
Abstract
The major histocompatibility complex (MHC) has long served as a model for the evolution of adaptive genetic diversity in wild populations. Pathogen-mediated selection is thought to be a main driver of MHC diversity, but it remains elusive to what degree selection shapes MHC diversity in complex biogeographical scenarios where other evolutionary processes (e.g. genetic drift and introgression) may also be acting. Here we focus on two closely related green lizard species, Lacerta trilineata and L. viridis, to address the evolutionary forces acting on MHC diversity in populations with different biogeographic structure. We characterized MHC class I exon 2 and exon 3, and neutral diversity (microsatellites), to study the relative importance of selection, drift, and introgression in shaping MHC diversity. As expected, positive selection was a significant force shaping the high diversity of MHC genes in both species. Moreover, introgression significantly increased MHC diversity in mainland populations, with a primary direction of gene flow from L. viridis to L. trilineata. Finally, we found significantly fewer MHC alleles in island populations, but maintained MHC sequence and functional diversity, suggesting that positive selection counteracted the effect of drift. Overall, our data support that different evolutionary processes govern MHC diversity in different biogeographical scenarios: positive selection occurs broadly while introgression acts in sympatry and drift when the population sizes decrease.
Similar content being viewed by others
Introduction
A long-standing goal in evolutionary ecology and conservation genetics is to understand the mechanisms that shape the genetic variation of wild populations (Hersch-Green et al. 2011). Even though putatively neutral markers are often used to capture the extent of this variation, neutral variation is not appropriate to understand all mechanisms that shape genetic variation (Kirk and Freeland 2011; Eizaguirre and Baltazar-Soares 2014). To understand how adaptive genetic variation is shaped in the wild, e.g., to which degree selection counteracts genetic drift, comparisons of selected and neutral markers across a range of populations with different biogeographic settings are needed. The major histocompatibility complex (MHC) is frequently found to be influenced by selection (e.g. Sommer 2005) and is therefore ideal to assess how populations respond to selection in different biogeographic settings.
The genes of the MHC loci encode cell-surface glycoproteins that are involved in the adaptive immune response of jawed vertebrates, by presenting self- and pathogen-derived antigens to T cells (Klein 1986; Murphy and Weaver 2016). As a consequence of multiple gene duplication events, MHC polymorphism within an individual can define a carrier’s resistance/susceptibility to parasitic diseases (Murphy and Weaver 2016). Experiments have shown that the MHC responds directly to parasite mediated selection (Lohm et al. 2002; Eizaguirre et al. 2012a, 2012b; Cornwall et al. 2018) and that populations adapt to local pathogen composition through differential shifts in MHC allele frequency (Eizaguirre et al. 2012a, 2012b; Cornwall et al. 2018). MHC allele frequency shifts therefore have an adaptive value, and the identification of the genetic mechanisms and processes shaping MHC diversity is directly relevant to the potential for conservation of adaptive genetic variation (Miller et al. 2010; Radwan et al. 2010; Ejsmond and Radwan 2011).
Although parasite-mediated selective forces are important for preserving MHC functional variation in the wild, other processes, including recombination, gene conversion, gene flow, introgression, and genetic drift also affect MHC variation and interact with selection (Hedrick 2002; Spurgin et al. 2011; Nadachowska-Brzyska et al. 2012; Hedrick 2013). Isolated populations with small effective population sizes are particularly susceptible to stochastic processes such as genetic drift (Jordan and Snell 2008; Hurston et al. 2009). Genetic drift can decrease neutral genetic diversity, but whether and under what conditions drift affects adaptive/functional diversity in the wild is not well understood. Whereas some studies have found that drift influences MHC functional diversity (e.g. Miller et al. 2010; Strand et al. 2012), others fail to find such an effect in spite of strong bottlenecks (e.g. Aguilar et al. 2004; Oliver and Piertney 2012; Vlček et al. 2016). Introgression, which may occur when closely related, occasionally hybridizing, species occur in sympatry, also affects MHC diversity (Nadachowska-Brzyska et al. 2012; Hedrick 2013). To gain an understanding of how selection, drift, and species interactions affect adaptive diversity, studies of a wide variety of populations with different biogeographic settings and population sizes are needed. A combination of mainland and island populations are particularly promising since they experience different types of ecological conditions (e.g. barriers and habitat area) that differentially influence the equilibrium between genetic drift and gene flow. Strong drift and little gene flow lower the genetic diversity of insular populations and promotes population genetic divergence (Jordan and Snell 2008; Hurston et al. 2009; Runemark et al. 2013). Because MHC diversity is linked to the capacity of species and populations to buffer against pathogen challenges (Sommer 2005), evaluating the relative role of selective forces and other evolutionary processes, such as genetic drift and introgression, in shaping MHC variability is of particular importance.
