
Abstract
Theory predicts that when populations are established by few individuals, random founder effects can facilitate rapid phenotypic divergence even in the absence of selective processes. However, empirical evidence from historically documented colonisations suggest that, in most cases, drift alone is not sufficient to explain the rate of morphological divergence. Here, using the human-mediated introduction of the silvereye (Zosterops lateralis) to French Polynesia, which represents a potentially extreme example of population founding, we reassess the potential for morphological shifts to arise via drift alone. Despite only 80 years of separation from their New Zealand ancestors, French Polynesian silvereyes displayed significant changes in body and bill size and shape, most of which could be accounted for by drift, without the need to invoke selection. However, signatures of selection at genes previously identified as candidates for bill size and body shape differences in a range of bird species, also suggests a role for selective processes in driving morphological shifts within this population. Twenty-four SNPs in our RAD-Seq dataset were also found to be strongly associated with phenotypic variation. Hence, even under population founding extremes, when it is difficult to reject drift as the sole mechanism based on rate tests of phenotypic shifts, the additional role of divergent natural selection in novel environments can be revealed at the level of the genome.
Similar content being viewed by others


Introduction
The speed at which divergent evolution proceeds is highly variable. However, the underlying processes driving differences in evolutionary rates are not well understood (Provine 1989; Barton 1998; Emerson et al. 2001; Price 2008; Emerson and Gillespie 2008). Instances of rapid phenotypic divergence following the establishment of new populations are often attributed to the strong selective pressures provided by novel biotic and abiotic environments (Millien 2006; Mathys and Lockwood 2011; Jensen et al. 2017). However, rapid divergence of phenotypic traits does not necessarily need to involve selection. Theory predicts that when new populations are established, the random sampling effect of drift during the founding event has the potential to facilitate rapid phenotypic divergence (Mayr 1942; Carson and Templeton 1984). Such rapid phenotypic changes could reflect the phenotypic attributes of founders themselves (Grant and Grant 1995; Berry 1998; Kliber and Eckert 2005; Baker et al. 2006) or arise via more complex means in which population founding results in the reorganisation of quantitative genetic variation, catalysing divergence (Mayr 1954; Carson and Templeton 1984). Despite this, studies that attribute phenotypic divergence in naturally established populations to drift and founder effects are rarer (e.g. Armstrong et al. 2018), than the numerous publications that invoke divergent natural selection (e.g. Clegg et al. 2002b; Balakrishnan and Edwards 2009; Illera et al. 2007; Rasner et al. 2004; Yeh 2004).
As the effects of drift are expected to be most pronounced when effective population sizes are small, recovery times are long and long-term effective population sizes are limited (Wright 1931; Nei et al. 1975; Clegg 2010), the opportunity for founder-induced phenotypic divergence following natural colonisation events may be limited. For example, in the silvereye (Zosterops lateralis) the absence of founder-induced morphological divergence following the natural colonisation of islands across the South Pacific may be explained by: establishment by founding flocks in excess of hundreds of individuals (Clegg et al. 2002a; Estoup and Clegg 2003), rapid population recovery within two to three generations (Clegg 2010) and occasional immigration from the source population (Clegg and Phillimore 2010). In contrast to natural colonisation events, human-mediated introductions may be more likely to produce rapid phenotypic shifts via founder-induced divergence, as the number of founding individuals are often smaller, recovery times often longer, long-term effective population sizes are often more limited than in natural colonisations, and where introductions are geographically remote from source populations, gene flow does not occur (Blackburn et al. 2009). In this study we examine the potential for the more extreme founding conditions associated with human-mediated introductions to produce rapid morphological divergence.
The islands of French Polynesia contain a rich assemblage of introduced bird species following documented releases of at least fifty-nine species during the late 19th and early 20th centuries (Lockwood et al. 1999), with thirteen establishing breeding populations (Thibault and Cibois 2017). The silvereye was introduced from the South Island of New Zealand to the island of Tahiti in 1937 by aviculturist Eastham Guild (Guild 1938, 1940). Following introduction, the silvereye persisted in low numbers until the late 1950s after which it rapidly increased in population size and expanded into all habitat types on the island (Thibault and Monnet 1990; Monnet et al. 1993; Thibault and Cibois 2017). Following this, Tahitian silvereyes naturally dispersed to ten other island across French Polynesia, including islands within the Austral, Society and Tuamotu archipelagos (Thibault and Monnet 1990; Thibault and Cibois 2017) (Fig. 1). Although the exact number of silvereyes introduced to Tahiti is unknown, Guild’s wiritings in the Avicultural Magazine (Guild 1940), suggest the founding population likely consisted of only a handful of individuals (the individuals released were a gift from Mr G. Rowland Hutchinson, the President of the Avicultural Society of New Zealand). A small founding population size combined with a long population recovery and geographic isolation from source and neighbouring populations (the nearest silvereye population Z. l. flaviceps in Fiji is located over 3000 km away), provide the potential for rapid phenotypic divergence, such as changes in body/bill size and shape.

Here, we combine genome-wide tests for selection with morphological analyses to test the hypothesis that rapid phenotypic divergence can result from drift alone when populations establish under more extreme conditions than those associated with natural colonisation (very few founding individuals and slower population recovery). We address the following specific questions: (1) to what extent did the French Polynesian population experience a strong and sustained population bottleneck following introduction? (2) Can the rate of phenotypic divergence between French Polynesian and New Zealand silvereyes be explained by drift alone? and (3) What is the genomic evidence for selective processes? Commenserate with the introduction of a handful of individuals, we show that compared with natural colonisations by the silvereye, the French Polynesian population has undergone a more substantial population bottleneck followed by slower population recovery. In most instances we could not rule out drift as the sole cause of observed morphological shifts. However, the identification of outlier loci at genes previously associated with morphological divergence in birds, also suggests a strong role for directional selection.
Materials and methods
Sample collection
We collected blood samples and morphological data from 190 silvereyes across the islands of Tahiti, Mo’orea, Huahine, Raiatea and Maupiti (Society Archipelago) in April and May 2017. Birds were caught using mist nets and 20–40 μl of blood collected from the brachial wing vein, stored in 1 ml of lysis buffer (0.01M Tris-HCl; 0.01M NaCl; 0.01M EDTA; 1% n-lauroylsarcosine, pH 8.0) and frozen at −20 °C (Seutin et al. 1991). Following this, six morphological traits were measured. Wing length (mm) was measured as maximum flattened cord of the longest primary feather using a butted metal rule. Dial callipers (accuracy ±0.1 mm) were used to measure metatarsal length (tarsus length), mandible length to proximal edge of the nasal opening (Bill length) and mandible width and depth at the distal edge of the nasal opening (Bill width and Bill depth) (mm). Tail length (mm) of central tail feathers was measured from base to tip using either dividers or a tail rule. Wing and tail measurements were excluded from subsequent analyses if the individual was moulting. Bill length measures were excluded if the mandible was broken or overgrown. In addition, blood samples and morphological data previously collected from South Island (New Zealand) were used in the study. New Zealand samples were collected for Clegg et al. (2002a) and were collected in the same manner as French Polynesian samples. Sampling locations and the number of samples in analyses are shown in Table 1. French Polynesian birds were measured by A.T.S.P. and S.M.C., whereas New Zealand birds were measured by S.M.C. only. A comparison of morphological measurements conducted by A.T.S.P. and S.M.C. of the same six individuals showed no significant effect of data collector on any of the morphological traits measured (all two-sample t-test P values > 0.05) and therefore suggesting no strong effect attributable to different observers.
