
Abstract
Genetic diversity of major histocompatibility complex (MHC) genes is linked to reduced pathogen susceptibility in amphibians, but few studies also examine broad spatial and temporal patterns of MHC and neutral genetic diversity. Here, we characterized range-wide MHC diversity in the Northern leopard frog, Rana pipiens, a species found throughout North America that is experiencing disease-related declines. We used previously sequenced neutral markers (mitochondrial DNA and microsatellites), sequenced an expressed MHC class IIß gene fragment, and measured infection prevalence and intensity of the global fungal pathogen Batrachochytrium dendrobatidis (Bd) across 14 populations. Four populations were sampled across two decades, enabling temporal comparisons of selection and demography. We recovered 37 unique MHC alleles, including 17 that were shared across populations. Phylogenetic and population genetic patterns between MHC and neutral markers were incongruent, and five MHC codon positions associated with peptide binding were under positive selection. MHC heterozygosity, but not neutral marker heterozygosity, was a significant factor explaining spatial patterns of Bd prevalence, whereas only environmental variables predicted Bd intensity. MHC allelic richness (AR) decreased significantly over time but microsatellite-based AR did not, highlighting a loss of functional immunogenetic diversity that may be associated with Bd selective pressures. MHC supertype 4 was significantly associated with an elevated risk of Bd infection, whereas one supertype 2 allele was associated with a nearly significant reduced risk of Bd. Taken together, these results provide evidence that positive selection contributes to MHC class IIß evolution in R. pipiens and suggest that functional MHC differences across populations may contribute to disease adaptation.
Similar content being viewed by others

Introduction
Infectious diseases in wildlife populations are increasing globally and are linked to species declines, threatening biodiversity and conservation (Daszak et al. 2000; Blanchong et al. 2016). Because standing genetic diversity provides the immediate foundation for populations to adapt, allelic polymorphism can mitigate negative impacts brought on by emerging infectious diseases (McKnight et al. 2017). Specifically, immune gene diversity within the major histocompatibility complex (MHC) is a key component of adaptive variation that can reduce disease impacts in natural populations (e.g., Wegner et al. 2003; Oliver et al. 2009). MHC loci occur in all jawed vertebrates and encode a diverse suite of proteins with a central role in immune defense, particularly acquired immune function (Kaufman 2018). The MHC includes two major classes (I and II) that encode cell-surface glycoproteins that recognize and bind to pathogens at their peptide-binding regions (PBR) and present them to T cells, initiating an acquired immune response (e.g., Bernatchez and Landry 2003). Classical class I MHC proteins are expressed on the surface of most cells and bind peptides from intracellular pathogens, such as viruses, whereas class II MHC genes are expressed on antigen presenting cells and primarily bind and present extracellular pathogen peptides, such as bacteria and fungi (Braciale et al. 1987). Many studies have found that higher diversity at MHC loci is linked to reduced pathogen susceptibility (e.g., Radwan et al. 2010; Blanchong et al. 2016) and underlies disease adaptation in numerous vertebrates (e.g., Bernatchez and Landry 2003; Acevedo-Whitehouse and Cunningham 2006; Blanchong et al. 2016).
MHC genes typically exhibit high allelic polymorphism and sequence diversity and harbor multiple functional gene copies within classes I and II (Kaufman 2018). In general, more MHC alleles and gene copies are associated with increased immunity because hosts with more diverse peptide-binding capabilities can eliminate a broader suite of pathogens (Bernatchez and Landry 2003; Spurgin and Richardson 2010; Radwan et al. 2020). However, excessive MHC diversity can act to dampen immune responses (Migalska et al. 2019). Three nonexclusive and empirically supported hypotheses can explain the extreme polymorphism of MHC genes: (1) heterozygote advantage, (2) negative frequency-dependent selection (sometimes termed rare-allele advantage), and (3) fluctuating selection in time and space (Spurgin and Richardson 2010). MHC diversity is beneficial under the heterozygote advantage hypothesis because heterozygous individuals can recognize more antigens and protect against a broader array of pathogens. Indeed, MHC heterozygotes often (but not always) have higher resistance to infections (e.g., Penn et al. 2002; but, see Ilmonen et al. 2007). Under the negative frequency-dependent selection hypothesis, novel or rare alleles have a selective advantage because pathogen populations rapidly evolve resistance to common MHC alleles (Takahata and Nei 1990; Bernatchez and Landry 2003; Phillips et al. 2018). Lastly, fluctuating selection can also explain the maintenance of high MHC allelic diversity if there are differences in pathogen strains and abundances based on environment, location, and time (Hedrick 2002; Osborne et al. 2017). Under this scenario, different MHC alleles confer a selective advantage and increase in frequency in different populations, preserving high MHC diversity across populations (e.g., Teacher et al. 2009). Because all three modes can act to shape overall MHC diversity, identifying the specific selective processes driving adaptation to a particular pathogen is challenging and requires extensive data on genetic structure, MHC diversity, ecology, and disease in natural populations.
Understanding disease resistance is particularly important in amphibians because emerging infectious diseases have led to drastic population declines (Echaubard et al. 2015; Lips 2016; McKnight et al. 2017). Globally, the most pervasive disease threat is from the infectious disease chytridiomycosis, caused by the fungi Batrachochytrium dendrobatidis (Bd) and Batrachochytrium salamandrivorans (Berger et al. 1998; Martel et al. 2013), which impacts over 500 species (Scheele et al. 2019). Although the number of functional MHC class II loci and the extent of copy number variation remain uncharacterized in most frogs, class II diversity is significantly associated with Bd susceptibility in several taxonomically distinct species (Bataille et al. 2015; Savage and Zamudio 2016; Fu and Waldman 2017). Several frogs in the genus Rana show MHC class II disease associations in natural populations. For example, Rana yavapaiensis experimentally exposed to Bd show significant associations between Bd survival, MHC class II heterozygosity, and specific MHC class II alleles (Savage and Zamudio 2011) at a single, constitutively expressed class II locus across frog species (Kiemnec-Tyburczy et al. 2010). Additionally, MHC class II genotypes at this same locus are significantly associated with overwinter survival in Rana chiricahuensis released into Bd-endemic ponds (Savage et al. 2018). This relationship between MHC diversity and disease susceptibility is not limited to Bd, as Rana sylvatica larvae show significant differences in Ranavirus infection intensities across MHC genotypes (Savage et al. 2019). In addition to MHC diversity, higher genetic variation at neutral markers has also been associated with lower disease risk in R. yavapaiensis (Savage et al. 2015) and other natural anuran populations (Allentoft and O’Brien 2010; Kosch et al. 2016; Horner et al. 2017). However, few studies have simultaneously examined MHC and neutral genetic patterns across space and time.
Leopard frogs are ideal study organisms to examine landscape-scale questions integrating population genetics, immunity, and disease because they are widespread, well-studied, and extensively affected by emerging infectious diseases (Woodhams et al. 2008; Echaubard et al. 2015). The northern leopard frog (Rana pipiens) is broadly distributed, ranging throughout southern Canadian provinces and most of the continental United States. A range-wide mitochondrial DNA (mtDNA) study investigating phylogeographic patterns identified that R. pipiens was split into populations containing discrete eastern and western haplotypes demarcated by the Mississippi River (Hoffman and Blouin 2004a). Moreover, western populations exhibited reduced mtDNA diversity relative to eastern populations, a pattern confirmed with microsatellites (Phillipsen et al. 2011). Although relatively mobile, R. pipiens exhibited distinct patterns of isolation-by-distance (Hoffman and Blouin 2004a; Hoffman et al. 2004; Waraniak et al. 2019) and temporal stability of genetic structure in eastern populations, suggesting that at least those populations were not undergoing contemporary extinction and recolonization events (Hoffman et al. 2004). This baseline of information on genetic structure and evolutionary history of R. pipiens provides a robust framework for determining relationships between recent demographic processes, disease, and adaptive genetic diversity.
