
Abstract
Even though populations of many large carnivores are expanding throughout Europe, the Eurasian lynx population in the Western Carpathians seems unable to spread beyond the western boundaries of its current distributional range. Many factors, both extrinsic and intrinsic, can influence the potential for range expansion: landscape fragmentation, natal philopatry, low natural fecundity and high mortality, and low and sex-biased dispersal rates. In this study we used non-invasive genetic sampling to determine population size fluctuation, sub-structuring and social organisation of the peripheral lynx population at the Czech–Slovak border. Even though the population size has been relatively stable over the period studied (2010–2016), the individual inbreeding coefficients of residents at the end of the study were much higher than those of founders at the beginning of the study. While non-resident individuals (predominantly males) occurred regularly in the study population, only resident individuals with well-established home ranges participated in breeding and produced offspring. Almost half the offspring detected in the study (predominantly females) settled in or near the natal area. Subsequent incestuous mating resulted in production of inbred individuals, reduction of effective population size of the population, and sub-structuring of the population through formation of two distinct family lineages. Our study illustrates how social constraints, such as territoriality, breeding of residents and natal philopatry of females, lead to incestuous mating in small-sized populations, especially at the periphery of their distribution. This threat should be taken into account in planning of conservation and population recovery of species with similar social structure.
Similar content being viewed by others


Introduction
The centre−periphery hypothesis predicts a decrease in genetic variation and demographic performance of a species from the centre to the periphery of its distribution range associated with the decrease in environmental suitability (Eckert et al. 2008). However, in the present human-dominated landscape the range boundaries are often not the result of historical species distribution, but rather a consequence of anthropogenic extinction forces, e.g. landscape fragmentation and/or human persecution (Channell and Lomolino 2000). This can disrupt the general pattern predicted by the centre−periphery hypothesis, but, in general, peripheral populations still tend to be more fragmented, genetically impoverished and differentiated due to isolation, low gene flow and small population sizes, often making them more sensitive to stochastic changes than populations in the core of the species’ range (Hampe and Petit 2005; Holt et al. 2005; Hardie and Hutchinson 2010; Janečka et al. 2011). A probability of their extinction is directly influenced by their variability, size and immigration rate (Channell and Lomolino 2000; Pironon et al. 2016).
Although the Eurasian lynx appears to be a highly mobile species that frequently makes long distance movements (up to 450 km) which are expected to facilitate gene flow among subpopulations (e.g. Schmidt 1998; Andersen et al. 2005), lynx also seem to show a high tendency towards local loss of genetic diversity (Schmidt et al. 2011). Due to its dietary specialisation as a stalking predator of medium-sized ungulates, lynxes occur in low population densities and are highly dependent on sufficient vegetation cover while hunting and dispersing (Sunquist and Sunquist 2002). This makes them extremely sensitive to habitat loss and landscape fragmentation (Podgórski et al. 2008; Schmidt et al. 2011).
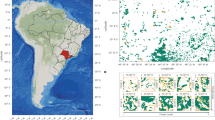
While some large carnivores are expanding across Europe, the native Eurasian lynx (Lynx lynx) population in the Carpathians seems to remain stable (von Arx et al. 2004; Ratkiewicz et al. 2012; Kaczensky et al. 2013; Chapron et al. 2014). The Czech–Slovak border represents the western edge of the native distribution of lynx in the Carpathians (Fig. 1). This area is fragmented into several discrete mountain ranges and partially isolated from the rest of the Western Carpathians by the highly urbanised valleys along the Slovak rivers Váh and Kysuca. Lynx were exterminated from the area at the turn of the nineteenth and twentieth century (Kratochvíl 1968; Kratochvíl and Vala 1968; Hell et al. 2004), such that in the 1930s, the western range edge of the Carpathian population was situated in the central part of Slovakia (Hell et al. 2004). After World War II lynx abundance in the Carpathians started to rise, and through natural migration, the distribution expanded up to the eastern (Moravian) part of the Czech Republic (Moravskoslezské Beskydy Mts, Jeseníky Mts). Up until the 1980s several colonisation−extinction cycles occurred in that area, significantly affected by legal and illegal hunting that influenced lynx abundance in the entire Western Carpathians (Kunc 1996). Thanks to the full protection of the lynx in the Czech Republic and later also in Slovakia (Hell and Slamečka 1996), they finally recolonised the Czech–Slovak border region in the second half of the 1980s (Anděra and Červený 2009).

Overview of samples collected within the study area from 1 May 2009 to 18 May 2016. Permanent occurrence (white colour) and sporadic occurrence (light grey colour) of Eurasian lynx in Europe is delineated on the smaller figure (right) according to Chapron et al. (2014). The star at the smaller figure represents the study area
Gathering data on the distribution and abundance of endangered species (or populations) is essential for their successful conservation. Understanding of population dynamics and dispersal processes of rare species is important for development of suitable management and conservation plans (e.g. Linnell et al. 2008). Such information is especially difficult to collect for elusive species like the Eurasian lynx (Linnell et al. 1989). Fortunately, identification of individuals by unique multi-locus DNA genotyping allow populations to be surveyed through non-invasive genetic sampling (Waits and Paetkau 2005; Taberlet et al. 1999). Besides estimates of population size this approach can also provide an insight into population sub-structuring, sex ratio, relatedness of individuals, and various population-level metrics like genetic diversity, gene flow, and dispersal rate (e.g. Ritland 1996; Schwartz et al. 2007), and has been successfully used for estimation of density and spatial activity of a variety of elusive carnivores, including the Eurasian lynx (e.g. De Barba et al. 2010; Ebert et al. 2010; Davoli et al. 2013; Sindičić et al. 2013; Müller et al. 2014).
There is in practice no information about the genetic structure of the Carpathian lynx population. In addition, there is little understanding of the social structure of lynx or other felids at a small geographic scale, even though the probability of extinction of peripheral or small isolated populations is directly influenced by their population size and dispersal rate. In this study, we analysed the genetic composition of the Carpathian lynx population at the western periphery of its continuous distribution in order to assess (1) population size, sex structure and relatedness among resident individuals; (2) variability in reproductive success and the rate of dispersal (immigration); and (3) the effect of social structure on population sub-structuring and effective population size. Insights into the lynx population structure, reproductive strategies and mechanisms of dispersal may help to define genetic or other possible constraints of population expansion and are relevant for the species’ successful conservation or reintroduction into new areas.
Material and methods
Study area
Our study area at the Czech–Slovak border covered two protected landscape areas (PLA), PLA Beskydy (Czech side, 1160 km2) and PLA Kysuce (Slovak side; 340 km2) which themselves embrace three mountain ranges: the Moravskoslezské (MS) Beskydy Mts, the Vsetínské Beskydy Mts and the Javorníky Mts (Fig. 1). The altitude varies between 350 and 1323 m a.s.l. and the landscape is characterised by a high diversity of habitats—various types of forest, ridge-top meadows and pastures together with hamlets scattered throughout the area (Weissmannová 2004). The majority of the area is forested (72%), characterised by rugged terrain with deep valleys and steep slopes, and with scattered settlements ranging up to mountain tops mainly on the Czech side. Extensive forest complexes representing suitable habitat for lynx are formed predominantly by Norway spruce monocultures, while natural beech-fir forests cover only a small proportion of the area (2.6%). Three large carnivores occur in this area at least occasionally—the Eurasian lynx (Lynx lynx), the grey wolf (Canis lupus) and the brown bear (Ursus arctos); however, only lynx is present in a stable and regularly breeding population (Červený and Koubek 2003; Anděra and Červený 2009).