RAD-PE sequencing and SNP calling
For 96 French Polynesian samples, genomic DNA was extracted using Qiagen DNEasy blood and tissue extraction kits (Qiagen, Venlo, Netherlands) and restriction site-associated DNA paired-end (RAD-PE) sequencing libraries constructed at the University of California, Los Angeles following the protocol outlined in Supplementary Material. The resulting libraries were sequenced on a single Illumina HiSeq4000 lane (Illumina, San Diego, CA, USA) at the UC Davis Genome Center using paired-end 150-bp sequence reads. Following sequencing, the quality of sequencing reads was checked visually using FASTQC (Andrews 2010). Sequence reads were assigned to individuals using the process_radtags script in the STACKS version 1.4 software pipeline (Catchen et al. 2013). Reads containing uncalled bases and/or bases of low quality were discarded in this step using default quality thresholds (an average Phred score of 10 in sliding windows of 15% of the length of the read). Sequences with possible adaptor contamination and/or missing the Sbf1 restriction site were also discarded. Reads were filtered to remove PCR duplicates using the STACKS clone_filter script. The remaining reads were then mapped to the Zosterops lateralis melanops genome assembly version 1 (NCBI Assembly GCA_001281735.1) (Cornetti et al. 2015) with BOWTIE2 version 2.2.6. (Langmead and Salzberg 2012) using end-to-end alignment and default settings (allowing for a maximum of two mismatches in the seed (-n 2)). Individual sample bam files were merged with existing RAD-PE sequencing reads for 23 New Zealand samples (unpublished raw data) to form a single bam file using Picard Tools version 2.7 (http://broadinstitute.github.io/picard/). Single Nucleotide Polymorphisms (SNPs) were identified using the HaplotypeCaller tool from the Genome Analysis Toolkit (GATK) nightly build version 2016-12-05-ga159770 (McKenna et al. 2010) and the resulting output filtered using VCFtools (Danecek et al. 2011) to remove indels and only include biallelic SNPs where the minor allele frequency > 0.01, minimum genotype quality = 30, minimum depth = 8, and where SNPs were called in at least 50% of individuals. Although more PCR duplicates would have been removed had duplicate filtering been applied following mapping of reads, given that in other widely used protocols such as double-digest RAD-sequencing (Peterson et al. 2012) duplicates cannot be removed at all, and also given that a recent study suggests PCR duplicate removal has little effect on genotyping (Euclide et al. 2020), remaining PCR duplicates are not likely to provide a substantial source of bias in our dataset.
To determine the optimum number of SNPs and individuals to retain for downstream analyses, data missingness was visualised (Fig. S1) using genoscapeRtools (https://github.com/eriqande/genoscapeRtools; https://doi.org/10.5281/zenodo.848279). The VCF file was then further filtered using VCFtools to include the optimum number of SNPs and only those individuals where less than 30% of data were missing.
As the Z. l. melanops genome is only assembled to the scaffold level (Cornetti et al. 2015), we mapped Z. l. melanops scaffolds to chromosomes of the Taeniopygia guttata genome assembly version 3.2.4 (NCBI Assembly GCA_000151805.2) using Satsuma Synteny (Grabherr et al. 2010). Output from Satsuma Synteny was then used to assign scaffolds to chromosomes and determine order, location and orientation using custom R scripts from Van Doren et al. (2017). Further custom scripts (see ‘Data availability’) were used to reorder the GATK outputted VCF file accordingly.
Population structure
To investigate genetic structure among samples we conducted a Principle Component Analysis (PCA) using the full SNP dataset. We also examined patterns of population structure by performing maximum likelihood estimation of individual admixture proportions using the program ADMIXTURE (Alexander et al. 2009), testing K values 1–6. For each value of K, we conducted 20 independent runs and summarised runs using CLUMPP v.1.1.2 (Jakobsson and Rosenberg 2007). As the ADMIXTURE manual recommends avoiding SNPs with high linkage disequilibrium, we used the ‘–indep-pairwise 100 kb 1 0’ command in PLINK (Purcell et al. 2007) to remove one of every pair of SNPs with r2 > 0 within 100 kb sliding windows.
Demographic history inference
In order to infer effective population size changes in French Polynesia, we estimated demographic parameters from the joint Site Frequency Spectrum (SFS) using the composite-likelihood and coalescent simulation approach implemented in fastsimcoal v. 2.6 (Excoffier and Foll 2011; Excoffier et al. 2013). As outgroup sequences were unavailable, demographic inference used the distribution of minor allele frequencies (folded-SFS). Based on findings from population structure analysis (see Fig. 2) we tested two different demographic models: a two-population demographic model which treated French Polynesia as a single population (Fig. 3a) and a three-population demographic model which incorporated a within French Polynesia population split in which Maupiti was treated as a separate population (Fig. 3b). Parameter search ranges were selected based on historical records. Assuming a generation time of 2.5 years (Kikkawa and Degnan 1998), and a known introduction time of 80 years ago, tintro was fixed to 32 generations. Population size following introduction (Nintro) was fixed between 2 and 100 individuals (within reasonable bounds for a single event, human-mediated introduction). Population recovery following introduction (texp) was fixed between 19 and 26 generations ago (in line with timing of documented population expansion within French Polynesia). For the three-population model the establishment of the Maupiti population (tcol) was fixed between 11 and 14 generations ago (in line with the documented colonisation timeframe) and founding population size of the Maupiti population (Ncol) fixed between 2 and 500 individuals. The three-population demographic model also incorporated a population size change within the Maupiti population (tchange) which was fixed between 2 and 11 generations ago. Contemporary population sizes were estimated for each population with initial search ranges for SNZ (NSNZ), all French Polynesia (NAll FP) and FP cluster 1 (NFP1) set to between 10,000 and 10,000,000 individuals. Given the very small geographic size of the island of Maupiti, search ranges for FP cluster 2 (Maupiti only) were bounded between 5 and 1,000 individuals. For each demographic model, we performed 100 independent runs (100 expectation/conditional maximisation cycles, 1,000,000 simulations per run), and chose the run with the highest likelihood as the best parameter estimates. As mutation rates for silvereyes are unknown, parameter estimates were based on estimated mutation rates for collared flycatchers (Ficedula albicollis): 4.6 × 10−9 mutations per nucleotide site per generation (Smeds et al. 2016). As fastsimcoal requires the use of unlinked SNPs and is sensitive to missing data, we further filtered the LD-pruned dataset (see ‘Population structure’ section) to include only SNPs present in all individuals. VCFs were then converted to SFS format using the python script EasySFS.py (https://github.com/isaacovercast/easySFS) using flag-a to include all sites.

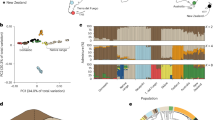
a Principle component analysis of genetic variation based on 5,414 LD-filtered SNPs. The variance explained by PC1 and PC2 is 5.53% and 2.84%, respectively. b Maximum likelihood estimation of individual ancestries calculated with ADMIXTURE (Alexander et al. 2009) for all populations analysed, testing K values from 2 to 3 and based on 5,414 LD-filtered SNPs. Mean cross-validation error: K2 = 0.613; K3 = 0.617. SNZ South Island, New Zealand; TAH Tahiti; MOO Mo’orea; HUA Huahine; RAI Raiatea; MAU Maupiti.