Contemporary populations of R. pipiens have experienced significant declines in the western part of the species range (Leonard et al. 1999; Hoffman and Blouin 2004b), especially in the southwestern USA (Clarkson and Rorabaug 1989; Rogers and Peacock 2012). Bd has negatively impacted R. pipiens (Parris et al. 2006; Woodhams et al. 2008; Voordouw et al. 2010) as well as other frogs in the southwestern USA (e.g., Savage and Zamudio 2011, 2016; Savage et al. 2018). Arizona shows a striking pattern where most R. pipiens populations have severely declined or been extirpated, but one metapopulation is thriving, possibly owing to introgression of eastern R. pipiens genes into this area (O’Donnell et al. 2017). O’Donnell et al. (2017) suggest that Bd resistance explains the success of this metapopulation, and that genetic resistance may occur due to eastern R. pipiens introgression of MHC variants or skin peptides that are not typically found in western populations. However, investigations of immune gene variation have not been performed across eastern or western R. pipiens populations.
Here, we characterized evolutionary history and population genetic diversity of an expressed MHC class IIß gene in natural populations of R. pipiens across the species range and across two decades to evaluate neutral, selective, and disease-associated processes shaping immune gene diversity. First, we conducted phylogenetic analysis and functional supertyping of MHC alleles to examine spatial and functional distribution of MHC clades. Second, we examined site- and branch-specific patterns of molecular evolution to identify individual codons under positive selection. Third, we compared MHC-derived population genetic parameters to values previously characterized at neutral genetic loci to assess the role of demography compared to selection in driving MHC diversification across populations. Fourth, for a subset of populations with temporal sampling spanning 1971–2018, we compared population genetic parameters from MHC and neutral loci to test whether significant genetic change had occurred over time. Finally, we measured Bd infection prevalence and intensity from all sampled individuals and tested the relative importance of neutral genetic diversity, functional immunogenetic diversity, and environmental factors in explaining spatial patterns of Bd. Taken together, these analyses help elucidate the demographic, environmental, spatial, and pathogen-associated factors that have shaped immunogenetic diversity in a widespread, declining amphibian species.
Methods
Field sampling and DNA extraction
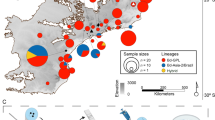
We sampled tissues from adult R. pipiens hand captured from 14 natural populations throughout North America (Fig. 1). Most populations were sampled in 1999–2001 for Hoffman and Blouin (2004a). Additionally, we used previously unpublished samples from Ohio, USA (Table 1, PLK) and samples collected during two different time periods at localities in Arizona, USA (1999 and new samples collected for this study in 2017–2018), Ontario, CAN (1979 and 2001), and New York, USA (1971 and 2001). At each site, we preserved a toe clip in ethanol from each captured individual. We extracted genomic DNA from all toe clips using either phenol-chloroform extractions or Qiagen DNeasy extraction kits. For sample sites characterized in previously published studies, we standardized population abbreviations for clarity (Table S1).

Circle size is proportional to sample size, with white fill showing the proportion uninfected and red showing the proportion infected with Bd. Population abbreviations follow Table 1 and the year of sampling is in parentheses for each population (color figure online).
MHC genotyping
Using newly collected samples and a subset of tissues from Hoffman and Blouin (2004a), we amplified a 272 bp fragment of a single MHC class IIβ locus (hereafter, MHC). We used a fusion primer approach designed by Mulder et al. (2017) to amplify and sequence exon 2 of a single MHC locus by targeting the flanking introns. First, we used the MHC primers ForN and RevA (Table S2; Mulder et al. 2017) in a 20 µL reaction using 5–50 ng of template DNA, 0.9X of OneTaq PCR buffer, 1 mM dNTPs, 0.8 µg/µL BSA, 2% DMSO, 0.16 µM of each primer, and 0.25 U OneTaq DNA polymerase (New England Biolabs). PCR conditions included initial denaturation of 95 °C for 3 min, 35 cycles of 95 °C for 30 s, annealing at 52 °C for 30 s, and 72 °C extension for 1 min, followed by 72 °C for 5 min. We then performed a second PCR using Nextera-style Illumina fusion primers combining Illumina adapters, an 8 bp barcode, a primer pad and linker, and the MHC primers used in the first PCR (Table S2). We used unique barcode combinations across forward and reverse fusion primers so that each individual had a unique molecular tag. Fusion primer PCRs consisted of 25 µL reactions using 4 µL of the MHC PCR product, 1X OneTaq PCR buffer, 0.2 mM dNTPs, 0.4 µM of each primer, and 0.625 U OneTaq with an initial denaturation of 95 °C for 3 min, 15 cycles of 95 °C for 30 s, annealing at 52 °C for 30 s, and 72 °C for 1 min, followed by a final extension period at 72 °C for 5 min. All samples were visualized on 2% agarose gels and grouped by eye into three pools of strong, medium, and weak PCR band intensity. Pools were cleaned with 1.0X SpeedBeads (Thermo Fisher Scientific, Waltham, MA) and quantified using KAPA qPCR kits (Roche Sequencing Systems, Pleasanton, CA). Quantified pools were equimolarly combined into sequencing libraries. Sequencing was accomplished across two Illumina MiSeq 2 × 250 bp runs, with 77 samples in the first run and 229 in the second. We conducted two independent PCR reactions for two individuals and sequenced them on two different runs to account for possible PCR and sequencing biases.
After demultiplexing, paired-end reads were joined using PEAR v 0.9.8 (Zhang et al. 2014), then clustered, filtered, and genotyped with AmpliSAS (Sebastian et al. 2016). We set the maximum number of alleles per amplicon at two because these MHC primers amplify a single locus (Kiemnec-Tyburczy et al. 2010; Mulder et al. 2017). We set the minimum amplicon depth at 200, and our clustering parameters followed recommendations for Illumina data (Sebastian et al. 2016): 1% substitution errors, 0.001% indel errors, and 25% minimum dominant frequency. For filtering, we set the minimum amplicon frequency to 10%. To further ensure that we only analyzed real MHC alleles, we only retained alleles that were recovered in at least two different individuals.
MHC evolutionary relationships and functional supertypes
We used MEGA7 (Kumar et al. 2016) to create an alignment, translate it, and identify any stop codons. We ran PartitionFinder v2.1.1 (Lanfear et al. 2017) to determine the best model of evolution for the nucleotide alignment. Next, we reconstructed a genealogy in MrBayes v3.2.6 (Ronquist et al. 2012) using Nanorana parkeri (Accession Number: KR535970.1) and Xenopus laevis (Accession Number: D13688.1) MHC as outgroups. We ran two MrBayes runs of 5 × 106 generations each with the first 10,000 trees discarded as burn-in. We analyzed the output in Tracer v1.7 (Rambaut et al. 2018) to confirm stationarity and sufficient posterior sampling. We also built a maximum likelihood genealogy with IQ-TREE v1.6.9 (Nguyen et al. 2014). Additionally, we created a 95% parsimony MHC haplotype network using the TCS algorithm (Clement et al. 2000) implemented in PopArt v1.7 (Leigh and Bryant 2015). Finally, we created a genus-wide MHC genealogy based on an alignment of all Rana (after Yuan et al. 2016) class II alleles in GenBank (identified with the search term “MHC class II Ranidae”) using the same alignment, evolutionary model, and phylogenetic reconstruction methodologies described above, with the same X. laevis MHC allele as the outgroup.