The aim of our study was to investigate the effect of different biogeographic settings and population size on MHC class I variation, using two closely related green lizard species (Lacerta trilineata and Lacerta viridis) from mainland and insular Greece, for which biogeography and evolutionary history are well studied (details are available in SI Appendix, SI Materials and Methods, Section SI.1) (Böhme et al. 2007; Sagonas et al. 2014). This pair of green lizards is an excellent study system as there are replica populations from mainland and insular settings with varied population size, different degree of isolation, and co-existence with the other species. To address our questions, we used MHC data in combination with neutral microsatellite markers. We expected higher levels of shared MHC alleles and MHC introgression in sympatric populations as this is where the species meet and may hybridize, but also due to advantage selection imposed by shared pathogen assemblies (Nadachowska-Brzyska et al. 2012; Phillips et al. 2018). When determining the level of MHC introgression in sympatric populations, we are also evaluating the degree of shared ancestral polymorphism (so-called trans-species polymorphism; Klein 1987) as well as evidence for potentially convergent evolution (Kriener et al. 2000; Srithayakumar et al. 2012). Furthermore, we predicted that the geographically isolated island populations will have lower MHC diversity and greater MHC structure compared to mainland populations due to restricted gene flow. Lower MHC allele polymorphism on islands may also be the result of weaker pathogen-mediated selection pressure due to the lower pathogen diversity prevailing on islands (Wikelski et al. 2004; Roca et al. 2009; Vlček et al. 2016). To address this, we directly compared the effects of insularity on the diversity of the adaptively and neutrally evolving loci.
Materials and methods
Samples
We analyzed 237 specimens, covering the whole distribution range of L. trilineata and L. viridis in Greece (Fig. 1) as well as 13 specimens (5 and 8, respectively) from the northernmost range of L. trilineata distribution in the Balkans. Approximately 20 specimens used in the study were not successfully genotyped, either for MHC or microsatellites, and were excluded from the respective datasets (SI Appendix, Table S1). This yielded 4–23 individuals from each population (see Table 1 for number of specimens per population). Specimens were classified as belonging to L. trilineata or L. viridis based on morphological data (Nettman 2001). When this was not possible, given the subtle morphological differences observed between individuals of the two species (Rykena et al. 2002), molecular data [two mitochondrial (cytochrome b and 16S rRNA) and one nuclear (NKTR) genes] and phylogenetic analyses were used (Sagonas et al. 2014).

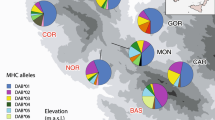
Sampling localities for Lacerta viridis and Lacerta trilineata. Black filled circles indicate the total number of MHC class I alleles found in each population of L. trilineata (i.e. 100%). Red filled circles denote the percent of shared alleles between L. trilineata and L. viridis. MHC allele similarity between species is differentiated along a north–south gradient, with northern populations of the two species sharing more MHC alleles compared to southern and insular ones, where L. viridis is present in low numbers or even absent
Neutral nuclear markers
Microsatellite genotyping
All samples were genotyped for 11 lacertid microsatellite loci (Boudjemadi et al. 1999; Nembrini and Oppliger 2003; Pinho et al. 2004; Poulakakis et al. 2005; Huyghe et al. 2009; Richard et al. 2012). Each locus was amplified separately, and then pooled for genotyping (SI Appendix, Table S2 for locus-specific annealing temperature and multiplexing schemes). STRand software v.2.4.110 (Toonen and Hughes 2001) was used to determine genotypes, while the binning of alleles was accomplished using FLEXIBIN 2 (Amos et al. 2007) in an attempt to minimize microsatellite allele miscalling.