a Two-population split where FP is treated as a single population. b Three-population split where FP is treated as two separate populations (FP1 = Tahiti, Mo’orea, Huahine and Raiatea; FP2 = Maupiti only). tintro = time of introduction to Tahiti; texp = time of population expansion following introduction; tcol = time of colonisation of Maupiti; tchange = time of switch to long-term population size of Maupiti population; NSNZ = current SNZ population size; NAll FP = current population size of French Polynesian population when treated as a single population; NFP1 = current population size of cluster 1 (Tahiti, Mo’orea, Raiatea and Huahine); NFP2 = current population size of cluster 2 (Maupiti); Nintro = population size following introduction; Ncol = population size following colonisation of Maupiti. Values in parentheses indicate parameter estimates from best model runs.
Morphological divergence
To summarise morphological divergence between the French Polynesian silvereye population(s) and representatives of its New Zealand ancestor, we conducted a PCA of morphological data collected from 189 silvereyes sampled across the Society Archipelago (French Polynesia) and 91 from Dunedin (South Island, New Zealand). PCA was conducted on log-transformed data for wing length, tail length, bill length, depth and width. Differences in PC scores between South Island and French Polynesian silvereyes were tested for significance using analysis of variance (ANOVA).
Morphological shifts between New Zealand and French Polynesia were calculated for each morphological trait measured. Morphological data were log-transformed, and shifts expressed as standard deviation units, using pooled standard deviation estimates. To determine if random processes alone could account for observed shifts in morphological traits, we calculated Lande’s (1976) statistic \(N_e^ \ast\), the effective population size required to explain morphological shifts by drift alone. \(N_e^ \ast\) was calculated for each morphological trait as follows:
where h2 = narrow-sense heritability of trait in question, t = number of generations, z = mean morphological shift and σ = phenotypic standard deviation of the colonised population. Calculations of heritability (h2) were based on parent offspring, full-sibling comparisons and cross-fostering experiments in the Heron Island silvereye (Z. l. chlorocephalus) population. Heritability (h2) estimates for morphological traits, ranged from 0.2 and 0.6 (Clegg et al. 2002b). Based on the mean breeding age of the Heron Island silvereye, estimated from a 15-year demographic study (Kikkawa, unpublished data, as cited in Clegg et al. (2002b)), generation time was estimated at between 2 and 3 years. Using these ranges in heritability and generation time we calculated a lower estimate (2-year generation time and h2 = 0.2) and upper estimate (3-year generation time and h2 = 0.6) of \(N_e^ \ast\) for each morphological trait. \(N_e^ \ast\) estimates were compared with estimates of Ne for French Polynesia calculated as harmonic means referred to as Ne(h)). The harmonic mean is always lower than the arithmetic mean and is especially sensitive to low values of Ne. Use of the harmonic mean is therefore relevant when two related demographic scenarios are suspected, an in situ population bottleneck or a founder event (Wright 1938). Where Ne(h) > \(N_e^ \ast\), drift cannot be the sole mechanism of divergence for a morphological trait and therefore selection must be invoked (Lande 1976). Both \(N_e^ \ast\) and Ne(h) were calculated when treating French Polynesia as a single population (All FP), and treating French Polynesia as two separate populations (FP1 and FP2).
Identifying signatures of selection and candidate gene analysis
To detect signatures of selection at the genomic level, we scanned for outlier loci using PCAdapt, a principal components-based method of outlier detection with a low rate of false-positive detection (Luu et al. 2017). PCAdapt requires the choice of K principal components, based on inspection of a scree plot, where K is the number of PCs with eigenvalues that depart from a straight line. PCAdapt then computes a test statistic based on Mahalanobis distance and controls for inflation of test statistics and false discovery rate (FDR). Outlier SNPs were identified using the following settings comparing each French Polynesian island individually to the New Zealand population: K = 2, MinMAF = 0.1 and FDR = 0.01. As PCAdapt does not included the option to assign individuals to populations, outlier detection was conducted using individual French Polynesian islands to ensure that identified outliers reflected differences between New Zealand and French Polynesia rather than within French Polynesia differences.
Known, novel or predicted genes of the T. guttata genome occurring within 10,000 bp of outlier SNPs were identified using the Ensembl BioMart database (https://www.ensembl.org/biomart). Genes that had been previously associated with morphological variation in birds were determined to be candidate genes underlying the observed divergence in body size and beak shape in silvereyes. Genes known to be involved in craniofacial variation/disease in non-avian species were also considered candidates.
Genetic associations with morphological traits
To associated variation at SNPs with PC1 scores, we used a Bayesian sparse linear‐mixed model (BSLMM) as implemented in the software package GEMMA (Zhou and Stephens 2012). As BSLMM combines linear-mixed models with sparse regression models, this method is well suited to situations where the underlying genetic architecture of the trait is unknown (Zhou et al. 2013). This genome-wide association approach controls for population structure by incorporating a relatedness matrix as a covariate in the mixed model. The model was run for 20 million iterations with a burn-in of 5 million iterations. This was repeated ten times, and the resulting hyperparameters averaged across runs. As a conservative approach to identify SNPs that were significantly associated with PC1 scores, we filtered for candidate SNPs using a strict posterior inclusion probability (PIP) ≥ 0.05. This strict cut-off is five times higher than the widely used level of 0.01. T. guttata genes occurring within 10,000 bp of associated SNPs were identified using BioMart, and those previously associated with morphological variation in birds, or where a strong case could be made based on associations in other taxa, were considered candidate genes.
Results
RAD-PE sequencing and bioinformatics
Overall, RAD-PE sequencing resulted in an average of 227,092 paired-end reads per sample (±16,784) covering 2,128,552 variable sites. Subsequent quality filtering (removal of indels and only including biallelic SNPs where: the minor allele count was ≥2; minimum genotype quality = 30; minimum depth = 8 and SNPs were called in at least 50% of individuals) reduced the total number of SNPs to 215,543. Of the 119 samples sequenced, 72 were retained after removing individuals where ≥30% of data were missing across a subsample of 64,663 SNPs. The number of individuals retained per location ranged from 8 to 21 (Table 1). Reordering of Zosterops scaffolds onto T. guttata chromosomes, based on output from Satsuma Synteny and removal of unmapped/unoriented scaffolds, resulted in 63,849 SNPs (full SNP dataset). The LD-filtered dataset contained 5,414 unlinked SNPs for admixture analysis, and subsequent removal of SNPs with missing data resulted in 1587 SNPs for demographic modelling.
Population structure
A PCA showed clear separation of individuals into three distinct clusters when plotted against PC1 and PC2 (Fig. 2a). The three clusters consisted of: (1) New Zealand only; (2) Tahiti, Mo’orea, Huahine and Raiatea combined (subsequently referred to as FP cluster 1) and (3) Maupiti only (subsequently referred to as FP cluster 2). Maximum likelihood estimation of individual ancestries calculated with ADMIXTURE (Alexander et al. 2009), consistently provided the lowest cross-validation error for K = 2 (mean cross-validation error across runs = 0.613) with New Zealand and French Polynesia forming distinct groups. At K = 3 (mean cross-validation error across runs = 0.617) admixture results were consistent with those of the PCA (Fig. 2b).
Demographic history
Based on demographic modelling with fastsimcoal2, we estimated the effective population size immediately following introduction to be between 2 and 6.5 individuals, and population recovery estimated to take between 8 and 13 generations depending on the Model used. For model A (two-population split), contemporary effective population sizes for New Zealand and French Polynesia were estimated as 3.73 million and 1.22 million individuals, respectively (Fig. 3a). For model B (three-population split) effective population sizes for New Zealand, FP cluster 1 (Tahiti, Mo’orea, Huahine and Raiatea) and FP cluster 2 (Maupiti) were estimated as 2.91 million, ~5100 and 10 individuals, respectively (Fig. 3b). Based on parameter estimates, the Maupiti population (which forms its own distinct population cluster) diverged from other French Polynesia islands ~13 generations ago. The effective population size of the Maupiti population immediately following colonisation was estimated as ~494 individuals.