Sampling and DNA extraction
We collected 446 non-invasive lynx samples (157 faecal, 248 hair, 41 urine) during the period from 1 May 2009 to 18 May 2016. The study covered seven “lynx” years; each lasting from 1 May to 30 April, except for the final year when the period ended by 18 May. Lynx generation time is estimated to be 4 years (Spong and Hellborg 2002). The dataset was complemented by eight invasively collected samples: four from individuals live-captured for radio-tracking (one blood, three hair samples), three tissue samples (two carcasses found after car accidents, one dead kitten) and one blood sample found after poaching. The whole dataset consists of 454 lynx samples (Fig. 1).
Non-invasive samples were collected during monitoring of signs of lynx presence, which covered the entire study area with a focus on suitable lynx habitats. Detailed information about intensity of monitoring during particular years is given in Supplementary materials (Fig. S1A, Fig. S2, Table S1). On the Czech side we also used hair traps from January 2011 and up to June 2014 (Fig. S1B). Hair traps consisted of a carpet (10 × 20 cm) and a band of Velcro. We used catnip oil mixed with synthetic beaver castoreum (1:32) as an olfactory attractant according to McDaniel et al. (2000) and Schmidt and Kowalczyk (2006). Over the period of the study, camera traps were also used for lynx monitoring in the whole area, in several cases installed in the front of hair traps or marking sites thus enabling to join photos with individual genotypes.
Hair and faecal samples were conserved by desiccation with silica gel, stored in 96% ethanol or frozen; urine and blood samples were frozen. The Genomic DNA Mini kit Tissue (Geneaid Biotech Ltd., New Taipei City, Taiwan) was used to extract DNA from blood, hair and urine samples according to the manufacturers’ protocols. The QIAamp DNA Stool Mini Kit (Qiagen GmbH, Hilden, Germany) was used to extract DNA from faecal samples. We used dedicated laboratories for DNA extraction and PCR setup for non-invasive samples, and enforced strict rules and procedures to prevent contamination.
Genotyping
We genotyped 15 nuclear microsatellite loci and the amelogenin marker for sex identification (Table S2). Each sample was amplified using PCR (polymerase chain reaction) in a 10–12 μl reaction volume, containing 1–4 μl DNA template (the default 2 μl of DNA template was adjusted in subsequent PCRs based on the peak height of successfully amplified loci), 5 μl of the Qiagen Multiplex PCR Kit and appropriate primers. Forward primers were labelled at the 5′ end with fluorescent dyes (Applied Biosystems—see Table S2 for more details about primer multiplexes and PCR conditions). PCR was performed in 35–45 cycles of denaturation for 60 s at 94 °C, annealing for 90 s at the multiplex-specific temperature (50–61 °C), extension for 60 s at 72 °C, preceded by 15 min of initial denaturation/polymerase activation at 95 °C and followed by a 10-min final extension at 72 °C. Then, 1 μl of the PCR products was added to a mixture of GeneScan™ 600 LIZ® Size Standard (Applied Biosystems) and formamide, and the mixture was denatured and run on the ABI Prism 3130 Genetic Analyzer (Applied Biosystems). The DNA fragment sizes were scored using GENEMAPPER 3.7 software (Applied Biosystems).
PCR amplification for non-invasive DNA samples was repeated in multiple parallels according to the quality and quantity of extracted DNA. We followed an adjusted multiple-tubes approach (Taberlet et al. 1996), with requirements of a minimum of four positive PCRs for homozygotes and two for heterozygotes; however, up to nine PCRs were required in some cases to confirm the genotype in samples with a significant level of allelic dropout. Each electropherogram was checked independently by two people and we did not accept an allele in a consensus genotype until we saw it at least twice. We conducted a two-phase screening of the PCR products. Firstly, eight loci in two independent PCR multiplexes were amplified, and all samples which produced readable genotypes at less than three loci were discarded. Samples that were successfully genotyped in the first phase were then amplified for all loci according to the multiple-tube approach, consensus genotypes produced and matched to identify which samples belonged to the same animals. All up to five mismatches (MM) between genotypes were again re-amplified in separate PCRs at least twice to confirm (or refute) the mismatch. In the final dataset all individual genotypes differed at least in 3-MM pairs except for two subadults, which differed in 2-MM pairs. This approach has been proved to determine the individuals correctly (Paetkau 2005).
While 309 out of 454 samples were analysed directly using this procedure at the Institute of Vertebrate Biology CAS, Brno, Czech Republic (IVB), 145 samples were first analysed in the laboratory at University of Ljubljana, Slovenia, using the panel of 20 microsatellite loci described by Sindičić et al. (2013). Due to a low overlap of the two marker panels (only three loci), the best sample of each individual identified in the Slovenian laboratory was re-genotyped at IVB laboratory using the 15 microsatellite loci (Table S2) to obtain the full and comparable dataset of identified individuals and their genotypes. The complete dataset was used in all analyses based on individual genotypes, while the calculations of allelic dropout and frequency of false alleles were based only on the 309 samples originally genotyped at 15 loci plus amelogenin at IVB.
For population size estimation using non-invasive genetic sampling, it is essential to distinguish between individuals with at least 99% certainty (Mills et al. 2000). Therefore, the probability of identity (PID) and the probability of identity among siblings (PID-sib) were calculated for each locus and cumulative for the entire panel according to Paetkau and Strobeck (1994) and Waits et al. (2001) using Gimlet v.1.3.1 (Valiére 2002). Due to high probability of occurrence of closely related individuals in our dataset, PID-sib was used as a more conservative value for assessing the number of loci required to reliably distinguish lynx individuals.
Genetic diversity
For the dataset of 40 unique genotypes the number of alleles (NA), and the observed (HO) and expected (HE) heterozygosity were estimated for each locus using FSTAT 2.9.3.2 (Goudet 2001). Departure from the Hardy−Weinberg equilibrium was analysed using the exact probability test in GENEPOP version 3.4 (Raymond and Rousset 1995). The P values for multiple testing were corrected using the Bonferroni correction (Rice 1989). GENEPOP (Raymond and Rousset 1995) was also used to calculate inbreeding coefficient (FIS). Nevertheless, in a small population FIS can be zero even when the population is highly inbred (Keller and Waller 2002), hence mean individual inbreeding coefficient (F) for the population was estimated using TrioML (Wang 2007) and DyadML (Milligan 2003), as implemented in the software package COANCESTRY 1.0 (Wang 2011). Allelic dropout and false alleles were estimated for the dataset of 309 originally genotyped field samples according to Broquet and Petit (2004).