Neutral markers: genetic diversity and population structure analysis
For each population, we calculated measures of genetic diversity in the form of expected heterozygosity (He), observed heterozygosity (Ho), and allelic richness (Ar) using GENETIX (Belkhir et al. 2000). Furthermore, we estimated the pairwise population differentiation (FST) between sampling localities in Arlequin v.3.5 (Excoffier and Lischer 2010) using 10 000 permutations to assess the statistical significance of the estimated values.
To assess population structure based on microsatellite markers of L. trilineata and L. viridis we used Bayesian clustering approach implemented in STRUCTURE 2.3.4 (Pritchard et al. 2000). Analyses were run assuming admixture with correlated allele frequencies, and the number of genetic clusters in data, K between 1 and 23 were evaluated. We used 100 000 Markov chain Monte Carlo (MCMC) repeats and discarded the first 50 000 as burn-in while five separate runs for each K value were used to ensure stationarity. Evanno’s test (Evanno et al. 2005) was applied to infer the most likely number of genetically homogeneous clusters in the dataset. STRUCTURE was run for the complete dataset and only for sympatric mainland populations of the two species in order to detect hybrids, since island populations are differentiated due to their isolation and this would make STRUCTURE discriminate them first. Moreover, we conducted an analysis of molecular variance (AMOVA) in Arlequin v.3.5 (Excoffier and Lischer 2010) to partition microsatellite genetic variation into hierarchical structure (populations nested within species). To directly compare microsatellite with MHC data, microsatellite data were binary-encoded (Herdegen et al. 2014). Significance of AMOVA components was tested with 10 000 permutations.
Effects of sympatry on neutral diversity
We performed Mann-Whitney U test to compare He, Ho, and Ar between mainland populations of the two species using both the dataset with all specimens and the one after removing hybrids to exclude gene flow effects. To explore whether geographic distance explains populations structure, we run Mantel’s test using the FST (SI Appendix, Tables S3 A and B). Under neutral evolution, genetic differentiation among continuous populations is expected to increase linearly with geographic distances (i.e. isolation by distance) (Wright 1943). However, to control for nonlinear relationship and the irregular distribution of populations across space (Borcard and Legendre 2012; Diniz-Filho et al. 2013), we also conducted a Mantel correlogram and investigated the relationship between genetic and geographic distances across space. Mantel correlogram was performed using the function mantel.correlog() of the vegan package (Oksanen et al. 2013) in R v.3.4 (R Development Core Team 2015), with Pearson correlation, fdr correction, and 1000 permutations.
To test for evidence of gene flow, we applied multiple approaches. First, we used t-test and compare the average pairwise FST values between sympatric (northern) and allopatric L. trilineata populations with northern L. viridis populations. Second, we assign individuals (“real” genotypes) from simulated data doing the following: we addressed the possibility of inter-specific hybridization in mainland sympatric populations in Greece (Epirus, Macedonia, Thessaly, and Thrace) and Serbia (Belgrade), a model-based Bayesian approach was used as implemented in NEWHYBRIDS v.1.1 (Anderson and Thompson 2002). We used a threshold value (q) of 0.8 for assignment of each specimen to a certain hybrid category (i.e. two pure parental populations corresponding to the two species that hybridize, their F1 hybrids as well as the F2 hybrids and backcrosses of F1 hybrids with either parental population). Furthermore, based on STRUCTURE results when only mainland populations were considered, “pure”, non-hybridized L. trilineata and L. viridis individuals were defined only those with q > 0.80, and these were used to simulate pure parental and hybrid classes of the two species in HYBRIDLAB (Nielsen et al. 2006). With HYBRIDLAB we simulated a dataset of 500 of each of the classes L. trilineata-parental, L. vidiris-parental, F1 hybrids, F2 hybrids, backcrosses with L. trilineata, and backcrosses with L. viridis. We then ran NEWHYBRIDS again using the same settings as before to estimate the posterior probability of an individual falling into one of the following categories: (i and ii) pure parental type (P1 and P2, respectively for each species), (iii) first-generation hybrids (F1), (iv) second-generation hybrids (F2), and (v and vi) backcrosses to either parental-type direction (BC-P1 and BC-P2). The analysis was performed using default genotype frequency classes with 100 000 iterations as a burn-in phase and 600 000 iterations post burn-in. Two independent runs with different starting points were performed for both for “Jeffreys-like” prior and the Uniform prior.