Patterns and rates of morphological divergence
Compared with their New Zealand ancestors, French Polynesian silvereyes had significantly longer tail lengths, longer and deeper, but narrower bills, and shorter wings (all Wilcoxon P values < 0.05; see Fig. 4a). Differences in tarsus length were non-significant (W = 7723.5, P = 0.189). These differences were also maintained when treating French Polynesia as two populations, with the exception wing length for FP2 (Maupiti only) which was not significantly different from New Zealand (W = 1192.5, P = 0.074; see Fig. 4a).

a The percent difference between New Zealand and French Polynesian morphological trait means. A negative percent difference in this case refers to a trait being smaller in the French Polynesian population compared with the New Zealand population, whereas a positive percent difference refers to a trait being larger in the French Polynesian population. Bars around means represent 95% confidence intervals. Asterisks indicate significant difference as determined using Wilcoxon rank sum tests: P < 0.001 (***); P < 0.01 (**); P < 0.05 (*). b Principal components analysis of morphological variation. Based on six morphological traits (wing length, tail length, tarsus length, bill length to posterior nasal opening and bill width/depth at anterior nasal opening) across 189 individuals of Z. lateralis from five islands in the Society Archipelago (French Polynesia) and 91 individuals from the South Island (New Zealand) source population. The variance explained by PC1 and PC2 is 29.4% and 21.8%, respectively (see Table S1). c PC1 scores (mean ± SE) and PC2 scores (mean ± SE); both calculated for New Zealand, French Polynesia combined and individual French Polynesian clusters, respectively. PC1 broadly summarises body size (all factor loadings had the same sign). PC2 broadly summarises variation in body shape (primarily contrasting bill size and body size traits).
The first two principle components (PCs) accounted for 29.38% and 21.75% of variance, respectively. PC1 broadly summarised body/bill size (all factor loadings had the same sign, although they were not entirely even), whereas PC2 summarised body/bill shape (primarily contrasting bill size and body size traits) (see Table S1 for factor loading). Differentiation between New Zealand and French Polynesian silvereyes along PC1 and PC2 is visible in the PCA summary plot (Fig. 4b). An ANOVA indicated significant variation between population units (PC1 scores: F(6,441) = 28.59, P < 0.001; PC2 scores: F(6,441) = 10.66, P < 0.001). PC1 and PC2 scores differed significantly between New Zealand and French Polynesian populations, with French Polynesian silvereyes showing a trend towards larger PC1 scores (increased body/bill size) and lower PC2 scores (larger longer/deeper bills, and shorter wings) (post hoc Tukey tests, P < 0.001 in both cases) (Fig. 4c).
Rate tests indicated that, in most instances, drift alone could not be dismissed as the sole cause of observed morphological shifts. For example, the effective population size required to explain the observed shift in wing length when treating French Polynesia as a single population (\(N_e^ \ast\) = 101–456) was well above the harmonic mean of effective population sizes at each generation (Ne(h) = 10.39). However, when using the least conservative (i.e. smallest) estimates of \(N_e^ \ast\) and treating French Polynesia as a single population, drift could be rejected as the sole mechanism driving differences in bill depth and PC1 scores. The minimum effective population sizes required to explain shifts in bill length (\(N_e^ \ast\) = 7) and PC1 scores (\(N_e^ \ast\) = 9) were both lower than the harmonic mean of effective population sizes (Ne(h) = 10.39). When treating French Polynesia as two populations, in no case was Ne(h) > \(N_e^ \ast\) (Table 2).
Signatures of selection and candidate genes
Using PCAdapt we identified between 15 and 509 outlier SNPs, putatively under selection (Fig. 5a). One hundred and sixty-two known, novel or predicted genes occurred within 10 kb of outlier SNPs (Table S2), of which eleven (E2F4, FREM2, NFIA, OSR2, PBX3, PTDSS1, RALGPS1, TMC6, VPS13B, VPS50 and ZMYND11) have been previously associated with bill/body size differences in birds or craniofacial morphogenesis in non-avian species (Table S3). The BSLMM identified 24 SNPs with a strong association with PC1 scores (PIP ≥ 0.5) (Fig. 5b). These highly associated SNPs were located within 10,000 bp of 15 genes, of which six (CDK14, OBSL1, IGF1R, INPP4B, RUNX3 and ZMYND11) were previously associated with morphological variation or craniofacial defects in vertebrates, including birds (Table S4). Genes not previously associated with morphological variation included: ATXN7, FNIP2, INHA, LDLRAP1, NOS1, SGK3, THEMIS and TOM1L2. A single SNP (Chr 2: 10,960,228 bp) was identified as both a PCAdapt outlier and strongly associated with PC1 scores in the BSLMM. This SNP was located within the ZMYND11 (Zinc Finger MYND-Type Containing 11).

a Manhattan plot of negative log10 (P values) estimated using PCAdapt. Points above the dashed line indicate outlier SNPs identified using FDR = 0.01. Genes containing outlier SNPs or within 10,000 bp of outlier SNPs and thought to be associated with morphological divergence are labelled. The distance of outlier SNPs from candidate genes is reported in Table S3. b Manhattan plot of BSLMM analysis of PC1 (body/bill size) scores. The dashed line indicates the threshold of posterior inclusion probability (PIP) = 0.5. Genes within 10,000 bp of SNPs with PIP ≥ 0.5 are labelled. Genes previously associated with morphological variation in birds, or where a strong case for inclusion as candidates can be made, are underlined. Chromosomes are numbered according to the T. guttata nomenclature. SNZ South Island, New Zealand; TAH Tahiti; MOO Mo’orea; HUA Huahine; RAI Raiatea; MAU Maupiti.
Discussion
The introduction of the silvereye to French Polynesia provides a contemporary case in which to assess the potential for founder-induced drift to facilitate morphological divergence under more extreme founding conditions than those observed in natural colonisations by this species. Consistent with the silvereye’s capacity for rapid differentiation (Frentiu et al. 2007; Clegg et al. 2002b; Clegg et al. 2008), we observed morphological shifts of significant magnitude, and rate tests suggested these shifts could be explained solely by drift in most cases. This result demonstrates the role of stochastic effects of population founding extremes in facilitating rapid morphological shifts at the very early stage of divergence. However, a role for divergent natural selection of ecologically relevant traits was also implicated from the genomic analysis, highlighting the joint role of the two microevolutionary processes, at the early stage of divergence.
Demographic history
While the exact date of introduction of silvereyes to French Polynesia was historically documented (Guild 1938; Thibault and Monnet 1990; Monnet et al. 1993; Thibault and Cibois 2017), Guild did not record the number of individuals he released in 1937, and as such population size immediately following founding is unknown. Combining historical records with demographic inference using the observed SFS, we were able to infer that the French Polynesian population was founded by few individuals and following introduction the population remained bottlenecked for several generations, and recovery relatively slow. This finding confirms that, compared with natural island colonisations by the silvereye which are thought to most often be established by flocks in excess of 100 individuals (Clegg et al. 2002a; Estoup and Clegg 2003) and population recovery thought to occur within 2–3 generations (Clegg 2010), the silvereye’s introduction to French Polynesia offers a more extreme case in which to assess the potential for founder-induced drift to facilitate morphological divergence. Such a small founding population size (<10 effective founders) and slow recovery (8–13 generations post introduction) is also in keeping with historical records—sightings of silvereyes were limited to around the release site until the late 1950s but becoming widespread across Tahiti by 1971 (Thibault and Cibois 2017). Although a more complete demographic model would provide further certainty regarding the demographic history of French Polynesian silvereyes, current sampling prevents this as only five of the eleven islands have been sampled. Nevertheless simple demographic models can be informative and have been used to effectively reconstruct recent known demographic histories (e.g. McCoy et al. (2014)).