Relatedness
Microsatellite profiles for the 15 loci were used to identify the most likely relationships among individuals. The software KinInfor v2.0 (Wang 2006) was used to calculate the informativeness of each locus to infer pairwise relationships and relatedness and to estimate the overall power of the set of microsatellite loci (see more details in Appendix 2). Individual parentage was first investigated using Bayesian framework for pedigree reconstruction in software FRANz v. 2.0.0 (Riester et al. 2009). FRANz was run using default settings except for specification of an approximate maximum number of females (Nfmax) and males (Nmmax), where all identified males and females were assigned as candidate parents and outputs in the form of maximum likelihood (ML) pedigree was used as initial pedigree draft for two main mountain ranges—MS Beskydy Mts and Javorníky Mts. After that, we used two computational assignments calculated within a likelihood framework: pairwise microsatellite-based relatedness coefficients and relationships among individuals conducted in the program ML-Relate (Kalinowski et al. 2006) and parentage analysis implemented in CERVUS 3.0.3 (Kalinowski et al. 2007). In CERVUS, initially, all males and all females were defined as candidate fathers and mothers; three individuals without identified sex were included in both father and mother files. Subsequently, the number of candidate parents was constrained using field monitoring data. When running simulations, the following parameters were used: 100,000 offspring, 1% mistyped loci and 98.7% genotyped loci (determined by CERVUS from our real dataset). Despite intensive fieldwork it is possible that we did not successfully sample all potential parents, so in simulations we applied 90% probability of sampling candidate mothers and fathers. Confidence intervals were calculated using LOD (Likelihood-of-Difference) scores at both 95% (strict) and 80% (relaxed) confidence levels. A minimum of the same eight genotyped loci was required for the analysis, what all individual genotypes fulfilled. Further, as an alternative approach, we conducted an analysis of relatedness using MOL_COANC 3.0 (Fernández and Toro 2006), which implies a simulated annealing algorithm to find the genealogy which maximises the correlation between the molecular coancestry matrix calculated from markers and the genealogical coancestry obtained for that particular pedigree. Thus the correlation between matrices is used as the driven parameter instead of likelihood used in previous methods. The parental relationships were estimated according to sib-ship relations (full/half-sibling) detected. Consequently, necessary field information from snow-tracking (date of sample collection, place, other complementary information as e.g. tracking of mother with kittens) and data from camera-trapping were used for evaluation of the most likely relationships and for discarding impossible couples even if compatible for genotypes. The most reliable relationships following the five steps mentioned above were used for reconstruction of final pedigrees.
Based on the genetic sampling, pedigree reconstructions and camera-trapping data we were able to split the individuals to adults and juveniles as well as to resident and non-resident individuals. The individuals that were present for at least 12 months in the study area, i.e. likely established home ranges, and/or the females that also bred in the study area (we detected their offspring) were assumed to be residents (territorial lynx). During the study we also detected a few unrelated individuals which immigrated into the study area, occupied the vacant territory (previously unoccupied or occupied by another individual) and became a resident. Except for offspring all other lynxes detected for shorter period than 12 months were assumed to be floaters (immigrants). This splitting was used for more reliable estimates of population density in the study area as well as for the detection of dispersal (immigration) rate. The combination of genetic data with field data recently helped to uncover the mating system of congeneric Iberian lynx and was recommended for parentage analysis of endangered felids worldwide (Lucena-Perez et al. 2018).
Population size and effective population size
Since our data fit the continuous (single-session) sampling design, programme CAPWIRE (Miller et al. 2005) was used for population size estimates. For each year separately, non-residents and offspring up to the age of 1 year were excluded from the analysis to meet the assumption of population closure. To reduce temporal and spatial autocorrelation between samples, multiple samples of the same individual collected during the same day and at the same area were considered to be a single capture. Likelihood ratio test incorporated in CAPWIRE was used to select the best model of capture probability. The equal capture model (ECM) assumes there is no capture heterogeneity in the dataset while the two innate rates model (TIRM) assigns individuals as having either a high or a low capture probability. Population estimates were derived using the selected model, and 95% confidence intervals estimated with parametric bootstrap of 10,000 replicates.
To determine effective population size (Ne) and to assess its temporal changes, we employed multiple ‘single-sample’ maximum-likelihood (linkage disequilibrium (Waples 2006), heterozygosity excess and molecular coancestry) and ‘two-sample’ temporal approaches (Nei and Tajima 1981; Pollak 1983; Jorde and Ryman 2007) implemented in NEESTIMATOR 2.01 (Do et al. 2014), and full-likelihood sib-ship assignment (Wang 2009) within COLONY V.2.0.4.5 (Jones and Wang 2010). Due to small number of sampled individuals detected yearly, the analysis was performed for pooled datasets of three periods (2009–2012, 2012–2014 and 2014–2016), and for the whole 7-year study period (2009–2016). As it was impossible to distinguish various cohorts in the studied population, our samples inevitably represented several overlapping generations. This can produce downward biased estimates due to the mini-Wahlund effect between generations (for more details see Waples et al. 2014). For linkage disequilibrium and heterozygosity excess approach, we calculated contemporary Ne using the lowest allele frequency cutoffs (Pcrit) based on the number of sampled individuals (S) according to formula “1/(2S) < Pcrit < 1/S” for S < 25 individuals and assuming random mating (males tend to mate with several females), and report results with 95% confidence intervals from the parametric method (Waples and Do 2010). A limitations of temporal methods of Ne estimation are given in Supplementary materials (Table S10).
Genetic sub-structuring
A Bayesian clustering procedure, implemented in STRUCTURE version 2.1 (Pritchard et al. 2000), was used to analyse genetic sub-structuring of the peripheral population. The program was run with 15 independent simulations for each value of K from 1 to 10, with 1.000,000 MCMC iterations with an initial burn-in of 100,000. In all simulations, an admixture ancestry model without using sampling locations as prior information and a correlated allele frequency model were used. The optimal K value was estimated using the Evanno’s calculation (Evanno et al. 2005), which is based on the second-order rate of change in the log probability of the data between successive values of K (∆K). The results of independent runs for each K were combined using STRUCTURE HARVESTER (Earl and von Holdt 2012) and the LargeKGreedy algorithm of CLUMPP 1.1.1 (Jakobsson and Rosenberg 2007). The averaged results were displayed graphically using DISTRUCT 1.1 (Rosenberg 2004).
The presence of closely related individuals within a sample set can lead to the false detection of population genetic structure when it is absent or to the overestimation of the number of clusters (Rodríguez-Ramilo and Wang 2012; Rodríguez-Ramilo et al. 2014). To confirm or reject the population structure revealed by STRUCTURE, we investigated the pattern of sample clustering using another clustering method in CLUSTER_DIST, which maximises the genetic distances between groups and thereby avoids the assumptions of Hardy−Weinberg and linkage equilibrium (Rodríguez-Ramilo et al. 2009, 2014; Velando et al. 2016) (for details see Appendix 3 in Supplementary materials). Further, the level of genetic difference between identified clusters as well as between two main mountain ranges was quantified by FST (Weir and Cockerham 1984) calculated in GENETIX 4.05.2 (Belkhir et al. 1996−2004).
Results
Collected samples and genotyping success
Lynx DNA was successfully amplified in 203 out of 454 samples (44.7%) (Fig. 1). Details about collected samples and genotyping success are provided in Supplementary materials within Appendix S1 (Fig. S3, Table S3). Allelic dropout was relatively low (on average 15.7% per locus) and false alleles were detected in low number of cases (on average 1.5%). We did not detect significant correlation between monitoring intensity expressed as number of visits/length of walked transects and the number of collected samples per lynx year (Pearson r = 0.698/0.691, P = 0.123/0.128).