We converted MHC alleles into functional supertypes based on quantifiable physiochemical properties of the amino acids comprising the mammalian-defined PBR of each allele (Brown et al. 1993). This analysis does not include phylogenetic relationships among alleles, instead grouping supertypes only based on functional similarity of PBR amino acids. We converted an alignment of these amino acid positions across all alleles into a matrix with five parameters for every amino acid, using the following physiochemical descriptor variables: z1 (hydrophobicity), z2 (steric bulk), z3 (polarity), z4, and z5 (electronic effects; Jombart et al. 2010). We then analyzed this matrix using discriminant analysis of principle components (DAPC) implemented in the R package adegenet 1.4-0 (Jombart and Ahmed 2011). We defined functional MHC supertypes as all alleles falling into distinct clusters after DAPC. The optimal number of clusters was determined using a change in the Bayesian Information Criterion (∆BIC) value ≤ 2.
MHC molecular evolution
We used our R. pipiens MHC alignment and the Bayesian output tree file to run HyPhy (Kosakovsky Pond et al. 2005) implemented on the Datamonkey server (Kosakovsky Pond and Frost 2005a). We ran model selection to determine the best model of evolution, then tested for intragenic recombination using SBP (single breakpoint) and Genetic Algorithms for Recombination Detection (GARD; Pond et al. 2006). Next, we tested for codon-based positive selection by estimating synonymous (dS) and non-synonymous (dN) substitution rate parameters using the following six methods: SLAC, FEL, iFEL, REL (Kosakovsky Pond and Frost 2005b), MEME (Murrell et al. 2012), and FUBAR (Murrell et al. 2013). We set the significance threshold at a P value ≤ 0.05 or a posterior probability ≥ 0.95, depending on the method. We considered codons to be significantly under positive selection if the majority of methods (at least four) found evidence of positive selection. We identified overlap between the codon positions we found to be under positive selection in our data set relative to the mammalian-defined PBR residues (Brown et al. 1993) and to other studies of codon-specific selection in frog MHC (Savage and Zamudio 2011; Bataille et al. 2015).
Spatial comparisons of MHC and neutral loci
The data we analyzed for mtDNA (Hoffman and Blouin 2004a) and microsatellites were derived from a previous study (Hoffman et al. 2006) and from two populations in Ohio, USA (PLK and CNRA) that were originally collected, extracted, and genotyped in 2001, but were previously unpublished. We generated MHC genotypes from a subset of the same samples (as well as the new 2017–2018 STLK sampling), thus we had microsatellite data, mtDNA data, or both for nearly every individual that we MHC genotyped. In this study, we report on seven microsatellite loci (Rpi100, Rpi101, Rpi103, Rpi106, Rpi108, RP193, and RP415) generated under the conditions in Hoffman et al. (2006). We tested for Hardy–Weinberg equilibrium (HWE) using exact tests in Genepop version 3.3 (Raymond and Rousset 1995) for the complete microsatellite data set (genotypes from previously published and unpublished populations). We calculated genetic diversity and differentiation estimates for MHC alleles, MHC supertypes, mtDNA haplotypes, and microsatellites. Although we did not have mtDNA and microsatellite data for all populations, we had at least one neutral marker type for all populations with MHC sequence data. To estimate expected heterozygosity (HE, a.k.a. gene diversity; Nei 1987), allelic richness (AR), and pairwise FST from MHC genotypes, MHC supertypes, and microsatellites, we used the program FSTAT v2.9.3 (Goudet 1995). To estimate observed heterozygosity (HO), HE, pairwise FST, and Tajima’s D from mtDNA haplotypes and MHC alleles, we used the program Arlequin v3.5 (Excoffier and Lischer 2010). To test whether HO and HE were significantly different from each other for the MHC locus and for microsatellite loci, we used Welch’s two sample t-tests (R Core Team 2013). To evaluate significant differences between MHC-derived and microsatellite-derived pairwise FST estimates, we conducted 5000 bootstrap replicates of all 78 pairwise population FST estimates for the MHC and microsatellite data sets and evaluated overlap between the means of the bootstrapped distributions (Code File S1). To test for correlations between MHC-derived, microsatellite-derived and mtDNA-derived FST, we conducted independent single Mantel tests using PASSaGE v. 2.0 (Rosenberg and Anderson 2011) for each pairwise combination of FST values, with Holm–Bonferroni sequential corrections to adjust for multiple comparison probabilities. To evaluate whether HO and HE derived from neutral loci and from MHC were significantly correlated, we measured Pearson’s correlation coefficients using the rcorr function in the R package Hmisc (R Core Team 2013). We also used effective population size (NE) previously estimated for a subset of our microsatellite data set (Phillipsen et al. 2011; NE(ABC, 10000)) and estimated Pearson’s correlation coefficients between NE, the number of MHC alleles, and the number of private MHC alleles.
To compare spatial connectivity among populations for MHC and neutral loci, we generated two connectivity indices: Euclidean distance (minimum straight-line distance between each population pair, using a least cost function to avoid lines crossing the Great Lakes) and a spatial metric of topographic resistance, estimated by employing our topographic complexity raster as a resistance grid using CIRCUITSCAPE v. 4.0 (McRae et al. 2013). We employed a cell connection scheme linking each node to four neighbors. Spatial connectivity analyses weighed all possible paths between population pairs and produced pairwise resistance matrices and summary connectivity rasters. To avoid the likely bias of multicollinearity in partial mantel tests (Cushman et al. 2013; Guillot and Rousset 2013), we used independent single Mantel tests to correlate Euclidean distance and topographic resistance with pairwise FST values (calculated separately for MHC, microsatellites, and mtDNA) using PASSaGE v. 2.0 (Rosenberg and Anderson 2011). We used Holm–Bonferroni sequential corrections to adjust for multiple comparison probabilities.
Temporal comparisons of MHC and neutral loci
To evaluate temporal shifts in MHC and neutral marker diversity metrics among the four populations sampled two decades apart, we tested whether MHC AR, HE, and HO changed significantly over time using paired t-tests. Similarly, for the three populations with temporal microsatellite data, we tested whether microsatellite AR, HE, and HO changed significantly over time using paired t-tests. We did not conduct this test for mtDNA diversity metrics because we lacked mtDNA data for at least one timepoint from all four populations with temporal MHC data.
Genetic and environmental predictors of Bd infection
We tested for Bd following qPCR protocols from Boyle et al. (2004) using the CFX96 Real-Time system (Bio-Rad). We performed our amplifications in a 25 µL reaction using 0.64X Supermix (Bio-Rad), 0.8 µM of each primer (ITS1-3 Chytr: 5′-CCTTGATATAATACAGTGTGCCATATGTC-3′ and 5.8 S Chytr: 5′-TCGGTTCTCTAGGCAACAGTTT-3′), 0.2 µM of probe (Chytr MGB2: 5′-CGAGTCGAAC-3′), 3 µL molecular-grade H2O, and 10–100 ng of template DNA. Standard curves were generated using serially diluted gBlocks (Integrated DNA Technologies) and run in duplicate ranging from 2 × 106 to 2 × 10−1 gene copies/reaction (Horner et al. 2017). The qPCR conditions included an initial denaturation of 95 °C for 5 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Negative and positive controls were run in duplicate with each run. Positives were run at least twice, including a third run if the first two runs were inconsistent by at least an order of magnitude. All values were log transformed to normalize the data and then averaged to obtain infection intensity.
We performed generalized linear model (GLM) selection procedures to identify which genetic diversity metrics and environmental variables best predicted Bd prevalence (binomial distribution/logit link) and log-transformed Bd infection intensity (normal distribution/identity link) throughout sampled R. pipiens populations. We only included population sampling from 1998 to 2001 for GLMs to ensure we were not confounding spatial Bd dynamics with temporal variation in pathogen exposure.