Estimate migration rates
To investigate the rates of gene flow among sympatric populations of the two species (Epeirus, Thessaly, Macedonia, Thrace, and Serbia) we used Migrate-n (Beerli and Felsenstein 2001). Four replications were performed for each run. The priors of the parameters θ and Μ were the same for all models, while a variable mutation rate amongst loci was considered. Migrate-n was run with the Brownian motion microsatellite model to estimate the parameters using Bayesian inference (BI). The uniform prior of θ ranged between 0 and 200, with mean 100 and delta equals to 20. The uniform prior for Μ remained as default. The running parameters included a long chain of 10 000 recorded steps, with an increment every step of 100. Parameter space was searched using static heating (temperatures: 1.0, 1.5, 3.0, 1 000 000).
Effects of insularity on neutral diversity
We used Mann-Whitney U test for the whole dataset and after excluding hybrids to assess the effects of genetic drift on neutral diversity measures (He, Ho, and Ar) between island (Andros, Crete, Corfu, Evvoia, Kythira, Lesvos, Milos, Rhodes, and Skyros) and mainland (Peloponnese, Thessaly, Sterea Ellada, Epirus, Macedonia, Thrace, and Serbia) L. trilineata populations. Likewise, we compared pairwise FST values between mainland and island populations using Mann-Whitney U test. We also performed linear models to assess the significance of population size and island isolation on neutral genetic diversity, using the lme4 package (Bates et al. 2015) in R v.3.4 (R Development Core Team 2015). To do so, we used the size of island (i.e. area, a proxy of population size; log-transformed) and Euclidean distance from the mainland (a proxy of gene flow barrier; log-transformed) as fixed effects (Table 1). As response variables, we used the estimates of genetic diversity (Ar, Ho, and He). To measure collinearity between island size and distance from mainland we used the variance inflation factor (VIF), which assesses the magnitude of variance increase when predictors are correlated. Our findings indicate that no correlation exists between these two factors (VIF = 1.11).
Adaptive (MHC class I) nuclear markers
DNA extraction, primer design, amplification, sequencing, and filtering of the MHC class I
Ethanol-preserved tongue or tail tip tissue samples were extracted with an ammonium acetate protocol (Richardson et al. 2001). We developed, using 11 lacertid lizards, degenerate F- and R-pyrosequencing primers that amplify MHC alleles for a variety of lacertid taxa (see SI Appendix, SI Materials and Methods, Section SI.2 for primers design information). We sequenced the target fragments of MHC I exons 2 (228 bp) and 3 (200 bp) that contain parts of the α1 and α2 domains, respectively, of the antigen-binding region of the MHC class I molecule (Miller et al. 2006; Wallny et al. 2006) using 454 GS Titanium technology. The target-specific primers used in PCR amplifications and the size of the amplicons are given in SI Appendix, Table S3. To estimate genotyping repeatability, a sub-sample of 107 samples out of the 233 amplified specimens (Supplementary Table S1) for the MHC class I genes were sequenced in two independent PCR reactions (SI Appendix, SI Results, Section SI.1) (c.f. Zagalska-Neubauer et al. 2010).
We used AmpliSAS (Sebastian et al. 2015) to de-multiplex the 454 sequences, screen for artifacts, and to assign sequence variants to individuals. The output from AmpliSAS was then manually evaluated to further remove low-quality variants (Phred quality score lower than 30), false MHC sequences due to PCR or sequencing artifacts (point mutations due to Taq polymerase and PCR chimeras), and potentially non-functional variants (see SI Appendix, SI Materials and Methods, Section SI.3 for further details). To investigate the differences in MHC diversity between species and populations, all unique MHC sequence variants detected were treated as different alleles, even though they may stem from different loci. Variants of exon 2 and exon 3 were aligned with tuatara (Sphenodon punctatus; Sppu-U*01, GenBank accession number DQ145788) (Miller et al. 2006) and a chicken species (Gallus gallus; B-F alpha chain, GenBank accession number Z54321) (Wallny et al. 2006) sequence to identify antigen-binding and non-antigen-binding sites (SI Appendix, Figure S1).