Genetic diversity
Among 203 genotyped samples, we identified 40 unique multi-locus genotypes (i.e. individuals) (Table 1). All 15 loci plus amelogenin were successfully genotyped in 33 individuals. Five individuals were genotyped at 15 loci, two individuals at 14 and 12 loci, respectively. However, the most informative loci were amplified in all individuals (see cumulative PID-sib in Table S4). The power of the entire panel of markers to accurately distinguish between closely related individuals was relatively high, PID-sib = 1.47910−4 (Table S4). Ten most informative loci thus seem to be sufficient for individual identification of close relatives (cumulative PID-sib under 0.001; Waits et al. 2001).
All identified lynx genotypes were used to calculate population-level metrics. Assuming a single population and following a Bonferroni adjustment (P = 0.000476) for multiple testing, no linkage disequilibrium was found between any pair of loci. All analysed loci were polymorphic, with the number of alleles varying from 2 to 7 (Table S5), even though locus FCA232 was almost monomorphic (the second allele was detected only in a single individual). We did not detect any significant deviations from Hardy–Weinberg expectations (adjusted P after Bonferroni correction was 0.0033, but none of the calculated P values were even below the 0.05 threshold; the test was not conducted for FCA232).
A total of 53 different alleles were found among 40 multi-locus genotypes, with the mean number of alleles per locus NA = 3.53. Expected heterozygosity was HE = 0.531. Detailed values of genetic variation at particular loci are presented in Table S5.
Identified individuals and their relatedness
The estimated power of our set of 15 neutral loci (PWR) was 86% for distinguishing full-sibling from unrelated pair and 63% for distinguishing parent-offspring from full-sibling (the strictest level for inferring pairwise relationships). These values confirmed a suitability of the set of markers for analysis of relatedness and pairwise relationships estimation. The calculated informativeness of each locus is provided in Table S6.
In a total sample of the population we detected a balanced sex ratio: 19 males and 18 females; the sex of three individuals was not identified. The number of genotyped samples belonging to males and females was 108 and 92 respectively; thus, the results did not confirm the higher capture probability of samples relating to males suggested in previous studies of felids (Augustine et al. 1996; Sugimoto et al. 2014). Twenty-two individuals were detected in MS Beskydy, 17 individuals in Javorníky Mts, and one in Vsetínské Beskydy Mts. + Javorníky Mts. Each multi-locus genotype was observed from 1 to 39 times (Table 1), with a mean of 5.08 ± 7.87 (SE) times. Number of samples per individual was ranging from one to 17 per lynx year. The average number of detections per individual per year varied from 1.5 (2009/10) to 4.2 (2012/13), the mean across all years being 1.7 per individual. The number of detected individuals per lynx year was not influenced by monitoring intensity (number of visits: Pearson r = 0.653/0.612; length of walked transects: P = 0.160/0.197).
Maximum likelihood pedigrees reconstructed using FRANz enabled us to construct an initial draft of family structure in our dataset (Fig. S4). This picture was then compared and completed with outputs from ML-Relate (Table S7), CERVUS, MOL_COANC, and additional camera-trapping and snow-tracking data. Final most-likely parent pairs obtained by multiple methods are shown in Table S8. Using this approach, we reconstructed two pedigrees, the first one for MS Beskydy with 16 individuals and the second one for Javorníky Mts with 13 individuals (Fig. 2). CERVUS and FRANz identified and supported 80.5% (33 of 41), MOL_COANC 82.9% (34 of 41), and ML-Relate 85.4% (35 of 41) of relationships in the final pedigrees.

Pedigree of lynx individuals from Moravskoslezské Beskydy Mts (a) and Javorníky Mts (b). Sex of the individual is marked as male = square, female = circle, and unknown sex = diamond; crossing = dead individual; labelling corresponds with Table 1. Individual inbreeding coefficient F (TrioML estimate) is given below for all individuals, highly inbred individuals (F > 0.1) are depicted in bold. Relatedness among founders was not detected
Mean individual inbreeding coefficients were 0.088 (TrioML) and 0.100 (DyadML), which indicates mating of related animals (parentage analysis detected a mating between grandfather−granddaughter (coming also from an inbred mating), half-siblings, father−daughter, Fig. 2). Nine individuals were highly inbred (F ≥ 0.1, Fig. 2, Table S9).
Estimation of actual number of individuals and effective population size
The number of individuals detected varied among study years between two and 16 individuals (Table 1). In total, we detected 17 adult individuals and 23 juveniles of which nine subsequently settled and matured in the study area. From 40 detected individuals 31 were identified as resident and nine as non-resident (Table 1). During the study ten males, two females and one individual without identified sex immigrated into the study area. Only three of these immigrants, all males, settled within the study area (BE010, BE016 and BE019), and only one produced offspring (BE019-Ota).
Population size estimated using CAPWIRE varied between 9 and 26 if all individuals were included, and between 4 and 10 if only resident individuals were included in the dataset (Table 2).
Absolute Ne estimates varied according to the method used for calculation. However, maximum-likelihood methods as well as temporal methods estimates except for heterozygosity excess suggested significant decline of Ne during the study (Table 3, Table S10).
Population sub-structuring
The Bayesian clustering analysis for all 40 lynx individuals performed in STRUCTURE detected the maximum value of likelihood for the model expecting three clusters (K = 3). The ΔK distribution (Evanno et al. 2005) also showed the highest peak at K = 3 (Fig. S5A, B). The same pattern of the best model for K = 3 was supported also by clustering analysis based on maximum genetic distance using CLUSTER_DIST (Fig. S6), while the pairwise Fst analysis confirmed that the three clusters identified represent statistically supported groups (Table S11).
The clustering procedure revealed a separate cluster in Javorníky Mts (white cluster; Fig. 3), formed by the male BE007 (Ludvík) and his inbred offspring. Ludvík was mating with his daughter BE018 and also with his granddaughter BE035. Similarly, in the MS Beskydy, where the number of breeding individuals was also low (two females and one male), juveniles together with their parents formed a second cluster/family group (dark grey cluster; Fig. 3). Non-resident individuals and several breeding individuals in both mountain ranges (probably all originating from the source Carpathian lynx population in Slovakia) seem to be forming a separate cluster (light grey cluster; Fig. 3)

Graphical output of Bayesian clustering analysis of 40 lynx individuals performed using STRUCTURE for K = 2–4. Each vertical bar corresponds to a single individual. The colours indicate the membership probability of the individual in a certain cluster. Labelling of individuals corresponds with Table 1, detected breeding individuals are outlined, and settled offspring involved in breeding are marked with a star
Discussion
Population size and dispersal at the periphery
Based on population size estimation using CAPWIRE (the estimated number of 8 (7–12) resident adults per 1500 km2 of the study area), population density at the Czech−Slovak border seems to be lower than in other areas of lynx occurrence (central West Carpathians: 0.58–0.82/100 km2, Kubala et al. 2017; Swiss Alps: 1.9–2.1 ind./100 km2, Pesenti and Zimmermann 2013; Bohemian Forest: 0.9 ind./100 km2, Weingarth et al. 2012; Eastern Europe: 2–6 ind./100 km2, Jedrzejewski et al. 1996). Low density at the periphery may have influenced the high levels of natal philopatry detected within the study. Lynx female offspring sampled during this study were predominantly philopatric. Such behaviour generally supports the resident fitness hypothesis, when related females tolerate the costs of increased resource competition due to the benefits they gain from inclusive fitness (Anderson 1989; Lambin et al. 2001). From 14 juvenile females detected during the study, two were found dead (car crashes) and eight (57%) settled near or within the maternal home range. Even though natal philopatry of females and the fact that they can take over the maternal home ranges is known from lynx as well as other felids (Zimmermann et al. 2005; Janečka et al. 2006; Fattebert et al. 2015b), at the Czech−Slovak border this behaviour might be facilitated by poaching. In Javorníky Mts three closely related females established their home ranges within maternal home ranges after poaching of two resident females (one case confirmed, the second one strongly suspected).