To include environmental data, we obtained the following information for each sampling location at 1 km resolution: normalized difference vegetation index (NDVI), 19 bioclimatic variables of temperature and precipitation from Worldclim/Bioclim extrapolated from a global network of climatic stations collecting data from 1970 to 2000 throughout the world (Fick and Hijmans 2017), and elevation from the Consultative Group on International Agricultural Research Consortium for Spatial Information (Jarvis et al. 2009). We generated a topographic complexity raster for North America by employing a moving-window approach, calculating the elevation variance for each pixel based on the eight neighboring 1 km pixels with high values denoting more complex topography. We used principal components analysis to consolidate cross-correlated temperature and rainfall variables and used the first two PC scores of temperature variables and the first two PC scores of consolidated rainfall variables in our model selection procedure. Additionally, because of high cross-correlation among MHC genetic diversity metrics (Fig. S2) and among neutral genetic diversity metrics (Fig. S3), we only included one MHC variable and one neutral variable in our models. Specifically, we used MHC HE (estimated from FSTAT) and microsatellite HE (except for the populations lacking microsatellite data, in which case we used mtDNA-based HE). Thus, our two model selection procedures (one for Bd prevalence and one for intensity) included the following 11 explanatory variables: MHC HE, neutral HE, latitude, longitude, elevation, topographic complexity, NDVI, temperature PC1, temperature PC2, rainfall PC1, and rainfall PC2. Competing models were ranked based on corrected Akaike Information Criterion (AICc) and we reported one of the most parsimonious models (based on ΔAICc) for each of our response variables. We tested residuals in our most parsimonious models including log-transformed Bd prevalence and intensity data using Shapiro–Wilk W test and confirmed normal distribution of residuals (Bd prevalence: W = 0.936, P = 0.248; Bd intensity: W = 0.947, P = 0.390).
To test whether MHC HE, MHC supertypes, or MHC alleles were associated with the likelihood of acquiring a Bd infection, we calculated the relative risk of infection for each supertype and allele that occurred at sufficient frequency (N ≥ 10), and for MHC heterozygotes compared to homozygotes. Specifically, we calculated relative risk of Bd infection as the ratio of the probability of infected individuals with a certain genotype versus individuals without that genotype, using the R package epitools (Aragon 2008) and two-sided Fisher’s exact tests to infer significance.
Results
MHC genotyping
We obtained high-quality MHC sequence data from 272 individuals including 50 sampled in 1971, 14 sampled in 1979, 182 sampled in 1998–2001, and 26 sampled in 2017–2018 (Fig. 1 and Table 1). The two individuals sequenced in both runs had identical MHC genotypes across runs. MHC amplified consistently across all individuals, and we found no evidence of more than two alleles per individual, suggesting minimal PCR artefacts arising from null alleles or amplifying multiple gene copies. We recovered a total of 37 unique MHC alleles based on complete exon 2 sequences and partial flanking introns (GenBank Accession Numbers: MN984182–984218). We recovered an average of 4.5 (±2.6) alleles per population, ranging from 1 to 10 alleles.
MHC evolutionary relationships and functional supertypes
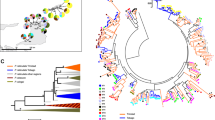
JC + I + G (Jukes and Cantor 1969) was the best model of evolution. Bayesian and maximum likelihood analyses produced similar topologies, therefore we only present the Bayesian genealogy (Fig. 2). MHC alleles did not group into clades based on population or geographic region (Fig. 2). All MHC alleles clustered into four functional supertypes (Fig. S1), and supertype 1 included only Rapi*32 and Rapi*33, whereas other supertypes included at least nine alleles (Fig. 2). Supertypes 1 and 3 formed monophyletic groups, but supertypes 2 and 4 were distributed throughout the genealogy (Fig. 2). MHC alleles were polyphyletic relative to mtDNA haplotype groups: four alleles (Rapi*03, Rapi*10, Rapi*32, and Rapi*33) were shared across eastern and western mtDNA haplotypes (Fig. 2). The haplotype network further illustrates that MHC alleles do not cluster by geographic region (Fig. 3). For example, allele Rapi*03 occurred in frogs from Arizona, Ontario, and New York, and alleles Rapi*32 and Rapi*33 were recovered from Ohio, Idaho, Nevada, Minnesota, and Arizona. Overall, 17 alleles were shared among multiple populations and 20 alleles occurred in only one population. All populations harbored at least two MHC alleles, except for DOCM (Prince Edward Island), which was fixed for a single, private MHC allele (Rapi*17), and WS (Idaho), which was fixed for a single allele (Rapi*04) present in three additional populations. Both of these alleles belong to supertype 4. Phylogenetic analysis including all other Rana sp. MHC alleles in GenBank (Fig. S4) demonstrates that R. pipiens MHC alleles show extensive trans-species polymorphism (TSP), clustering with alleles from nine other species that occur in North America, Central America, Europe, and Asia.

Node posterior probabilities are reported when P > 0.7. The colored squares indicate previously defined mtDNA clades (see text) and supertypes are depicted with colored stars. Abbreviations of states/provinces where each allele was found is listed in parentheses (color figure online).

Circles (and names outside circles) represent distinct haplotypes and pie sizes depict number of samples per haplotype. Colors indicate location of each allele. Black dots represent inferred haplotypes and dashes represent mutations between haplotypes (color figure online).
MHC molecular evolution
Model F81 was the best fit model of evolution for our MHC codons, and neither SBP nor GARD analyses found evidence of intragenic recombination. Significant positive selection occurred in five MHC codons based on at least four of the six methods we implemented (Table S3). All five of these codons align to mammalian-defined PBR residues (Brown et al. 1993; Fig. S5).
Spatial comparisons of MHC and neutral loci
The mtDNA data came from 94 individuals, including 84 that we MHC genotyped (all collected from 1998 to 2001), and the microsatellite data came from 428 individuals (96 sampled in 1971, 39 sampled in 1979, and 293 sampled in 1998–2001; Table 1), including 216 that we MHC genotyped. For microsatellites, exact tests revealed that only 3/91 comparisons deviated from HWE expectations after a sequential Bonferroni correction (Rice 1989). Specifically, Rpi108 deviated significantly from HWE in the 1971 populations from HPV, and both time points for MONT. Because of the rarity of loci out of HWE, we included all microsatellite loci from all populations in downstream analyses. Across sampled populations, HO and HE were not significantly different for microsatellite loci (t = −1.83, N = 13, P = 0.093), but HO was significantly lower than HE for the MHC locus (t = −4.59, N = 18, P = 0.00029; Table 2). Tajima’s D estimated from MHC was significantly positive in six populations and not significant in all other populations, whereas Tajima’s D estimated from mtDNA was significantly negative in three populations and all other values were nonsignificant (Table 2). Pairwise FST values estimated from mtDNA versus microsatellite data were not significantly correlated, despite showing a positive trend (r = 0.296, N = 10, P = 0.048, adjusted Holm–Bonferroni threshold for significance = 0.012; Table S5). Likewise, pairwise FST values estimated from mtDNA versus MHC were not significantly correlated (r = 0.374, N = 28, P = 0.077; Fig. 4A and Table S5) nor were pairwise FST values estimated from microsatellite versus MHC (r = 0.331, N = 78, P = 0.075; Fig. 4B and Table S5). HE measured from MHC versus mtDNA data was also not significantly correlated (r = 0.470, N = 9, P = 0.200), but HE estimated from MHC versus microsatellites showed a significant, positive correlation (r = 0.710, N = 13, P = 0.007). In contrast, HO estimated from MHC was not significantly correlated with HO estimated from microsatellites (r = 0.252, N = 13, P = 0.407). Overall, MHC-derived FST estimates were significantly larger than microsatellite-derived FST estimates (no overlap across 5000 bootstrapped mean pairwise FST estimates; Fig. 4C). However, specific population comparisons showed that MHC-derived FST was equal to or less than microsatellite-derived FST for some comparisons (Fig. 4D). For populations with microsatellite-based NE estimates (Tables 2 and S4), there were no significant correlations between NE and the number of MHC alleles (r = 0.194, N = 10, P = 0.591) or private alleles (r = −0.102, N = 10, P = 0.779). However, the two populations with the smallest NE estimates also had the fewest MHC alleles (Table S4).