The role of drift
We observed rapid shifts in multiple morphological traits, with French Polynesian silvereyes overall showing significant increases in body and bill size and changes in body shape (longer and deeper bills, longer tails and shorter wings). Unlike previous studies of natural island colonisations by birds, in most instances we were unable to dismiss drift as the sole mechanism through which morphological shifts may have arisen using rate tests. Whereas natural island colonisations by birds may have reduced potential for founder-induced morphological divergence due to the establishment by large flocks (Vincek et al. 1997; Clegg et al. 2002a; Estoup and Clegg 2003; Illera et al. 2007), rapid population recovery following founding (Clegg 2010) and continued immigration from the source population (Clegg and Phillimore 2010), our results suggest that founder-induced morphological divergence may occur under more extreme founding conditions. This finding is at odds with other studies of avian introductions (Lima et al. 2012; Valentin et al. 2018; Gleditsch and Sperry 2019), all of which were unable to explain observed morphological shifts by drift alone despite small founding population sizes.
Non-random founding population
While a combination of founder-induced drift and strong selective pressures associated with island-living provides the most likely explanation for morphological shifts in French Polynesian silvereyes, there is the possibility that the founding individuals were a non-random phenotypic subset of the source population. An alternative explanation for increased body size is size biased dispersal, in which only the largest of individuals survive long-range colonisation across water barriers (Clegg et al. 2002a). Parallel ‘size selective’ mechanisms could operate in human-mediated introductions; for example, Guild may have exerted size bias when releasing birds to Tahiti, or only the largest birds survived to be released. Unfortunately, such bias cannot be tested for.
The role of natural selection
The pattern of morphological divergence we observed is broadly consistent with the island syndrome in which, amongst other repeated patterns of change, birds on islands exhibit larger body size and have more robust bills than their mainland conspecifics (Clegg and Owens 2002; Leisler and Winkler 2015). While we can only speculate on the proximate causes of selection in French Polynesia, the island syndrome is thought to extend from exposure to a common suite of biotic conditions on islands; a combination of reduced predation (Losos and Ricklefs 2009), and a shift in the balance of inter- versus intra-specific competition (Blondel 2000), features that fundamentally change selective pressures in predictable ways (Grant 1998). Given the paucity of endemic land birds on oceanic islands, the absence of competitors in French Polynesia may favour the exploitation of a greater breadth of resources, which may be facilitated by a larger body size (Grant 1968; Lack 1971). The role of interspecific competition in shaping morphology is well-evidenced (Schluter et al. 1985; Kirschel et al. 2009) and can occur remarkably rapidly, even within timescales as short as a couple of decades (Grant and Grant 2006). Alternatively, larger body size may confer an advantage in high-density island populations where aggression is expected to be increased and territoriality expected to be year-round (Kikkawa 1980a, b; Robinson-Wolrath and Owens 2003). Finally, as small size usually makes it easier to escape or hide from predators, larger body size may also result from relaxed risk of predation on islands (Lomolino 1985). Tests of these hypotheses in the Capricorn silvereye (Z. l. chlorocephalus) on Heron Island suggest that, in this species, increased intraspecific competition is the prevailing mechanism driving increased body size of insular forms (Kikkawa 1980b; Scott et al. 2003; Robinson-Wolrath and Owens 2003).
Despite French Polynesian silvereyes having increased PC1 scores (larger overall size) compared with their New Zealand ancestor, their wing lengths were shorter and tarsus lengths unchanged. Smaller wing size may suggest reduced reliance on flight and/or increase use of densely vegetated habitat (Savile 1957; Winkler and Leisler 1985; Leisler and Winkler 2015). However, as decreased wing length was not accompanied by an increase in tarsus length—as would be expected to aid terrestrial movement (Zeffer and Norberg 2003; Zeffer et al. 2003), this result is difficult to interpret. Interestingly, decreased wing length but no change in tarsus length has recently been reported for non-native frugivores on the Hawaiian island of O’ahu (Gleditsch and Sperry 2019).
As the observed morphological shifts have taken place within a maximum of 80 years (~32 generations), our study supports the hypothesis that the bulk of differences can develop rapidly, potentially within the first couple of hundred generations after colonisation as proposed for the Capricorn silvereye (Clegg et al. 2008). It would therefore be valuable to capture the divergence process at an even earlier stage e.g. during population establishment, as has been done for Eurasian blackbirds on the island of Heligoland (Engler et al. 2019). This may be possible in this system as the silvereye continues to expand its range in French Polynesia, for example a population has established on Rimatara (Austral archipelago) as recently as 2014 (Thibault and Cibois 2017).
Candidate genes
Using genome-wide sequencing data we are able to identify signatures of selective processes operating within French Polynesia. Outlier SNP detection allowed us to identify 12 candidate genes potentially underlying morphological differences, eight of which were mapped to directly by SNPs putatively under directional selection and four which occurred within 10 kb of outliers. These included: VPS50—previously associated with bill length in Berthelot’s pipit (Anthus berthelotii) (Armstrong et al. 2018); VPS13B—identified as under directional selection between species of Darwin’s finches (Lawson and Petren 2017); NFIA—associated with bill length in the house sparrow (Passer domesticus) (Lundregan et al. 2018) and craniofacial abnormality in humans (Rao et al. 2014); PTDSSI—which is under directional selection in birds of paradise (Prost et al. 2018); OSR2—which has been experimentally demonstrated to play a role in beak development in birds (Brugmann et al. 2010) and craniofacial defects in mice (Stanier and Moore 2004); and E2F4, FREM2, PBX3, RALGPS1, TMC6 and ZMYND11, which are all associated with craniofacial variation/disorders in a range of non-avian species including house mouse (Mus musculus), baboons (genus: Papio), European glass eels (Anguilla anguilla) and humans (Humbert et al. 2000; Cobben et al. 2014; Amaya 2015; Joganic 2016; De Meyer et al. 2017). The low overlap of genes identified across French Polynesian islands could reflect different selective pressures operating between islands, or alternatively may reflect differences in our power to detect outliers between sub-populations.
By performing a genome-wide association analysis we aimed to link phenotypic variation across French Polynesian and New Zealand silvereyes to the genomic variation in our SNP dataset. We identified several SNPs with strong associations with PC1 (body size) scores. Although not all strongly associated SNPs occurred within gene coding regions, a finding which is perhaps not surprising given the density of our marker set, several did occur within or close to gene coding regions including genes that have previously been associated with morphological variation in birds, non-avian vertebrates or implicated in craniofacial disease in humans. This included: OBSL1—a cytoskeletal adaptor protein previously associated with body size in humans (Hanson et al. 2009) and cetaceans (Sun et al. 2019); and IGF1R—a transmembrane receptor of insulin-like growth factor 1 (IGF1). The IGF1 pathway has a well-known role in body size scaling in a range of taxa, including chickens (Beccavin et al. 2001; Beckman et al. 2003), cattle (Grossi et al. 2015), brown house snakes (Sparkman et al. 2010) and dogs (Sutter et al. 2007); and has been previously associated with bill size in the black-bellied seedcracker (onHoldt et al. 2018). RUNX3—which, in the zebrafish, modulates bone morphogenetic protein (BMP) signalling during craniofacial development (Dalcq et al. 2012). BMPs such as BMP4 have been strongly associated with morphological variation of beaks in Darwin’s finches (Abzhanov et al. 2004). Finally, CDK14, ZMYND11 and INPP4B, which have been associated with craniofacial morphology in finches, humans and house mouse, respectively (Cobben et al. 2014; Amaya 2015; Pallares et al. 2015; Lawson and Petren 2017). ZMYND11 was the only candidate gene to be both associated with PC1 scores and also contain outlier SNPs.