Amongst males we detected natal philopatry in one single male (out of 7), who settled within the father’s large home range (580 km2 based on GPS telemetry; Krojerová et al. 2014). Opportunistic philopatry of males has been documented in other felids prior to population saturation (Balme et al. 2009; Fattebert et al. 2015a, 2015b).
Landscape fragmentation, e.g. by highly urbanised valleys, may represent a significant factor affecting local connectivity among lynx populations in this part of the Carpathians, further contributing to the high levels of inbreeding observed. It seems that landscape fragmentation might represent the barrier for lynx dispersal between the two mountain ranges in the study area, MS Beskydy and Javorníky Mts. During the study we did not detect any movements between these two ranges and the analysis of genetic structure revealed the formation of separate family groups in these two different mountain ranges. The reduced ability of subadults to cross physical barriers may contribute to circular dispersal and a return to the vicinity of their natal areas. Such circular dispersal behaviour was previously observed in the Alpine lynx population (Breitenmoser-Würsten et al. 2007) and in the congeneric Iberian lynx (Ferreras et al. 2004) and has also been reported for the cougar population in Florida (Maehr et al. 2002).
On the other hand, 13 lynxes immigrated to the study area from outside. Their occurrence at the Czech−Slovak border confirmed that this peripheral population is still connected with the source Western Carpathian population. Males clearly dominated (ten males vs. two females) among the immigrants—a finding consistent with the general pattern of male-biased dispersal described for polygynous mammals, in which evolution favours males to disperse because of intra-sexual competition for mates (Greenwood 1980; Dobson 1982). A similar pattern of sex-biased dispersal, favouring males has previously been reported in other felids, e.g. leopard (Fattebert et al. 2015a), jaguar (Wultsch et al. 2016), bobcat (Janečka et al. 2006, 2007) or tiger (Gour et al. 2013). Of lynx arriving in our study area only four males established home ranges but just one successfully produced offspring. Other “floaters” did not participate in breeding, even though they occurred during the mating season and probably were looking for females. Even if dispersal is still ongoing, the gene flow is strongly limited because of social structure in the peripheral population.
Successful breeding was ensured every year only by the few residents, often related to each other. The same breeding strategy (breeding of residents and the existence of floaters) was observed also amongst lynx in Białowieża Primeval forest at the range edge of the Baltic lynx population (Schmidt et al. 2016). Similarly, importance of established home ranges for mating success is known from other felids (Janečka et al. 2006, 2007; Fattebert et al. 2015a).
The risk of inbreeding at the range periphery and its consequences
As an inevitable consequence of low density, high female philopatry and the deficit of unrelated females due to male-biased dispersal, we found high levels of inbreeding within both distinct subpopulations in the MS Beskydy and Javorníky Mts. During the study we detected 23 individuals born within the area that were offspring of eight different males. However, 15 juveniles (65%) were fathered just by two males (Ota in MS Beskydy—8 offspring, Ludvík in Javorníky Mts—7 offspring), 13 of them were born during the last 3 years (Fig. 2). Together with high female philopatry and lack of new unrelated females available for mating (because of significantly male-biased immigration), this leads to incestuous mating and increase of inbreeding. Inbreeding depression is a well-known negative factor for both short-term and long-term survival of small-sized isolated populations, manifested in felids e.g. by sperm abnormalities or low birth weights in offspring (Roelke et al. 1993; Barone et al. 1994, Johnson et al. 2010). Although it is difficult to disentangle the relative importance of the different causes of dispersal, the pattern observed in felids suggests that inbreeding avoidance is more likely a consequence of dispersal resulting from mate competition rather than the ultimate cause for male dispersal (Pusey and Wolf 1996; Fattebert et al. 2015a). It is hypothesised that in stable felid populations with no vacant territories, natal philopatry is low and offspring dispersal is high; thus unrelated males and females regularly mate. In disturbed small-sized populations, when the stability is regularly disturbed by human activity (poaching, car accidents, etc.) natal philopatry is high, younger individuals participate in breeding, and the risk of inbreeding is higher. In disturbed (over-harvested) felid populations, e.g. cougar or leopard, it was observed that juveniles remained in their natal ranges or nearby whenever it was possible even though this increased the likelihood of breeding with relatives (Balme et al. 2009; Onorato et al. 2011; Fattebert et al. 2015a).
The effective population size Ne is one of the most important parameters in wildlife management and conservation, because it can help to predict the extinction risk of the population (Newman and Pilson 1997; Frankham 2005). It is notoriously difficult to estimate Ne due to stochasticity and several biases influencing the computation. Each Ne estimation method makes a number of assumptions and, for example, low number of alleles per locus, low sample size, and immigration could bias Ne estimations. However, the aim of our study was to explore the temporal variation in Ne values of the peripheral population, rather than quantify absolute values of Ne. The results suggested a short-term significant reduction of effective population size of the peripheral population due to increasing relatedness of sampled individuals and inbreeding (Table 3).
The combination of persistent low density, high turnover of residents and female philopatry might inevitably hinder any significant range expansion at the distributional periphery of Carpathian lynx in Central Europe. In contrast to wolves and bears, which can disperse over long distances (Swenson et al. 1998; Stratman et al. 2001), subadult lynxes (similarly as other felids) seem to be more conservative dispersers which maintain close contact to conspecifics and if it is possible (vacant territories available) they settle close to the maternal home range, especially in the case of females (Zimmermann et al. 2005). High natal philopatry is specific for felids prior to population saturation. Once the estimated population density reached capacity, the socio-spatial organisation in adults stabilised with less turnover and few territorial vacancies, and emigration usually increased (Balme et al. 2009; Fattebert et al. 2015a, 2015b). The relatively high level of natal philopatry supports the idea of a pre-saturation phase of habitat occupation at the Czech–Slovak border. This conclusion of a pre-saturation phase is supported also by non-occupation or short-term occupation of other suitable parts of the study area (e.g. Vsetínské Beskydy Mts; Fig. 1). However, further sampling in the central part of the Western Carpathians is necessary to compare survival rate and dispersal pattern between central and peripheral parts of the Carpathian lynx population.
Conservation implications
The results of this study showed the importance of connectivity among local Eurasian lynx populations as the main factor affecting their genetic architecture. The lynx population we studied at the edge of the current distributional range represents an unstable, small-size population in the pre-saturation phase that cannot persist in the long term without frequent gene flow from the core population. Highly urbanised valleys, still increasing landscape fragmentation and poaching in the Western Carpathians represent serious threats for its viability. This makes conservation and maintenance of landscape permeability crucial for successful conservation of this species in the landscapes of the Western Carpathians. Our study also pointed out the necessity of long-term, continuous monitoring of lynx populations. Time is the critical component, and only extended monitoring programmes allow collection of longitudinal data needed to gain deep insights into population dynamics.