Pairwise Fst estimates derived from MHC class IIβ allele frequencies are plotted against pairwise Fst estimate derived from mitochondrial haplotype frequencies (panel (A)) and from microsatellite allele frequencies (panel (B)). Mantel test r values and P values are shown for each comparison. Panel (C) shows the distribution of 5000 bootstrapped replicates of mean pairwise FST estimates derived from microsatellites (left) and the MHC locus (right). Panel (D) depicts comparisons of Fst estimates of neutral loci (microsatellites) to MHC class IIB for each pairwise comparison of 11 of our populations. Error bars indicate 95% confidence intervals and were estimated using bootstrapping.
Mantel tests showed significant positive correlations between Euclidean distance and topographic resistance compared to microsatellite-derived FST, and for Euclidian distance compared to mtDNA FST (Table S5). In contrast, neither Euclidean distance nor topographic resistance was significantly correlated with MHC-derived FST (Table S5). Furthermore, the slopes of associations were considerably steeper for microsatellite data compared to both mtDNA and MHC data (Fig. S6).
Temporal comparisons of MHC and neutral loci
Comparing the populations sampled approximately two decades apart, we did not detect significant temporal change in MHC HE (t = −0.236, N = 4, P = 0.828) or MHC HO (t = −1.072, N = 4, P = 0.456), but we did detect a significant decrease in MHC AR over time (t = −3.854, N = 4, P = 0.031), with the average number of alleles decreasing from 6.25 to 4.75 (Fig. 5). Notably, this included the three northeastern populations sampled in the 1970s versus 1998–2001, as well as the Arizona population sampled in 1999 versus 2017–2018, suggesting a temporal effect of allele loss that is not specific to a particular location or time frame. We did not detect a significant change in microsatellite HE (t = −1.281, N = 3, P = 0.328), microsatellite HO (t = −1.191, N = 3, P = 0.307), or microsatellite AR (t = −0.443, N = 3, P = 0.701) over time. Only allele Rapi*03 occurred in all four temporally sampled populations (Fig. 5). This allele had a very low frequency (below 0.1) in the New York populations, but was present at moderate to high frequencies (0.2–0.8) in Ontario and Arizona.

(A) HV, NewYork, USA in 1971 vs. 2001; (B) MONT, New York, USA in 1971 vs. 2001; (C) FAIR, Ontario, CAN, 1979 vs. 2001; and (D) STLK, Arizona, USA in 2001 vs. 2017-18. Vertical axes denote the frequency of each allele (along X-axis) in temporally sampled populations (population abbreviations follow Table 1).
Genetic and environmental predictors of Bd infection
Eight of 14 sites were infected with Bd during 1998–2001 sampling (Fig. 1 and Table S6). Average Bd prevalence ranged from 0.05 (MONT) to 1.0 (DOCM) with a mean of 0.24. Average infection intensity ranged from 40.7 (OS) to 35,706 (DOCM) genome equivalents with a mean of 12,498 (±12,575 SD). Average intensity across all positive individuals was 16,497 (±20,349 SD). Model selection indicated that higher MHC HE predicted lower Bd prevalence (β = −4.574, χ2 = 15.466, P < 0.0001; Tables S7 and S8), along with temperature and precipitation variables. Bd infection intensity, however, was best explained by environmental variables only (i.e., rainfall, temperature, and latitude; Tables S7 and S8). Topographic complexity was a positive predictor of Bd prevalence (β = 0.001, χ2 = 16.725, P < 0.0001) and latitude was a positive predictor of both Bd prevalence (β = 1.128, χ2 = 41.786, P < 0.0001) and intensity (β = 1.011, t = 5.890, P < 0.0001). Among MHC alleles that occurred in at least ten individuals, none showed significantly elevated or reduced risk of Bd infection. However, allele Rapi*03 showed a threefold reduced risk of Bd infection (relative risk = 0.32) at P = 0.088, and none of the 31 individuals with Rapi*03 were infected with Bd. Among supertypes occurring in at least ten individuals, supertype 4 homozygotes showed a significantly elevated risk of Bd infection relative to all other supertype genotypes (relative risk = 2.3, P = 0.029).
Discussion
Multiple studies in anurans have compared differentiation at MHC genes to neutral loci to establish whether selection contributes to contemporary immunogenetic patterns. The majority of these studies find discordant patterns between MHC and neutral loci (e.g., Savage and Zamudio 2016; Savage et al. 2018; Belasen et al. 2019), confirming that neutral demographic processes alone are insufficient to explain MHC genetic structure. However, these studies typically examine a limited number of natural populations within a species, and none examine MHC and neutral genetic patterns over time. Here, we demonstrate discordant patterns of MHC relative to neutral genetic diversity across the species range, as well as significant temporal changes in MHC but not neutral AR. These changes coincide with North American chytridiomycosis emergence (Skerratt et al. 2007), suggesting that Bd may have contributed to MHC evolution over the past 50 years. The distribution of our 37 R. pipiens MHC alleles across every well-supported major clade of the Rana-wide MHC genealogy (Fig. S4) demonstrates extensive TSP (Klein et al. 1998), which has been documented for MHC loci in diverse taxa including other amphibians (Bos and Dewoody 2005), fish (Ottova et al. 2005), and mammals (Cutrera and Lacey 2007). Because many R. pipiens alleles group with MHC alleles from species with South American and Asian distributions, these TSP are unlikely to have arisen from introgression, and instead represent ancestral polymorphisms maintained by balancing selection preventing the loss of rare alleles.
Spatial comparisons of MHC and neutral loci
Spatial analyses of MHC differentiation typically show contrasts between neutral- and MHC-derived genetic patterns to infer whether adaptive processes shape MHC evolution (reviewed in Bernatchez and Landry 2003; Radwan et al. 2010). A common trend found in a wide variety of taxa, including mammals (e.g., Cammen et al. 2011), birds (e.g., Strand et al. 2012), fish (Aguilar and Garza 2006), and amphibians (Li et al. 2016), is an overall pattern of neutral markers (e.g., microsatellites) exhibiting lower FST estimates than MHC-derived FST. These studies provide evidence that natural selection is favoring different MHC alleles across populations, increasing differentiation relative to levels produced from demographic processes alone. However, a few studies find the opposite pattern of lower MHC-derived FST estimates, and attribute low MHC differentiation as evidence of balancing selection or homogenizing directional selection (e.g., Evans et al. 2010; Fraser et al. 2010). In R. pipiens, mean MHC-derived FST estimates were consistently larger than microsatellite-derived FST estimates across 5000 bootstrapped replicates (Fig. 4C), but some population comparisons showed the opposite pattern (Fig. 4D), suggesting balancing selection as well as some homogenizing directional selection. We found a positive correlation between MHC- and microsatellite-derived HE estimates, suggesting that neutral processes partially contribute to MHC genetic structure. However, MHC FST estimates were not significantly correlated with Euclidean distance or topographic resistance, and the slopes of association for Mantel tests were steeper for neutral compared to MHC FST estimates, highlighting that selection on MHC alleles counteracts demographic processes.
Temporal comparisons of MHC and neutral loci
We had a unique opportunity to measure MHC evolution over time using repeat sampling from four populations, each across a 20-year span. Microsatellite-based AR, HE, and HO in the three northeastern populations remained stable over time, thus the significant loss of MHC-based AR over time provides direct observation of directional selection. To our knowledge, only one experimental study in sticklebacks has also demonstrated directional selection by showing that MHC alleles conferring pathogen resistance increased in frequency in a single generation (Eizaguirre et al. 2012). However, other temporal MHC studies recover different patterns. For example, Oliver and Piertney (2012) showed that water voles experiencing a severe population bottleneck recovered MHC (but not microsatellite) diversity within 2 years. Additional long-term studies comparing patterns of MHC genetic diversity over time in populations with and without selective pressure from pathogens may help to resolve the role of directional selection in shaping MHC diversity over ecological rather than evolutionary timescales.
