, Melbourne, pp 64–108
Scott JK, Bowran DG, Corey SA (1996) Emex australis—biology, management and research. Plant Prot Q 11:137–178
Shafer ABA, Peart CR, Tusso S, Maayan I, Brelsford A, Wheat CW et al. (2016) Bioinformatic processing of RAD-seq data dramatically impacts downstream population genetic inference. Methods Ecol Evol 8:907–917
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Stryjewski KF, Sorenson MD (2017) Mosaic genome evolution in a recent and rapid avian radiation. Nat Ecol Evol 1:1912
Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A (2018) Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4:vey016
Tallmon DA, Luikart G, Waples RS (2004) The alluring simplicity and complex reality of genetic rescue. Trends Ecol Evol 19:489–496
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Toews DP, Brelsford A (2012) The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol 21:3907–3930
Toon A, Hughes JM, Joseph L (2010) Multilocus analysis of honeyeaters (Aves: Meliphagidae) highlights spatio‐temporal heterogeneity in the influence of biogeographic barriers in the Australian monsoonal zone. Mol Ecol 19:2980–2994
Tria FDK, Landan G, Dagan T (2017) Phylogenetic rooting using minimal ancestor deviation. Nat Ecol Evol 1:0193
Weeks AR, Heinze D, Perrin L, Stoklosa J, Hoffmann AA, van Rooyen A et al. (2017) Genetic rescue increases fitness and aids rapid recovery of an endangered marsupial population. Nat Commun 8:1071
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
White NE, Phillips MJ, Gilbert MTP, Alfaro-Núñez A, Willerslev E, Mawson PR et al. (2011) The evolutionary history of cockatoos (Aves: Psittaciformes: Cacatuidae). Mol Phylogenet Evol 59:615–622
Whiteley AR, Fitzpatrick SW, Funk WC, Tallmon DA (2015) Genetic rescue to the rescue. Trends Ecol Evol 30:42–49
Whitfield JB, Lockhart PJ (2007) Deciphering ancient rapid radiations. Trends Ecol Evol 22:258–265
Willing EM, Dreyer C, van Oosterhout C (2012) Estimates of genetic differentiation measured by FST do not necessarily require large sample sizes when using many SNP markers. PLoS ONE 7:e42649
Younger JL, Clucas GV, Kao D, Rogers AD, Gharbi K, Hart T et al. (2017) The challenges of detecting subtle population structure and its importance for the conservation of emperor penguins. Mol Ecol 26:3883–3897
Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C et al. (2014) Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346:1311–1320
Acknowledgements
We are grateful to the University of Sydney Industry and Community Engagement Seed Fund, Birdlife Australia (Stuart Leslie Bird Research Award), Australian Museum Foundation, Australian Government Research Training Programme (RTP) Scholarship and University of Sydney Merit Award, and Michael Sverns, Glenn Sharp and Stephen Lavelle from the Department of Environment, Land, Water and Planning (State Government of Victoria) for funding. We thank the following people/organizations for their help in obtaining samples: Robert Palmer (Australian National Wildlife Collection), Scott Ginn and Leah Tsang (Australian Museum), Ron Johnstone and Rebecca Bray (Western Australian Museum), Claire Keely (Museum Victoria) and Jessica Lee (Wildlife Reserves Singapore). We also thank Frankie Sitam and Jeffrine Rovie-Ryan for providing access and training at the National Wildlife Forensic Laboratory (Kuala Lumpur, Malaysia), Linda Neaves for her assistance with the mtDNA PCRs and sequencing, Kate FitzGerald for her assistance with the graphics, and Ron Johnstone for his expert advice on red-tailed black-cockatoos in Western Australia. Collection of birds for research purposes was approved by the Australian Museum Animal Care and Ethics Committee (approval number 16‐02). Transfer of C. banksii DNA from Australia to Malaysia was authorized under CITES permit number PWS2018-AU-000019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Xiangjiang Zhan
Supplementary information
Rights and permissions
About this article

Cite this article
Ewart, K.M., Lo, N., Ogden, R. et al. Phylogeography of the iconic Australian red-tailed black-cockatoo (Calyptorhynchus banksii) and implications for its conservation. Heredity 125, 85–100 (2020). https://doi.org/10.1038/s41437-020-0315-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-0315-y
This article is cited by
-
Building meaningful collaboration in conservation genetics and genomics
Conservation Genetics (2024)
-
Pleistocene allopatric differentiation followed by recent range expansion explains the distribution and molecular diversity of two congeneric crustacean species in the Palaearctic
Scientific Reports (2021)