In Darwin’s finches bill size is thought to be modulated by the transcriptional regulator HMGA2: ‘high mobility group AT-hook 2’ (Lamichhaney et al. 2015) and beak shape strongly associated with ALX1: ‘ALX homeobox 1’ haplotypes (Lamichhaney et al. 2016). However, as SNPs within our RAD-Seq dataset did not overlap with the position of these genes, we are unable to speculate on the role of HMGA2 and ALX1 in modulating bill divergence in silvereyes. Whole genome analyses would likely provide the opportunity to identify further candidate genes and allow us to address the role of HMGA2 and ALX1 in silvereye morphological divergence. Although not addressed here, the French Polynesian silvereye’s ability to undergo rapid morphological divergence may be further modulated by epigenetic changes. For example, in the absence of genotypic changes, epigenetic variation has been shown to facilitate rapid change in the bill size and shape of Darwin’s finches in response to sudden environmental changes such as drought or human disturbance (Skinner et al. 2014; Lamichhaney et al. 2015; McNew et al. 2017). Future work should therefore consider the role of epigenetics in facilitating rapid differentiation.
Conclusion
Given that knowledge of contemporary natural colonisation events is limited, evolutionary insights into the early stages of divergence have been predominantly retrospective or laboratory based. However, contemporary introductions of birds to islands provide ideal systems in which to study the genomic and phenotypic changes that take place during the early stages of divergence. Taking advantage of the well-documented introduction of the silvereye to French Polynesia, we set out to assess the role of founder-induced drift in driving phenotypic divergence under more extreme founding conditions than provided by natural colonisation events in this species. While we were unable to dismiss drift as the sole cause of significant shifts in morphological traits, given that island-living is known to exert strong selective pressure on avian morphology and French Polynesian silvereyes show body size/shape changes consistent with the ‘island syndrome’, observed phenotypic shifts within the French Polynesian population likely result from a combination of drift and selective processes. Studies of colonisation in action would provide the opportunity to tease apart the roles of drift and selection in driving morphological shifts at different stages of divergence. Although, such events are extremely rare, the potential establishment of a silvereye population on the French Polynesian island Rimatara (Austral archipelago) may provide such an opportunity. Genetic and phenotypic changes associated with population founding could also be further investigated in other species introduced to French Polynesia, such as common waxbill (Estrilda astrild), red-browed firetail (Neochmia temporalis) and chestnut-breasted mannikin (Lonchura castaneothorax), allowing a contrast between a range of species.
Data availability
Resequencing data from this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena) under accession numbers PRJEB36361 and PRJEB25440. VCF files and custom scripts required to replicate analyses are available on dryad (https://doi.org/10.5061/dryad.7d7wm37rm).
References
Abzhanov A, Protas M, Grant BR, Grant PR, Tabin CJ (2004) Bmp4 and morphological variation of beaks in Darwin’s finches. Science 305:1462
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Amaya L (2015) Genetic architecture of craniofacial shape in the house mouse: a genetic and morphological perspective. PhD Thesis, University of Kiel
Andrews S (2010) FASTQC: a quality control tool for high throughput sequence data. www.bioinformatics.babraham.ac.uk/projects/fastqc/
Armstrong C, Richardson DS, Hipperson H, Horsburgh GJ, Küpper C, Percival-Alwyn L et al. (2018) Genomic associations with bill length and disease reveal drift and selection across island bird populations. Evol Lett 2:22–36
Baker MC, Baker MSA, Tilchman LM (2006) Differing effects of isolation on evolution of bird songs: examples from an island-mainland comparison of three species. Biol J Linn Soc 89:331–342
Balakrishnan CN, Edwards SV (2009) Nucleotide variation, linkage disequilibrium and founder-facilitated speciation in wild populations of the zebra finch (Taeniopygia guttata). Genetics 181:645
Barton NH (1998) Natural selection and random genetic drift as causes of evolution on islands. In: Grant PR (ed) Evolution on islands. Oxford University Press, Oxford, p 102–123
Beccavin C, Chevalier B, Cogburn LA, Simon J, Duclos MJ (2001) Insulin-like growth factors and body growth in chickens divergently selected for high or low growth rate. J Endocrinol 168:297–306
Beckman BR, Larsen DA, Dickhoff WW (2003) Life history plasticity in chinook salmon: relation of size and growth rate to autumnal smolting. Aquaculture 222:149–165
Berry R (1998) Evolution of small mammals. In: Grant PR (ed) Evolution on islands. Oxford University Press, Oxford, p 35–50
Blackburn TM, Lockwood JL, Cassey P (2009) Avian invasions: the ecology and evolution of exotic birds. Oxford University Press, Oxford
Blondel J (2000) Evolution and ecology of birds on islands: trends and prospects. Vie et Milieu 50:205–220
Brugmann SA, Powder KE, Young NM, Goodnough LH, Hahn SM, James AW et al. (2010) Comparative gene expression analysis of avian embryonic facial structures reveals new candidates for human craniofacial disorders. Hum Mol Genet 19:920–930
Carson HL, Templeton AR (1984) Genetic revolutions in relation to speciation phenomena: the founding of new populations. Annu Rev Ecol Syst 15:97–132
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Clegg SM (2010) Evolutionary changes following island colonisation in birds: empirical insights into the roles of microevolutionary processes. In: Losos JB, Ricklefs RE (eds) The theory of island biogeography revisited. Princeton University Press, New Jersey, p 293–325
Clegg DSM, Kikkawa J, Moritz C, Estoup A, Owens IPF (2002a) Genetic consequences of sequential founder events by an island-colonizing bird. PNAS 99:8127–8132
Clegg SM, Degnan SM, Moritz C, Estoup A, Kikkawa J, Owens IPF et al. (2002b) Microevolution in island forms: the roles of drift and directional selection in morphological divergence of a passerine bird. Evolution 56:2090–2099
Clegg SM, Frentiu FD, Kikkawa J, Tavecchia G, Owens IPF (2008) 4000 Years of phenotypic change in an island bird: heterogeneity of selection over three microevolutionary timescales. Evolution 62:2393–2410
Clegg S, Owens P (2002) The ‘island rule’ in birds: medium body size and its ecological explanation. Proc R Soc B Biol Sci 269:1359–1365
Clegg SM, Phillimore AB (2010) The influence of gene flow and drift on genetic and phenotypic divergence in two species of Zosterops in Vanuatu. Philos Trans R Soc B Biol Sci 365:1077–1092
Cobben J, Weiss M, Van Dijk F, De Reuver R, de Kruiff C, Pondaag W et al. (2014) A de novo mutation in ZMYND11, a candidate gene for 10p15.3 deletion syndrome, is associated with syndromic intellectual disability. Eur J Med Genet 57:636–638
Cornetti L, Valente LM, Dunning LT, Quan X, Black Ra, Hébert O et al. (2015) The genome of the “Great Speciator” provides insights into bird diversification. Genome Biol Evol 7:2680–2691