Genetic and environmental predictors of Bd infection
Previous studies documented Bd infections in R. pipiens (Woodhams et al. 2008; Voordouw et al. 2010), but genetic predictors of susceptibility were not investigated. Here, we found that higher MHC HE, but not neutral HE, predicted lower Bd prevalence (Tables S7 and S8), consistent with other Bd studies that find genetic associations with Bd prevalence but not intensity (e.g., Savage et al. 2015; Horner et al. 2017). We also found that individuals homozygous for supertype 4 had a significantly elevated risk of Bd infection, while individuals with allele Rapi*03 had reduced risk of Bd infection that was nearly significant. Furthermore, of the two sampled populations with no MHC variability (DOCM and WS, each fixed for a supertype 4 allele), one had 100% Bd prevalence and the other had Bd positive individuals (Fig. 1). Finally, Rapi*32 and Rapi*33 (the only supertype 1 alleles; Fig. 2) form a distinct, well-supported clade with two closely related MHC alleles from Rana catesbeiana (Fig. S4), a species with notoriously high tolerance to Bd infection (Garner et al. 2006). Rapi*32 and Rapi*33 were also the most widespread alleles we recovered, each occurring in the same six populations throughout North America. These patterns imply that supertype 1 may promote Bd tolerance, but further sampling of this supertype during cooler months (when Bd infections spike) is necessary to confirm this pattern. Overall, we suggest that MHC variation contributes to disease risk in R. pipiens, and that our focal MHC locus is evolving at least partially due to disease pressure.
MHC codon evolution in R. pipiens provides further evidence that selection acts to promote disease adaptation. We detected positive selection acting on five mammalian-defined PBR residues (Brown et al. 1993), consistent with other amphibian immunogenetic studies (Mulder et al. 2017; Savage et al. 2019). Moreover, one of these codons (position 49; Fig. S5) is a P9 pocket residue significantly associated with Bd resistance in the tree frog Litoria verreauxii alpina (Bataille et al. 2015) and is the same residue under positive selection in allele Q, an allele significantly associated with Bd resistance in R. yavapaiensis (Savage and Zamudio 2011, 2016). Additionally, allele Rapi*03, which is present in the stable eastern populations (Hoffman et al. 2004) and the thriving Arizona metapopulation (O’Donnell et al. 2017), across all years we sampled (1971–2018), and which is associated with reduced risk of Bd infection, shares the same residue at position 49 as allele Q. Taken together, these studies suggest Bd as a selective agent driving molecular evolution of the focal MHC locus.
Our data support the recent hypothesis that MHC-based tolerance against Bd via introgression from eastern populations may explain the success of the thriving Arizona R. pipiens metapopulation (O’Donnell et al. 2017). In the west, allele Rapi*03 was only recovered in the Arizona metapopulation. Rapi*03 is the only allele found at both time points for all temporally sampled populations, is associated with a reduced risk of Bd infection, and shares functionally important similarities with other Bd resistance alleles (see above). While this pattern is consistent with introgression of Rapi*03 from the east conferring higher survival in the Arizona metapopulation compared to other western populations, explicit evidence of introgression and experimental validation of the relationship between Rapi*03 and Bd resistance are needed. Eventually, such studies could validate the utility of implementing genetic rescue to restore declining western populations (reviewed in Ralls et al. 2020).
Multiple types of pathogen-mediated positive selection likely shape and maintain MHC diversity over the evolutionary history of a species (Takahata and Nei 1990). Determining and distinguishing among these modes of selection in natural populations remain challenging given the required breadth of spatial and temporal sampling (Spurgin and Richardson 2010; Radwan et al. 2020). Our study thus provides a unique opportunity to distinguish among the potential modes of Bd-mediated selection—heterozygote advantage, negative frequency-dependent selection, and fluctuating selection—that may be shaping MHC diversity in R. pipiens. We found no evidence for heterozygote advantage because MHC heterozygotes were just as likely to be infected with Bd as homozygotes. Although no concrete evidence supports negative frequency-dependent selection, it remains plausible based on our temporal comparisons, where many common alleles decreased in frequency over time (Fig. 5; e.g., Rapi*01 and Rapi*05) and many rare alleles increased (e.g., Rapi*07 increased in three populations). However, we do not have temporal Tajima’s D estimates for neutral loci so cannot interpret the role of selection in shaping MHC-based Tajima’s D changes over time. Finally, three lines of evidence suggest fluctuating selection may contribute to MHC diversity: (1) environmental variation combined with MHC HE (but not neutral HE) was a significant predictor of Bd prevalence in most of the top models, (2) pairwise FST was consistently higher and more variable for MHC compared to microsatellite loci, and (3) the majority of MHC alleles (20/37) were private alleles, and NE was not correlated to the number of MHC alleles or private alleles. Thus, unique disease dynamics within populations and environmental variation may favor different MHC alleles over time and across populations of varying sizes, contributing to high overall MHC diversity and some differentiation.
Conclusions
In summary, genetic diversity and differentiation at an expressed MHC class IIß locus differ significantly from neutral markers in R. pipiens across both spatial and temporal scales. Across sampled populations, we found that higher MHC HE predicted lower Bd prevalence, whereas neutral heterozygosity showed no relationship. MHC supertype 4 conferred a significantly elevated risk of Bd infection. In contrast, allele Rapi*03 was associated with lower Bd risk, and Arizona populations of R. pipiens may be doing well in the face of Bd due to the unique presence of Rapi*03, which is absent from all other western sites. Comparing modes of pathogen-mediated selection acting on MHC, we found evidence consistent with fluctuating selection and negative frequency-dependent selection, but no support for heterozygote advantage. Because disease can decimate population genetic diversity across both neutral and functional loci (O’Brien and Evermann 1988), establishing that MHC diversity fluctuations are distinct from overall genetic diversity loss is important for illustrating that frog populations are adapting immunogenetically to disease and other selective pressures.
Data availability
All new genetic data generated for this study has been submitted to GenBank (Accession Numbers: MN984182–984218). All other data used in this study are provided in the Supporting Information.