Genetic diversity and population structure of the MHC
MHC allele numbers and diversity were calculated separately for the different exons. For each population, four measures of MHC diversity were estimated using DnaSP v.5.10 (Librado and Rozas 2009). (1) Total number of unique alleles per population (Na), (2) average nucleotide diversity (π), (3) the average number of alleles per individual (NaI), and (4) the average functional diversity of MHC I as the mean intra-individual evolutionary distance between amino-acid variants (D) using a p-distance model in MEGA5 (Tamura et al. 2011). Furthermore, we calculated genetic distances for exon 2 and exon 3 alleles, in MEGA v 5.05 (Tamura et al. 2011), using the Tamura and Nei model of nucleotide substitution (Tamura and Nei 1993). Finally, given the positive association between the number of unique alleles and number of specimens sampled (exon 2: F1,21 = 9.82, r2 = 0.32, P = 0.005; exon 3: F1,21 = 3.67, r2 = 0.15, P = 0.069), we estimated MHC allele richness (MHCAr) using rarefaction with the hierfstat package (Goudet 2005) in R.
Signatures of selection on the MHC
To test for the mode of evolution, we estimated nonsynonymous (dN) and synonymous (dS) substitutions using DNAsp v.5.10 (Librado and Rozas 2009) and performed a Z-test implemented in MEGA5 (Tamura et al. 2011), using the Nei-Gojobori method of pairwise comparison and applying the Jukes-Cantor correction (Nei and Gojobori 1986). Given that sites encoding amino acids interacting with parasites are expected to be under positive selection, we performed those analyses separately for the antigen-binding region and the whole sequence.
We used the CODEML (codon-based approach) algorithm available in PAML 4.8 package (Yang 2007) to identify signs of codon-specific positive selection, using a likelihood ration test and the maximum likelihood (ML) phylogenetic trees of exon 2 and exon 3. Two pairs of site models, nomenclature as in CODEML, were used (Wong et al. 2004). The simplest one included models M1a (nearly neutral gene evolution) and M2a positive selection with ω ≥ 1 (dN/dS ratio) (Wong et al. 2004), while the more complex one included models M7, beta distributed variable selective pressure, and M8, beta plus ω (Yang et al. 2000). Positive selected sites in M2a and M8 models were inferred from posterior probabilities using the Bayes Empirical Bayes procedure (Yang et al. 2005). In addition, we used mixed-effects model of evolution (MEME) (Murrell et al. 2012) to test for sites that experience episodic events of selection, i.e. sites that are not subjected to pervasive selection or affecting only a small subset of lineages. This model allows the distribution of ω to vary between sites (fixed effects) and between branches (random effects).
MHC intra exon recombination
Historical recombination events often play significant role in MHC evolution and diversity (Reusch and Langefors 2005), but can significantly bias phylogenetic analyses (Schierup et al. 2001). We examined if there was evidence for intra-fragment recombination using the algorithms GeneConv (Padidam et al. 1999), MaxChi2 (Smith 1992), and RDP (Martin and Rybicki 2000), as implemented in rpd4 (Martin et al. 2015). Those methods were chosen first, because they have been shown to perform well in recognizing recombination events when present and not claiming recombination when absent, and second, because they detect different patterns (i.e. a single method cannot detect all recombination events). Because the number of tests during an exploratory recombination screen increases exponentially with the number of sequences (Martin et al. 2010), we used 30 subsets of 35 sequences randomly chosen from our initial dataset. Bonferroni corrections for multiple tests were applied. We took a conservative approach and considered an event as true recombination if more than one method supported it.
Trans-species polymorphism, introgression, and convergent evolution
To test for evidence of trans-species polymorphism (i.e. the sharing of functionally important MHC alleles between species due to preservation of ancient allelic lineages) (Klein 1987) for exon 2 and exon 3 variants, we constructed their phylogenies using ML and BI methods (details on phylogenetic analyses are available in SI Appendix, SI Materials and Methods, Section SI.4) after removing hybrid individuals and tested for phylogenetic inconsistencies between MHC gene trees and gene trees based on other part of the genome.
Following similar approaches as with neutral genetic markers we tested for MHC gene flow. An AMOVA with populations nested within species in Arlequin (Excoffier and Lischer 2010) using 10 000 permutations and calculated FST values between all pairs of populations was used. To assess populations’ MHC genetic structure, we ran STRUCTURE as described for neutral loci above and estimated the number of MHC genetic clusters amongst sampling locations, both for the complete dataset and for sympatric mainland populations only. Data were binary-encoded with the presence/absence of an allele coded for as 1⁄0 treating each allele as a separate dominant locus as for AFLP datasets (c.f. Nadachowska-Brzyska et al. 2012).