Non-invasive genetic sampling enhances our understanding of lynx demographics and population dynamics and as such represents a valuable approach complementary to other methods of lynx monitoring, e.g. camera-trapping or telemetry. Our results showed that dispersal alone does not necessarily guarantee gene flow from core areas to the periphery, especially in the long-term perspective. Although non-resident lynxes, predominantly males, regularly occur at the range edge, they rarely establish their home range there and they do not participate in breeding. However, connection of the periphery with the core population ensures occurrence of unrelated individuals from the source, which can potentially genetically refresh the peripheral population (source-sink hypothesis).
Nevertheless, male-biased dispersal, unavailability of unrelated females in the area, and the apparent lack of other inbreeding avoidance mechanisms in Eurasian lynx and probably also in other felids with similar social structure (e.g. cougar, Iberian lynx, leopard) can pose considerable threats to small and isolated populations, and should be taken into account in conservation management and reintroduction of these species into new areas. The success of reintroduction programmes may be greatly affected by the tendency of animals to return to natal areas or former established home ranges. Even though translocations may help to ensure gene flow among isolated small-sized felid populations, relationships among released individuals, together with philopatric behaviour and social structure should be taken into account when planning these conservation actions. It should also be expected that in disturbed populations affected by poaching or legal hunting, juveniles may remain in natal areas more frequently even though this subsequently leads to breeding with their relatives.
Data archiving
Genotype data available from Dryad: https://doi.org/10.5061/dryad.7m7p25k.
References
Anděra M, Červený J (2009) Large mammals in the Czech Republic. Distribution, history and protection. 2. Carnivores (Carnivora). National Museum, Prague
Anderson PK (1989) Dispersal in rodents: a resident fitness hypothesis. Spec Publ, Am Soc Mammal 9:viii+141
Andersen R, Odden J, Linnell JDC, Herfindal I, Panzacchi M, Høgseth Ø et al. (2005) Gaupe og rådyr i Sørøst-Norge. Oversikt over gjennomførte aktiviteter 1995–2004. NINA Rapport 29, Trondheim, Norway [in Norwegian with English summary]
Augustine J, Miquelle DG, Korkishko VG (1996) Preliminary results of the Far Eastern leopard ecology project: implications for conservation and management. Zov Taigi 4:6–11
Balme G, Slotow R, Hunter LTB (2009) Impact of conservation interventions on the dynamics and persistence of a persecuted leopard (Panthera pardus) population. Biol Conserv 142:2681–2690
Barone MA, Roelke ME, Howard JG, Brown JL, Anderson AE, Wildt DE (1994) Reproductive characteristics of male Florida panthers: comparative studies from Florida, Texas, Colorado, Latin America, and North American zoos. J Mammal 75:150–162
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (1996) GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Genome, Populations, Interactions, CNRS UMR 5000, Université de Montpellier II, Montpellier, France, 2004
Breitenmoser-Würsten Ch, Zimmermann F, Molinari-Jobin A, Molinari P, Capt S, Vandel JM et al. (2007) Spatial and social stability of Eurasian lynx Lynx lynx population: an assessment of 10 years of observation in the Jura Mountains. Wildl Biol 13:365–380
Broquet T, Petit E (2004) Quantifying genotyping errors in noninvasive population genetics. Mol Ecol 13:3601–3608
Červený J, Koubek P (2003) The brown bear in the Czech Republic. In: Kryštůfek B, Flajšman B, Griffiths HI (eds) Living with bears: a large European carnivore in a shrinking world. Ecological Forum of the Liberal Democracy of Slovenia, Ljubljana, pp. 245–257
Channell R, Lomolino MV (2000) Dynamic biogeography and conservation of endangered species. Nature 403:84–86
Chapron G, Kaczensky P, Linnell JD, von Arx M, Huber D, Andrén H et al. (2014) Recovery of large carnivores in Europe’s modern human-dominated landscapes. Science 346:1517–1519
Davoli F, Schmidt K, Kowalczyk R, Randi E (2013) Hair snaring and molecular genetic identification for reconstructing the spatial structure of Eurasian lynx populations. Mammal Biol 78:118–126
De Barba M, Waits LP, Garton EO, Genovesi P, Randi E, Mustoni A et al. (2010) The power of genetic monitoring for studying demography, ecology and genetics of a reintroduced brown bear population. Mol Ecol 19:3938–3951
Do C, Waples RS, Peel D, Macbeth GM, Tullett BJ, Ovenden JR (2014) NeEstimatorv2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Dobson FS (1982) Competition for mates and predominant juvenile male dispersal in mammals. Anim Behav 30:1183–1192
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ebert C, Knauer F, Storch I, Hohmann U (2010) Individual heterogeneity as a pitfall in population estimates based on non-invasive genetic sampling: a review and recommendations. Wildl Biol 16:225–240
Eckert CG, Samis KE, Lougheed SC (2008) Genetic variation across species’ geographical ranges: the central–marginal hypothesis and beyond. Mol Ecol 17:1170–1188
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fattebert J, Balme G, Dickerson T, Slotow R, Hunter L (2015a) Density-dependent natal dispersal patterns in a leopard population recovering from over-harvest. PLoS ONE 10(4):e0122355
Fattebert J, Robinson HS, Balme G, Slotow R, Hunter L (2015b) Structural habitat predicts functional dispersal habitat of a large carnivore: how leopards change spots. Ecol Appl 25:1911–1921
Fernández J, Toro MA (2006) A new method to estimate relatedness from molecular markers. Mol Ecol 15:657–667
Ferreras P, Delibes M, Palomares F, Fedriani JM, Calzada J, Revilla E (2004) Proximate and ultimate causes of dispersal in the Iberian lynx Lynx pardinus. Behav Ecol 15:31–40
Frankham R (2005) Genetics and extinction. Biol Conserv 126:131–140
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Updated from Goudet (1995).