References
Acevedo-Whitehouse K, Cunningham AA (2006) Is MHC enough for understanding wildlife immunogenetics? Trends Ecol Evol 21:433–438
Aguilar A, Garza JC (2006) A comparison of variability and population structure for major histocompatibility complex and microsatellite loci in California coastal steelhead (Oncorhynchus mykiss walbaum). Mol Ecol 15:923–937
Allentoft ME, O’Brien J (2010) Global amphibian declines, loss of genetic diversity and fitness: a review. Diversity 2:47–71
Aragon TJ (2008) Epitools: epidemiology tools. R package version 0.5-2, http://www.medepi.com
Bataille A, Cashins SD, Grogan L, Skerratt LF, Hunter D, McFadden M et al. (2015) Susceptibility of amphibians to chytridiomycosis is associated with MHC class II conformation. Proc R Soc B Biol Sci 282:1–9
Belasen AM, Bletz MC, Leite D, da S, Toledo LF, James TY (2019) Long-term habitat fragmentation is associated with reduced MHC IIB diversity and increased infections in amphibian hosts. Front Ecol Evol 6:1–12
Berger L, Speare R, Daszak P, Green DE, Cunningham AA, Goggin CL et al. (1998) Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proc Natl Acad Sci 95:9031–9036
Bernatchez L, Landry C (2003) MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol 16:363–377
Blanchong JA, Robinson SJ, Samuel MD, Foster JT (2016) Application of genetics and genomics to wildlife epidemiology. J Wildl Manag 80:593–608
Bos DH, Dewoody JA (2005) Molecular characterization of major histocompatibility complex class II alleles in wild tiger salamanders (Ambystoma tigrinum). Immunogenetics 57:775–781
Boyle DG, Boyle DB, Olsen V, Morgan JAT, Hyatt AD (2004) Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time Taqman PCR assay. Dis Aquat Organ 60:141–148
Braciale TJ, Morrison LA, Sweetser MT, Sambrook J, Gething M, Braciale VL (1987) Antigen presentation pathways to class I and class II MHC-restricted T lymphocytes. Immunol Rev 98:95–114
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL et al. (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39
Cammen K, Hoffman JI, Knapp LA, Harwood J, Amos W (2011) Geographic variation of the major histocompatibility complex in Eastern Atlantic grey seals (Halichoerus grypus). Mol Ecol 20:740–752
Clarkson RW, Rorabaug JC (1989) Status of leopard frogs (Rana pipiens complex: Ranidae) in Arizona and Southeastern California. Southwest Nat 34:531–538
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Cushman SA, Wasserman TN, Landguth EL, Shirk AJ (2013) Re-evaluating causal modeling with mantel tests in landscape genetics. Diversity 5:51–72
Cutrera AP, Lacey EA (2007) Trans-species polymorphism and evidence of selection on class II MHC loci in tuco-tucos (Rodentia: Ctenomyidae). Immunogenetics 59:937–948
Daszak P, Cunningham A, Hyatt A (2000) Emerging infectious diseases of wildlife—threats to biodiversity and human health. Science 287:443–449
Echaubard P, Pauli BD, Trudeau VL, Lesbarrères D (2015) Ranavirus infection in northern leopard frogs: the timing and number of exposures matter. J Zool 2015:1–7
Eizaguirre C, Lenz TL, Kalbe M, Milinski M (2012) Rapid and adaptive evolution of MHC genes under parasite selection in experimental vertebrate populations. Nat Commun 3:1–6
Evans ML, Neff BD, Heath DD (2010) MHC genetic structure and divergence across populations of chinook salmon (Oncorhynchus tshawytscha). Heredity 104:449–459
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fick SE, Hijmans RJ (2017) WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int J Climatol 37:4302–4315
Fraser BA, Ramnarine IW, Neff BD (2010) Selection at the MHC class IIB locus across guppy (Poecilia reticulata) populations. Heredity 104:155–167
Fu M, Waldman B (2017) Major histocompatibility complex variation and the evolution of resistance to amphibian chytridiomycosis. Immunogenetics 69:529–536
Garner TW, Perkins MW, Govindarajulu P, Seglie D, Walker S, Cunningham AA, Fisher MC (2006) The emerging amphibian pathogen Batrachochytrium dendrobatidis globally infects introduced populations of the North American bullfrog, Rana catesbeiana. Biol Lett 2:455–459
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Guillot G, Rousset F (2013) Dismantling the mantel tests. Methods Ecol Evol 4:336–344
Hedrick PW (2002) Pathogen resistance and genetic variation at MHC loci. Evolution 56:1902–1908
Hoffman EA, Blouin MS (2004a) Evolutionary history of the northern leopard frog: reconstruction of phylogeny, phylogeography, and historical changes in population demography from mitochondrial DNA. Evolution 58:145–159
Hoffman EA, Blouin MS (2004b) Historical data refute recent range contraction as cause of low genetic diversity in isolated frog populations. Mol Ecol 13:271–276
Hoffman EA, Schueler FW, Blouin MS (2004) Effective population sizes and temporal stability of genetic structure in Rana pipiens, the northern leopard frog. Evolution 58:2536–2545
Hoffman EA, Schueler FW, Jones AG, Blouin MS (2006) An analysis of selection on a colour polymorphism in the northern leopard frog. Mol Ecol 15:2627–2641
Horner AA, Hoffman EA, Tye MR, Hether TD, Savage AE (2017) Cryptic chytridiomycosis linked to climate and genetic variation in amphibian populations of the southeastern United States. PLoS ONE 12:e0175843
Ilmonen P, Penn DJ, Damjanovich K, Morrison L, Ghotbi L, Potts WK (2007) Major histocompatibility complex heterozygosity reduces fitness in experimentally infected mice. Genetics 176:2501–2508
Jarvis A, Reuter H, Nelson A, Guevara E (2009) Hole-filled seamless SRTM data V4. http://srtm.csi.cgiar.org
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:1–15
Jukes TH, Cantor CR (1969) Evolution of protein molecules. In: Munro HN (ed) Mammalian protein metabolism, III. Academic Press, New York, p 21–132
Kaufman J (2018) Unfinished business: evolution of the MHC and the adaptive immune system of jawed vertebrates. Annu Rev Immunol 36:383–409
Kiemnec-Tyburczy KM, Richmond JQ, Savage AE, Zamudio KR (2010) Selection, trans-species polymorphism, and locus identification of major histocompatibility complex class IIβ alleles of new world ranid frogs. Immunogenetics 62:741–751
Klein J, Sato A, Nagl S, Colm O (1998) Molecular trans-species polymorphism. Annu Rev Ecol Syst 29:1–21
Kosakovsky Pond SL, Frost SDW (2005a) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533
Kosakovsky Pond SL, Frost SDW (2005b) Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222
Kosakovsky Pond SL, Frost SDW, Muse SV (2005) HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679
Kosch TA, Bataille A, Didinger C, Eimes JA, Rodríguez-Brenes S, Ryan MJ, Waldman B (2016) Major histocompatibility complex selection dynamics in pathogen-infected túngara frog (Physalaemus pustulosus) populations. Biol Lett 12:20160345
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2017) Partitionfinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34:772–773
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Leonard WP, Mcallister KR, Friesz RC (1999) Survey and assessment of northern neopard frog (Rana pipiens) populations in Washington state. Northwest Nat 80:51–60
Li J, Shen H, Wang H, Zhao M, Luo Z, Wu H (2016) Diversifying selection is the dominant factor affecting the geographical variation of MHC class II genes in the omei tree frog. J Zool 300:197–204
Lips KR (2016) Overview of chytrid emergence and impacts on amphibians. Philos Trans R Soc B Biol Sci 371:1–9
Martel A, Spitzen-van der Sluijs A, Blooi M, Bert W, Ducatelle R, Fisher MC et al. (2013) Batrachochytrium salamandrivorans sp. nov. causes lethal chytridiomycosis in amphibians. Proc Natl Acad Sci 110:15325–15329
McKnight DT, Schwarzkopf L, Alford RA, Bower DS, Zenger KR (2017) Effects of emerging infectious diseases on host population genetics: a review. Conserv Genet 1–11
McRae BH, Shah VB, Mohapatra TK (2013) Circuitscape 4 user guide. The Nature Conservancy. http://www.circuitscape.org
Migalska M, Sebastian A, Radwan J (2019) Major histocompatibility complex class I diversity limits the repertoire of T cell receptors. Proc Natl Acad Sci 116:5021–5026
Mulder KP, Cortazar-Chinarro M, Harris DJ, Crottini A, Campbell Grant EH, Fleischer RC et al. (2017) Evolutionary dynamics of an expressed MHC class IIβ locus in the Ranidae (Anura) uncovered by genome walking and high-throughput amplicon sequencing. Dev Comp Immunol 76:177–188
Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL et al. (2013) FUBAR: a fast, unconstrained bayesian AppRoximation for inferring selection. Mol Biol Evol 30:1196–1205
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL(2012) Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York, USA
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ (2014) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274
O’Brien SJ, Evermann JF (1988) Interactive influence of infectious disease and genetic diversity in natural populations. Trends Ecol Evol 3:254–259
O’Donnell RP, Drost CA, Mock KE (2017) Cryptic invasion of northern leopard frogs (Rana pipiens) across phylogeographic boundaries and a dilemma for conservation of a declining amphibian. Biol Invasions 19:1039–1052
Oliver MK, Piertney SB (2012) Selection maintains MHC diversity through a natural population bottleneck. Mol Biol Evol 29:1713–1720
Oliver MK, Telfer S, Piertney SB (2009) Major histocompatibility complex (MHC) heterozygote superiority to natural multi-parasite infections in the water vole (Arvicola terrestris). Proc R Soc B Biol Sci 276:1119–1128
Osborne MJ, Pilger TJ, Lusk JD, Turner TF (2017) Spatio-temporal variation in parasite communities maintains diversity at the major histocompatibility complex class II b in the endangered Rio Grande silvery minnow. Mol Ecol 26:471–489
Ottova E, Andrea S, Martin J-F, Gouy de Bellocq J, Gelnar M, Allienne J-F et al. (2005) Evolution and trans-species polymorphism of MHC class IIb genes in cyprinid fish. Fish Shellfish Immunol 18:199–222
Parris MJ, Reese E, Storfer A (2006) Antipredator behavior of chytridiomycosis-infected northern leopard frog (Rana pipiens) tadpoles. Can J Zool 84:58–65
Penn DJ, Damjanovich K, Potts WK (2002) MHC heterozygosity confers a selective advantage against multiple-strain infections. Proc Natl Acad Sci 99:11260–11264
Phillips KP, Cable J, Mohammed RS, Herdegen-radwan M, Raubic J, Przesmycka KJ et al. (2018) Immunogenetic novelty confers a selective advantage in host—pathogen coevolution. Proc Natl Acad Sci USA 115:1552–1557
Phillipsen IC, Funk WC, Hoffman EA, Monsen KJ, Blouin MS (2011) Comparative analyses of effective population size within and among species: ranid frogs as a case study. Evolution 65:2927–2945
Pond SLK, Posada D, Gravenor MB, Woelk CH, Frost SDW (2006) Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol 23:1891–1901
R Core Team (2013) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/.