To investigate the differences of MHC allele pools amongst sympatric and allopatric populations of the study species, we compared the abundance of the different exon variants with analysis of similarity (ANOSIM) using the Bray-Curtis algorithm and 10 000 permutations. We used Bray-Curtis similarity matrix (SI Appendix, Table S2C) to test for isolation by distance in MHC similarity across populations using a Mantel’s test and Mantel correlogram with 1000 permutations as described before. Furthermore, we used Welch’s t-tests to compare adaptive diversity indices (MHCAr, NaI, π, and D) between mainland populations of the two species. Fisher’s exact test was used to test for differences in the number of shared MHC alleles between sympatric and allopatric populations of the two species. All analyses were carried out in R v.3.4 (R Development Core Team 2015).
Due to the similar pathogen assemblies that sympatric L. trilineata and L. viridis populations may experience, we tested whether convergent evolution could decrease the inconsistency between allelic genealogies. To test for convergence, we examined whether (i) identical codons or (ii) certain amino acids but not codons, are maintained at the antigen-binding region between sympatric populations of the two species. We compared the likelihood of two scenarios. The first scenario gives support for trans-species polymorphism and suggests that codon similarity of MHC I exon 2 and exon 3 between sympatric populations of L. trilineata and L. viridis is significantly higher than expected by chance. The second scenario supports convergent evolution, identified by highly similar amino acids and with codon identity of antigen-binding sites following the overall expected frequency. To test which of these two scenarios is most likely, we performed a random permutation sampling. Based on the fact that the genetic code is degenerated, we estimated the expected codon identity for each species and compared with the observed codon identity between species. We performed the analysis using Perl scripts provided by Lenz et al. (2013a, 2013b). This analysis was carried out, including sympatric populations of the two species and random combinations of allopatric populations.
Effects of insularity on adaptive diversity
Welch’s t-test was carried out to evaluate the effects of drift on genetic and functional diversity of MHC (MHCAr, NaI, π, and D) between mainland (Peloponnese, Thessaly, Sterea Ellada, Epirus, Macedonia, Thrace, and Serbia) and island (Andros, Crete, Corfu, Evvoia, Kythira, Lesvos, Milos, Rhodes, and Skyros) populations of L. trilineata. We used a re-sampling approach to estimate whether the observed nucleotide diversity (π) and amino-acid divergence (D) on islands was higher than expected. To achieve this, we performed re-sampling using data from the mainland populations. Twenty sequences were randomly selected to match the average number of unique MHC alleles found on islands (Table 1) and this was repeated to create 100 subsets. T-test was then conducted to examine differences between expected and observed π and D. To explore the effects of drift on MHC adaptive diversity we run Mantel’s test for the pairwise FST values and the pairwise Bray-Curtis similarity (SI Appendix, Tables S3). We also performed linear models as aforementioned to test the predictive value of population size and distance from the mainland on MHC variation (e.g. Vlček et al. 2016). Estimates of MHC genetic diversity were used as response variables and population of origin as a random effect. T-test was used to compare pairwise FST values between island–mainland and mainland–mainland L. trilineata populations. Lastly, to better indicate the differences of MHC allele composition among islands, we conducted a principal component analysis.
Results
Neutral nuclear markers
Genetic diversity and population structure for neutral markers
The comparison of neutral diversity indices between mainland populations of L. trilineata and L. viridis showed no differences (for the whole dataset: Ar: W = 24.5, P = 0.839; He: W = 25, P = 0.797; and Ho: W = 13, P = 0.240; and excluding hybrids: Ar: W = 24.5, P = 0.558; He: W = 20, P = 0.999; and Ho: W = 15, P = 0.524). In contrast, we found significantly lower microsatellite allelic richness (Ar: W = 12, P = 0.013; after correcting for hybrids: W = 14.5, P = 0.043) and expected heterozygosity (He: W = 12, P = 0.011 and after hybrid removal W = 14.5, P = 0.044), but not observed heterozygosity (Ho: W = 32, P = 0.48 or W = 24, P = 0.268), in island L. trilineata populations compared to their mainland conspecifics (Table 1).This loss of heterozygosity (expected and observed) on islands was related to distance from the mainland but not island size (Table 2). Microsatellite genotypes are available in SI Appendix, Table S5.