Danecek P, Auton A, Abecasis G, Albers CA, Banks E (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Dalcq J, Pasque V, Ghaye A, Larbuisson A, Motte P, Martial JA et al. (2012) RUNX3, EGR1 and SOX9B form a regulatory cascade required to modulate BMP-signaling during cranial cartilage development in zebrafish. PLoS ONE 7:e50140–e50140
De Meyer J, Maes GE, Dirks RP, Adriaens D (2017) Differential gene expression in narrow- and broad-headed European glass eels (Anguilla anguilla) points to a transcriptomic link of head shape dimorphism with growth rate and chemotaxis. Mol Ecol 26:3943–3953
Emerson BC, Gillespie RG (2008) Phylogenetic analysis of community assembly and structure over space and time. Trends Ecol Evol 23:619–630
Emerson BC, Paradis E, Thébaud C (2001) Revealing the demographic histories of species using DNA sequences. Trends Ecol Evol 16:707–716
Engler JO, Sacher T, Coppack T, Bairlein F (2019) Assortative mating frames establishment in a young island bird population. R Soc Open Sci 6:190050
Estoup A, Clegg SM (2003) Bayesian inferences on the recent island colonisation history by the bird Zosterops lateralis lateralis. Mol Ecol 12:657–674
Euclide PT, McKinney GJ, Bootsma M, Tarsa C, Meek MH, Larson WA (2020) Attack of the PCR clones: Rates of clonality have little effect on RAD-seq genotype calls. Mol Ecol Resour 20:66–78
Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M (2013) Robust demographic inference from genomic and SNP data. PLoS Genet 9:e1003905
Excoffier L, Foll M (2011) Fastsimcoal: a continuous-time coalescent simulator of genomic diversity under arbitrarily complex evolutionary scenarios. Bioinformatics 27:1332–1334
Frentiu FD, Clegg SM, Blows MW, Owens IPF (2007) Large body size in an island-dwelling bird: a microevolutionary analysis. J Evolut Biol 20:639–649
Gleditsch JM, Sperry JH (2019) Rapid morphological change of nonnative frugivores on the Hawaiian island of O’ahu. Evolution 73:1456–1465
Grabherr MG, Russell P, Meyer M, Mauceli E, Alföldi J, di Palma F et al. (2010) Genome-wide synteny through highly sensitive sequence alignment: Satsuma. Bioinformatics 26:1145–1151
Grant PR (1968) Bill size, body size, and the ecological adaptations of bird species to competitive situations on islands. Syst Biol 17:319–333
Grant PR (1998) Evolution on islands. Oxford University Press, New York, NY
Grant PR, Grant BR (1995) The founding of a new population of Darwin’s finches. Evolution 49:229–240
Grant PR, Grant BR (2006) Evolution of character displacement in Darwin’s finches. Science 313:224–226
Grossi D, do A, Buzanskas ME, Grupioni NV, de Paz CCP, Regitano LC, de A, de Alencar MM et al. (2015) Effect of IGF1, GH, and PIT1 markers on the genetic parameters of growth and reproduction traits in Canchim cattle. Mol Biol Rep 42:245–251
Guild E (1938) Tahitian aviculture: acclimation of foreign birds. Avic Mag 3:8–11
Guild E (1940) Western bluebirds in Tahiti. Avic Mag 5:284–285
Hanson D, Murray PG, Sud A, Temtamy SA, Aglan M, Superti-Furga A et al. (2009) The primordial growth disorder 3-M syndrome connects ubiquitination to the cytoskeletal adaptor OBSL1. Am J Hum Genet 84:801–806
Humbert PO, Rogers C, Ganiatsas S, Landsberg RL, Trimarchi JM, Dandapani S et al. (2000) E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol Cell 6:281–291
Illera JC, Emerson BC, Richardson DS (2007) Population history of Berthelot’s pipit: colonisation, gene flow and morphological divergence in Macaronesia. Mol Ecol 16:4599–4612
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jensen AJ, Hansen LP, Johnsen BO, Karlsson S (2017) Rapid evolution of genetic and phenotypic divergence in Atlantic salmon following the colonisation of two new branches of a watercourse. Genet Sel Evol 49:22
Joganic JL (2016) A quantitative genetic analysis of craniofacial variation in baboons. PhD Thesis, Washington University in St. Louis, Missouri
Kikkawa J (1980a) Weight changes in relation to social hierarchy in captive flocks of silvereyes, Zosterops lateralis. Behaviour 74:92–100
Kikkawa J (1980b) Winter survival in relation to dominance classes among silvereyes Zosterops lateralis chlorocephala of Heron Island, great barrier reef. Ibis 122:437–446
Kikkawa J, Degnan S (1998) Effective population size of an island passerine. In: Adams NJ, Slotow RH (eds) Proceedings of the 22nd international ornithological congress, Birdlife South Africa, Johannesburg, p 213
Kirschel AN, Blumstein DT, Smith TB (2009) Character displacement of song and morphology in African tinkerbirds. PNAS 106:8256–8261
Kliber A, Eckert CG (2005) Interaction between founder effect and selection during biological invasion in an aquatic plant. Evolution 59:1900–1913
Lack DL (1971) Ecological isolation in birds. Harvard University Press, Massachusetts
Lamichhaney S, Berglund J, Almén MS, Maqbool K, Grabherr M, Martinez-Barrio A et al. (2015) Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518:371–375
Lamichhaney S, Han F, Berglund J, Wang C, Almén MS, Webster MT et al. (2016) A beak size locus in Darwin’s finches facilitated character displacement during a drought. Science 352:470–474
Lande R (1976) Natural selection and random genetic drift in phenotypic evolution. Evolution 30:314–314
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Lawson LP, Petren K (2017) The adaptive genomic landscape of beak morphology in Darwin’s finches. Mol Ecol 19:4978–4989
Leisler B, Winkler H (2015) Evolution of island warblers: beyond bills and masses. J Avian Biol 46:236–244
Lima MR, Macedo RHF, Martins TLF, Schrey AW, Martin LB, Bensch S (2012) Genetic and morphometric divergence of an invasive bird: the introduced house sparrow (Passer domesticus) in Brazil. PLoS ONE 7:e53332
Lockwood JL, Moulton MP, Balet KL (1999) Introduced avifaunas as natural experiments in community assembly. In: Weiher E, Keddy P (eds) Ecological assembly: Advances, perspectives, retreats. Cambridge University Press, Cambridge, p 108–125
Lomolino MV (1985) Body size of mammals on islands: the island rule reexamined. Am Nat 125:310–316
Losos JB, Ricklefs RE (2009) The theory of island biogeography revisited. Princeton University Press, New Jersey
Lundregan SL, Hagen IJ, Gohli J, Niskanen AK, Kemppainen P, Ringsby TH et al. (2018) Inferences of genetic architecture of bill morphology in house sparrow using a high-density SNP array point to a polygenic basis. Mol Ecol 27:3498–3514
Luu K, Bazin E, Blum MGB (2017) pcadapt: an R package to perform genome scans for selection based on principal component analysis. Mol Ecol Resour 17:67–77
Mathys BA, Lockwood JL (2011) Contemporary morphological diversification of passerine birds introduced to the Hawaiian archipelago. Proc R Soc B Biol Sci 278:2392–2400
Mayr E (1942) Systematics and the origin of species, from the viewpoint of a zoologist. Harvard University Press, Massachusetts
Mayr E (1954) Change of genetic environment and evolution. In: Huxley J, Hardy AC, Ford EB (eds) Evolution as a process. Allen and Unwin, London, p 157–180
McCoy RC, Garud NR, Kelley JL, Boggs CL, Petrov DA (2014) Genomic inference accurately predicts the timing and severity of a recent bottleneck in a nonmodel insect population. Mol Ecol 23:136–150
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
McNew SM, Beck D, Sadler-Riggleman I, Knutie SA, Koop JA, Clayton DH et al. (2017) Epigenetic variation between urban and rural populations of Darwin’s finches. BMC Evol Biol 17:183