Radwan J, Biedrzycka A, Babik W (2010) Does reduced MHC diversity decrease viability of vertebrate populations? Biol Conserv 143:537–544
Radwan J, Babik W, Kaufman J, Lenz TL, Winternitz J (2020) Advances in the Evolutionary Understanding of MHC Polymorphism. Trends Genet 36:298–311
Ralls K, Sunnucks P, Lacy RC, Frankham R (2020) Genetic rescue: a critique of the evidence supports maximizing genetic diversity rather than minimizing the introduction of putatively harmful genetic variation. Biol Cons 251:108784
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904
Raymond M, Rousset F (1995) genepop, version 1.2: populationgenetics software for exact tests and ecumenicism. J Hered 86:248–249
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Rogers SD, Peacock MM (2012) The disappearing northern leopard frog (Lithobates pipiens): conservation genetics and implications for remnant populations in western Nevada. Ecol Evol 2:2040–2056
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S et al. (2012) Mrbayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Rosenberg MS, Anderson CD (2011) PASSaGE: pattern analysis, spatial statistics and geographic exegesis. Version 2. Methods Ecol Evol 2:229–232
Savage AE, Zamudio KR (2011) MHC genotypes associate with resistance to a frog-killing fungus. Proc Natl Acad Sci USA 108:16705–16710
Savage AE, Becker CG, Zamudio KR (2015) Linking genetic and environmental factors in amphibian disease risk. Evol Appl 8:560–572
Savage AE, Mulder KP, Torres T, Wells S (2018) Lost but not forgotten: MHC genotypes predict overwinter survival despite depauperate MHC diversity in a declining frog. Conserv Genet 19:309–322
Savage AE, Muletz-Wolz CR, Campbell Grant EH, Fleischer RC, Mulder KP (2019) Functional variation at an expressed MHC class II β locus associates with Ranavirus infection intensity in larval anuran populations. Immunogenetics 1–12
Savage AE, Zamudio KR(2016) Adaptive tolerance to a pathogenic fungus drives major histocompatibility complex evolution in natural amphibian populations. Proc R Soc B Biol Sci 283:20153115
Scheele BC, Pasmans F, Skerratt LF, Berger L, Martel A, Beukema W et al. (2019) Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science 363:1459–1463
Sebastian A, Herdegen M, Migalska M, Radwan J (2016) Amplisas: a web server for multilocus genotyping using next-generation amplicon sequencing data. Mol Ecol Resour 16:498–510
Skerratt LF, Berger L, Speare R, Cashins S, McDonald KR, Phillott AD et al. (2007) Spread of chytridiomycosis has caused the rapid global decline and extinction of frogs. Ecohealth 4:125–134
Spurgin LG, Richardson DS (2010) How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc R Soc B Biol Sci 277:979–988
Strand TM, Segelbacher G, Quintela M, Xiao L, Axelsson T, Hoglund J (2012) Can balancing selection on MHC loci counteract genetic drift in small fragmented populations of black grouse? Ecol Evol 2:341–353
Takahata N, Nei M (1990) Allelic genealogy under overdominant and frequency-dependent selection and polymorphism of major histocompatibility complex loci. Genetics 124:967–978
Teacher AGF, Garner TWJ, Nichols RA (2009) Evidence for directional selection at a novel major histocompatibility class I marker in wild common frogs (Rana temporaria) exposed to a viral pathogen (Ranavirus). PLoS ONE 4:1–6
Voordouw MJ, Adama D, Houston B, Govindarajulu P, Robinson J (2010) Prevalence of the pathogenic chytrid fungus, Batrachochytrium dendrobatidis, in an endangered population of northern leopard frogs, Rana pipiens. BMC Ecol 10:1–10
Waraniak JM, Fisher JDL, Purcell K, Mushet DM, Stockwell CA (2019) Landscape genetics reveal broad and fine-scale population structure due to landscape features and climate history in the northern leopard frog (Rana pipiens) in North Dakota. Ecol Evol 9:1041–1060
Wegner KM, Reusch TBH, Kalbe M (2003) Multiple parasites are driving major histocompatibility complex polymorphism in the wild. J Evol Biol 16:224–232
Woodhams DC, Hyatt AD, Boyle DG, Rollins-Smith LA (2008) The northern leopard frog Rana pipiens is a widespread reservoir species harboring Batrachochytrium dendrobatidis in North America. Herpetol Rev 39:66–68
Yuan ZY, Zhou WW, Chen X, Poyarkov NA, Chen HM, Jang-Liaw NH et al. (2016) Spatiotemporal diversification of the true frogs (Genus Rana): a historical framework for a widely studied group of model organisms. Syst Biol 65:824–842
Zhang J, Kobert K, Flouri T, Stamatakis A (2014) PEAR: a fast and accurate Illumina paired-end reAd mergeR. Bioinformatics 30:614–620
Acknowledgements
We thank Susi MacVean and the Arizona Game and Fish Department for facilitating field sampling, Matt Atkinson, Jacob LaFond, Veronica Urgiles, Kevin Mulder, Rachel Gutner, Noah Penney, and Eric Erb for assisting with frog sampling, the Savage Lab for helpful feedback on earlier drafts of this paper, and Johnny Konvalina and Dr. Pedro Quintana-Ascencio for providing R code for bootstrapping analyses. Thank you to M. Blouin for storing frog tissues in his freezer for the past 20 years and thereby facilitating this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Barbara Mable
Supplementary information
Rights and permissions
About this article

Cite this article
Trujillo, A.L., Hoffman, E.A., Becker, C.G. et al. Spatiotemporal adaptive evolution of an MHC immune gene in a frog-fungus disease system. Heredity 126, 640–655 (2021). https://doi.org/10.1038/s41437-020-00402-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-00402-9