Gour DS, Bhagavatula J, Bhavanishankar M, Reddy PA, Gupta JA, Sarkar MS et al. (2013) Philopatry and dispersal patterns in tiger (Panthera tigris). PLoS ONE 8(7):e66956
Greenwood PJ (1980) Mating systems, philopatry and dispersal in birds and mammals. Anim Behav 28:1140–1162
Hampe A, Petit RJ (2005) Conserving biodiversity under climate change: the rear edge matters. Ecol Lett 8:461–467
Hardie DC, Hutchinson JA (2010) Evolutionary ecology at the extremes of species’ ranges. Environ Rev 18:1–20
Hell P, Slamečka J (1996) Current status of the lynx (Lynx lynx) in Slovakia. In: Koubek P, Červený J (eds) Lynx in the Czech and Slovak Republics, Vol. 30. Acta Sc Nat, Brno, pp. 64–78
Hell P, Slamečka J, Gašparík J (2004) Rys a divá mačka v slovenských Karpatoch a vo svete. PaRPress, Bratislava (in Slovak)
Holt RD, Keitt TH, Lewis MA, Maurer BA, Taper ML (2005) Theoretical models of species’ borders: single species approaches. Oikos 108:18–27
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Janečka JE, Blankenship TL, Hirth DH, Kilpatrick CW, Tewes ME, Grassman LIJ (2007) Evidence for male-biased dispersal in bobcats Lynx rufus using relatedness analysis. Wildl Biol 13:38–47
Janečka JE, Blankenship TL, Hirth DH, Tewes ME, Kilpatrick CW, Grassman LIJ (2006) Kinship and social structure of bobcats (Lynx rufus) inferred from microsatellite and radio-telemetry data. J Zool 269:494–501
Janečka JE, Tewes ME, Laack LL, Caso A, Grassman Jr LI, Haines AM, Shindle DB, Davis BW, Murphy WJ, Honeycutt RL (2011) Reduced genetic diversity and isolation of remnant ocelot populations occupying a severely fragmented landscape in southern Texas. Anim Conserv 14:608–619
Jedrzejewski W, Jedrzejewska B, Okarma H, Schmidt K, Bunevich AN, Milkowski L (1996) Population dynamics (1869-1994), demography, and home ranges of the lynx in Białowieza primeval forest (Poland and Belarus). Ecography 19:122–138
Johnson WE, Onorato DP, Roelke ME et al. (2010) Genetic restoration of the Florida panther. Science 329:1641–1645
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Res 10:551–555
Jorde PE, Ryman N (2007) Unbiased estimator for genetic drift and effective population size. Genetics 177:927–935
Kaczensky P, Chapron G, von Arx M, Huber D, Andrén H, Linnell J (eds) (2013). Status, management and distribution of large carnivores—bear, lynx, wolf and wolverine—in Europe. Part 1, Report for the EU Commission, 72pp.
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106
Kalinowski ST, Wagner AP, Taper ML (2006) ML-Relate: a computer program for maximum likelihood estimation of relatedness and relationship. Mol Ecol Notes 6:576–579
Keller LF, Waller DM (2002) Inbreeding effects in wild populations. Trends Ecol Evol 17:230–241
Kunc L (1996) Lynx (Lynx lynx) in the Moravskoslezské Beskydy Mts. In: Koubek P, Červený J (eds), Lynx in the Czech and Slovak Republics, Vol. 30. Acta Sci Nat, Brno, pp. 58–63
Kratochvíl J (1968) Recent distribution of the lynx in Europe. Acta Sci Nat, Brno 5/6:1–74
Kratochvíl J, Vala F (1968) History occurrence of the lynx in Bohemia and Moravia. In: Kratochvíl J (ed) History of the distribution of the lynx in Europe. Acta Sci Nat, Brno, 50pp.
Krojerová J, Barančeková M, Homolka M, Koubek P (2014) Monitoring of large carnivores in SCI Beskydy. A final report for the State Conservation Agency, 152pp. (in Czech)
Kubala J, Smolko P, Zimmermann F, Rigg R, Tám B, Iľko T et al. (2017) Robust monitoring of the Eurasian lynx Lynx lynx in the Slovak Carpathians reveals lower numbers than officially reported. Oryx: 1–9. https://doi.org/10.1017/S003060531700076X
Lambin X, Aars J, Piertney SB (2001) Dispersal, intraspecific competition, kin competition and kin facilitation: a review of the empirical evidence. In: Clobert ED, Dhondt AA, Nichols JD (eds) Dispersal: individual, population, and community. Oxford University Press, Oxford, pp. 110–122
Linnell JDC, Salvatori V, Boitani L (2008). Guidelines for population level management plans for large carnivores. A Large Carnivore Initiative for Europe report prepared for the European Commission, 85pp.
Linnell JDC, Swenson JE, Landa A, Kvam T (1989) Methods for monitoring European large carnivores. A worldwide review of relevant experience. Nina Oppdragsmeld 549:1–38
Lucena-Perez M, Soriano L, López-Bao JV, Marmesat E, Fernández L, Palomares F, Godoy JA (2018) Reproductive biology and genealogy in the endangered Iberian lynx: Implications for conservation. Mammal Biol 89:7–13
Maehr DS, Land ED, Shindle DB, Bass OL, Hoctor TS (2002) Florida panther dispersal and conservation. Biol Conserv 106:187–97
McDaniel GW, McKelvey KS, Squires JR, Ruggiero LF (2000) Efficacy of lures and hair snares to detect lynx. Wildl Soc Bull 28:119–123
Miller CR, Joyce P, Waits LP (2005) A new method for estimating the size of small populations from genetic mark—recapture data. Mol Ecol 14:1991–2005
Milligan BG (2003) Maximum-likelihood estimation of relatedness. Genetics 163:1153–1167
Mills LS, Citta JJ, Lair KP, Schwartz MK, Tallmon DA (2000) Estimating animal abundance using noninvasive DNA sampling: promise and pitfalls. Ecol Appl 10:283–294
Müller J, Wölfl M, Wölfl S, Müller DWH, Hothorn T, Heurich M (2014) Protected areas shape the spatial distribution of a European lynx population more than 20 years after reintroduction. Biol Conserv 177:210–217
Nei M, Tajima F (1981) Genetic drift and estimation of effective population size. Genetics 98:625–640
Newman D, Pilson D (1997) Increased probability of extinction due to decreased genetic effective population size: experimental populations of Clarkia pulchella. Evolution 51:354–362
Onorato D, Desimone R, White C, Waits LP (2011) Genetic assessment of paternity and relatedness in a managed population of cougars. J Wildl Manag 75:378–384
Paetkau DW (2005) The optimal number of markers in genetic capture-mark-recapture studies. J Wildl Manag 68:449–452
Paetkau D, Strobeck C (1994) Microsatellite analysis of genetic variation in black bear populations. Mol Ecol 3:489–495
Pesenti E, Zimmermann F (2013) Density estimations of the Eurasian lynx (Lynx lynx) in the Swiss Alps. J Mammal 94:73–81
Pironon S, Papuga G, Villellas J, Angert AL, García MB, Thompson JD (2016) Geographic variation in genetic and demographic performance: new insights from an old biogeographical paradigm. Biol Rev 92:1877–1909
Podgórski T, Schmidt K, Kowalczyk R, Gulczyñska A (2008) Microhabitat selection by Eurasian lynx and its implications for species conservation. Acta Theriol 53:97–110
Pollak E (1983) A new method for estimating the effective population size from allele frequency changes. Genetics 1041:531–548
Pusey A, Wolf M (1996) Inbreeding avoidance in animals. Trends Ecol Evol 11:201–206
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Ratkiewicz M, Matosiuk M, Kowalczyk R, Konopin MK, Okarma H, Ozolins J (2012) High levels of population differentiation in Eurasian lynx at the edge of the species’ western range in Europe revealed by mitochondrial DNA analyses. Anim Conserv 15:603–612
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Riester M, Stadler PF, Klemm K (2009) FRANz: reconstruction of wild multi-generation pedigrees. Bioinformatics 25(16):2134–2139
Ritland K (1996) Estimators for pairwise relatedness and individual inbreeding coefficients. Genet Res 67:175–185
Rodríguez-Ramilo ST, Toro MA, Fernández J (2009) Assessing population genetic structure via the maximisation of the genetic distance. Genet Sel Evol 41:49