Millien V (2006) Morphological evolution is accelerated among island mammals. PLoS Biol 4:1863–1868
Monnet C, Thibault JC, Varney A (1993) Stability and changes during the twentieth century in the breeding landbirds of Tahiti (Polynesia). Bird Conserv Int 3:261–280
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10
Pallares LF, Carbonetto P, Gopalakrishnan S, Parker CC, Ackert-Bicknell CL, Palmer AA et al. (2015) Mapping of craniofacial traits in outbred mice identifies major developmental genes involved in shape determination. PLoS Genet 11:e1005607–e1005607
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135
Price T (2008) Speciation in birds. Roberts and Company, Colorado
Prost S, Armstrong EE, Nylander J, Thomas GW, Suh A, Petersen B et al. (2018) Genome analyses show strong selection on coloration, morphological and behavioral phenotypes in birds-of-paradise. https://doi.org/10.1101/287086
Provine W (1989) Founder effects and genetic revolutions in microevolution and speciation: an historical perspective. In: Giddings LV, Kaneshiro KY, Anderson WW (eds) Genetics, speciation and the founder principle. Oxford University Press, New York, NY, p 43–76
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Rao A, O’Donnell S, Bain N, Meldrum C, Shorter D, Goel H (2014) An intragenic deletion of the NFIA gene in a patient with a hypoplastic corpus callosum, craniofacial abnormalities and urinary tract defects. Eur J Med Genet 57:65–70
Rasner CA, Yeh P, Eggert LS, Hunt KE, Woodruff DS, Price TD (2004) Genetic and morphological evolution following a founder event in the dark-eyed junco, Junco hyemalis thurberi. Mol Ecol 13:671–681
Robinson-Wolrath SI, Owens IPF (2003) Large size in an island-dwelling bird: intraspecific competition and the dominance hypothesis. J Evol Biol 16:1106–1114
Savile OBO (1957) Adaptive evolution in the avian wing. Evolution 11:212–224
Schluter D, Price TD, Grant PR (1985) Ecological character displacement in Darwin’s finches. Science 227:1056–1059
Scott SN, Clegg SM, Blomberg SP, Kikkawa J, Owens IPF (2003) Morphological shifts in island-dwelling birds: the roles of generalist foraging and niche expansion. Evolution 57:2147–2156
Seutin G, White B, Boag PT (1991) Preservation of avian blood and tissue samples for DNA analyses. Can J Zool 69:82–90
Skinner MK, Gurerrero-Bosagna C, Haque MM, Nilsson EE, Koop JAH, Knutie SA et al. (2014) Epigenetics and the evolution of Darwin’s finches. Genome Biol Evol 6:1972–1989
Smeds L, Qvarnström A, Ellegren H (2016) Direct estimate of the rate of germline mutation in a bird. Genome Res 26:1211–1218
Sparkman AM, Byars D, Ford NB, Bronikowski AM (2010) The role of insulin-like growth factor-1 (IGF-1) in growth and reproduction in female brown house snakes (Lamprophis fuliginosus). Gen Comp Endocrinol 168:408–414
Stanier P, Moore GE (2004) Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet 13:R73–R81
Sun Y, Liu Y, Sun X, Lin Y, Yin D, Xu S et al. (2019) Insights into body size variation in cetaceans from the evolution of body-size-related genes. BMC Evolut Biol 19:157
Sutter NB, Bustamante CD, Chase K, Gray MM, Zhao K, Zhu L et al. (2007) A single IGF1 allele is a major determinant of small size in dogs. Science 316:112
Thibault JC, Cibois A (2017) Birds of eastern Polynesia. A biogeographic Atlas. Lynx Publishing, Ontario
Thibault JC, Monnet C (1990) Le Zosterops à poitrine grise Zosterops lateralis (Latham) en Polynésie: un oiseau introduit devenu colonisateur actif L’Oiseau et la Rev Francaise d’Ornithologie 60:233–240
Valentin RE, Lockwood JL, Mathys BA, Fonseca DM (2018) Influence of invasion history on rapid morphological divergence across island populations of an exotic bird. Ecol Evol 8:5291–5302
Van Doren BM, Campagna L, Helm B, Illera JC, Lovette IJ, Liedvogel M (2017) Correlated patterns of genetic diversity and differentiation across an avian family. Mol Ecol 26:3982–3997
Vincek V, O’Huigin C, Satta Y, Takahata N, Boag PT, Grant PR et al. (1997) How large was the founding population of Darwin’s finches? Proc R Soc Lond Ser B Biol Sci 264: 111–118
vonHoldt BM, Kartzinel RY, Huber CD, Le Underwood V, Zhen Y, Ruegg K et al. (2018) Growth factor gene IGF1 is associated with bill size in the black-bellied seedcracker Pyrenestes ostrinus. Nat Commun 9:4855–4855
Winkler H, Leisler B (1985) Morphological aspects of habitat selection in birds. In: Cody ML (ed) Habitat selection in birds. Academic Press Inc, Orlando, p 415–434
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97
Wright S (1938) Size of population and breeding structure in relation to evolution. Science 87:430–431
Yeh PJ (2004) Rapid evolution of a sexually selected trait following population establishment in a novel habitat. Evolution 58:166–174
Zeffer A, Johansson LC, Marmebro A (2003) Functional correlation between habitat use and leg morphology in birds (Aves). Biol J Linn Soc 79:461–484
Zeffer A, Norberg UM (2003) Leg morphology and locomotion in birds: requirements for force and speed during ankle flexion. J Exp Biol 206:1085–1097
Zhou X, Carbonetto P, Stephens M (2013) Polygenic Modelling with Bayesian sparse linear mixed models. PLOS Genet 9:e1003264
Zhou X, Stephens M (2012) Genome-wide efficient mixed-model analysis for association studies. Nat Genet 44:821
Acknowledgements
This work was funded by a Natural Environment Research Council (NERC) studentship and Genetics Society Heredity Fieldwork Grant awarded to ATSP and, a John Fell Fund grant awarded to SMC and Tim Coulson, and a Percy Sladen Memorial Fund grant to SMC. Sample collection was conducted with the permission of the governments of French Polynesia and New Zealand. We thank Neil Davies and Marie Fourdrigniez for logistical assistance and Jeffrey Tamati, Cael Sendell-Price and Olivia Grant for assistance during sample collection. The authors would like to acknowledge the use of the University of Oxford’s Advanced Research Computing (ARC) facility and the UC Davis Genome Center in carrying out this work. We thank Barbara Mable and three anonymous reviewers who provided helpful comments on earlier drafts of this manuscript.
Author information
Authors and Affiliations
Contributions
ATSP and SMC jointly conceived the project and SMC supervised the project. Fieldwork was conducted by ATSP and SMC. Bioinformatics and analyses were carried out by ATSP with assistance from SMC and KCR. The manuscript was written by ATSP and SMC, with input from KCR.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sendell-Price, A.T., Ruegg, K.C. & Clegg, S.M. Rapid morphological divergence following a human-mediated introduction: the role of drift and directional selection. Heredity 124, 535–549 (2020). https://doi.org/10.1038/s41437-020-0298-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-0298-8
This article is cited by
-
On the brink of explosion? Identifying the source and potential spread of introduced Zosterops white-eyes in North America
Biological Invasions (2024)
-
What can genetics tell us about the history of a human-mediated introduction of the golden-striped salamander south of its native range?
European Journal of Wildlife Research (2022)
-
Genetic structure in Orkney island mice: isolation promotes morphological diversification
Heredity (2021)