Rodríguez-Ramilo ST, Wang J (2012) The effect of close relatives on unsupervised Bayesian clustering algorithms in population genetic structure analysis. Mol Ecol Res 12:873–884
Rodríguez-Ramilo ST, Toro MA, Wang J, Fernández J (2014) Improving the inference of population genetic structure in the presence of related individuals. Genet Res 96:e003
Roelke ME, Martenson JS, O’Brien SJ (1993) The consequences of demographic reduction and genetic depletion in the endangered Florida panther. Curr Biol 3:340–350
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Schmidt K (1998) Maternal behaviour and juvenile dispersal in the Eurasian lynx. Acta Theriol 43:391–408
Schmidt K, Davoli F, Kowalczyk R, Randi E (2016) Does kinship affect spatial organization in a small and isolated population of a solitary felid: the Eurasian lynx? Integr Zool 11:334–349
Schmidt K, Kowalczyk R (2006) Using scent-marking stations to collect hair samples to monitor Eurasian lynx populations. Wildl Soc Bull 34:462–466
Schmidt K, Ratkiewicz M, Konopinski MK (2011) The importance of genetic variability and population differentiation in the Eurasian lynx Lynx lynx for conservation, in the context of habitat and climate change. Mammal Rev 41:112–124
Schwartz MK, Luikart G, Waples RS (2007) Genetic monitoring as a promising tool for conservation and management. Trends Ecol Evol 22:25–33
Sindičić M, Polanc P, Gomerčić T, Jelenčič M, Huber Đ, Trontelj P, Skrbinšek T (2013) Genetic data confirm critical status of the reintroduced Dinaric population of Eurasian lynx. Conserv Genet 14:1009–1018
Spong G, Hellborg L (2002) A near: extinction event in lynx: do microsatellite data tell the tale? Conserv Ecol 6:15
Stratman MR, Alden CD, Pelton MR, Sunquist ME (2001) Long distance movement of a Florida black bear in the southeastern coastal plain. Ursus 12:55–58
Sugimoto T, Aramilev VV, Kerley LL, Nagata J, Miquelle DG, McCullough DR (2014) Noninvasive genetic analyses for estimating population size and genetic diversity of the remaining Far Eastern leopard (Panthera pardus orientalis) population. Conserv Genet 15:521–532
Sunquist ME, Sunquist FC (2002) Wild cats of the world. The University of Chicago Press, Chicago and London, p. 452
Swenson JE, Sandegren F, Söderberg A (1998) Geographic expansion of an increasing brown bear population: evidence for presaturation dispersal. J Anim Ecol 67:819–826
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N et al. (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 26:3189–3194
Taberlet P, Waits LP, Luikart G (1999) Non-invasive genetic sampling: look before you leap. Trends Ecol Evol 14:323–327
Valiére N (2002) GIMLET: a computer program for analysing genetic individual identification data. Mol Ecol Notes 2:377–379
Velando A, Morán P, Romero R, Fernández J, Piorno V (2016). Invasion and eradication of the American mink in the Atlantic Islands National Park (NW Spain): a retrospective analysis. Biol Invasions. https://doi.org/10.1007/s10530-016-1326-8
von Arx M, Breitenmoser-Würsten Ch, Zimmermann F, Breitenmoser U (2004) Status and conservation of the Eurasian lynx (Lynx lynx) in Europe in 2001. KORA Bericht Nr. 19e. KORA, Muri, Switzerland
Waits LP, Luikart G, Taberlet P (2001) Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 10:249–256
Waits LP, Paetkau D (2005) Non-invasive genetic sampling tools for wildlife biologists: a review of applications and recommendations for accurate data collection. J Wildl Manag 69:1419–1433
Wang J (2006) Informativeness of genetic markers for pairwise relationship and relatedness inference. Theor Popul Biol 70:300–321
Wang J (2007) Triadic IBD coefficients and applications to estimating pairwise relatedness. Genet Res 89:135–153
Wang J (2009) A new method for estimating effective population sizes from a single sample of multilocus genotypes. Mol Ecol 18:2148–2164
Wang J (2011) Coancestry: a program for simulating, estimating and analysing relatedness and inbreeding coefficients. Mol Ecol Res 11:141–145
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7:167–184
Waples RS, Antao T, Luikart G (2014) Effects of overlapping generations on linkage disequilibrium estimates of effective population size. Genetics 197:769–780
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262
Weingarth K, Heibl C, Knauer F, Zimmermann F, Bufka L, Heurich M (2012) First estimation of Eurasian lynx (Lynx lynx) abundance and density using digital cameras and capture–recapture techniques in a German national park. Anim Biodivers Conserv 35:197–207
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Weissmannová H (2004) Ostravsko. In: Mackovčin P, Sedláček M (eds) Chráněná území ČR. AOPK ČR, EkoCentrum Brno, Praha (in Czech)
Wultsch C, Caragiulo A, Dias-Freedman I, Quigley H, Rabinowitz S, Amato G (2016) Genetic diversity and population structure of Mesoamerican Jaguars (Panthera onca): implications for conservation and management. PLoS ONE 11(10):e0162377
Zimmermann F, Breitenmoser-Würsten Ch, Breitenmoser U (2005) Natal dispersal of Eurasian lynx (Lynx lynx) in Switzerland. J Zool 267(4):381–395
Acknowledgements
The study was financially supported by the Operational Programme Environment through State Environmental Fund of the Czech Republic and by Nature Conservation Agency of the Czech Republic (project No. 9028766, 2011–2014), by EEA Grants (No. EHP-CZ02-OV-1-022-2015), Swiss-Czech Cooperation Programme, International Visegrad Fund, and by Institutional Research Plan (RVO: 68081766). We would like to thank PLA Administration Beskydy (especially Dana Bartošová and František Jaskula), ČSOP Salamandr (especially Petr Konupka) and to all Wolf patrols and other volunteers (namely Radek Červenka, Martina Dušková, Martin Gendiar, Lukáš Jonák, Martin Kraus, Leona Kutalová, Jiří Labuda, Beňadik Machciník, Barbora Telnarová, Vlado Trulík, Martin Váňa, Josefa Volfová) for their valuable help with sample collection, Miroslava Barančeková and Peter Vallo for help with DNA isolation and Anna Bryjová and Karel Janko for technical support in fragment analysis. Also we would like to thank Rory Putman for revision of English and all valuable comments and suggestions to the manuscript. Further, we thank the editor and three anonymous reviewers for their valuable suggestions that contributed to the improvement of final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Krojerová-Prokešová, J., Turbaková, B., Jelenčič, M. et al. Genetic constraints of population expansion of the Carpathian lynx at the western edge of its native distribution range in Central Europe. Heredity 122, 785–799 (2019). https://doi.org/10.1038/s41437-018-0167-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0167-x
This article is cited by
-
Connectivity patterns of bottlenose dolphins (Tursiops truncatus) in the north-east Mediterranean: implications for local conservation
Conservation Genetics (2024)
-
Ex situ versus in situ Eurasian lynx populations: implications for successful breeding and genetic rescue
Conservation Genetics (2023)
-
The role of the Caucasus, Carpathian, and Dinaric–Balkan regions in preserving wolf genetic diversity
Mammalian Biology (2023)
-
Radiographic pelvimetry in free-ranging Eurasian lynx (Lynx lynx carpathicus) from Switzerland
European Journal of Wildlife Research (2022)
-
Multi-seasonal systematic camera-trapping reveals fluctuating densities and high turnover rates of Carpathian lynx on the western edge of its native range
Scientific Reports (2021)