Pairwise FST between species ranged from 0.017 to 0.450 (SI Appendix, Table S4A), with pairwise differences for island–mainland comparisons (from 0.022 to 0.450, mean: 0.224) being higher (t = 4.34, df = 88, P < 0.001) than those for mainland–mainland comparisons (from 0.017 to 0.297, mean: 0.140). Interestingly, sympatric populations of L. trilineata and L. viridis had lower genetic differentiation (ranged from 0.017 to 0.297; mean: 0.141) compared to allopatric population pairs of the two species (0.017 to 0.450; mean: 0.204) (t = 3.29, df = 88, P = 0.001). A Mantel’s test indicated that this pattern of genetic differentiation between mainland populations (Peloponnese, Sterea Ellada, Thessaly, Epirus, Macedonia, Thrace, and Serbia) (Mantel’s test; r = 0.30, P = 0.012) but not between island populations (Mantel’s test; r = 0.06, P = 0.37) complied with isolation-by-distance expectations (SI Appendix, Table S4 A and B). Furthermore, and in agreement with the Mantel’s test, Mantel correlogram revealed a spatial correlation of genetic and geographic distances in almost all cases (Table 3).
AMOVA, with populations nested within species, for binary coded microsatellite data revealed that species and populations explained similar amounts of variance (12.3%; P < 0.001 in both cases) (Table 4). When sympatric and allopatric L. trilineata and L. viridis populations were used as the highest level of hierarchical clustering we obtained similar results (Table 4).
NEWHYBRIDS results suggested substantial genetic exchange between L. trilineata and L. viridis and patterns of unidirectional or asymmetrical introgression from L. viridis to L. trilineata, revealing several F2 hybrid individuals from Macedonian, Thracian, and Serbian populations (northern Greece; SI Appendix, Table S6), where both species occur in sympatry. However, the comparison of genetic diversity measures (Ar, He, and Ho) on both data (with or removing hybrids) between sympatric and allopatric L. trilineata populations showed that this genetic exchange did not lead to an increase in neutral genetic diversity (all P > 0.05, SI Appendix, Table S7).
STRUCTURE analyses (for the complete dataset and for sympatric mainland populations of the two species only; Fig. 2 and SI Appendix, Figure S2A) for K = 23 suggested the presence of two clusters, one comprising of L. trilineata and the second of L. trilineata and L. viridis (SI Appendix, Table S8 A-B and Figure S2 A).

Genetic structure of Lacerta trilineata and L. viridis (Lv on the map) populations inferred by STRUCTURE analysis for K = 2 (best K for microsatellite data; SI Appendix, Table S1). Pie charts on the map display the results of MHC class I structure for each population, based on the average ancestry proportions for all individuals in a population. Structure plot at the bottom displays STRUCTURE results for microsatellites, where the two clusters are highlighted (one comprising of L. trilineata and the second of L. trilineata and L. viridis). Population abbreviations as in Table 1. Pie charts that are not indicated as Lv refer to Lacerta trilineata
Consistent with the abovementioned results, Migrate-n revealed that migration events are quite high. In particular, the mean value of estimated migration rates from L. trilineata to L. viridis was 0.37 (Thessaly: 0.37; Epirus: 0.41; Macedonia: 0.32; Thrace: 0.35; and Serbia: 0.43), whereas the mean rate from L. viridis to L. trilineata was 0.39 (Thessaly: 0.39; Epirus: 0.39; Macedonia: 0.36; Thrace: 0.40; and Serbia: 0.40).
Adaptive (MHC class I) nuclear markers
Genetic diversity of the MHC
Pearson correlation revealed that the mean number of MHC class I alleles per individual (NaI) and the total number of MHC alleles per population (Na) were positively correlated with microsatellite allelic richness (Ar) and expected heterozygosity (He). However, neither π or D was significantly correlated with neutral genetic diversity indices (SI Appendix, Table S9).
Of the 228 aligned sites examined for exon 2, 162 were variables and 140 were parsimoniously informative, whereas for exon 3 we found 141 variable sites and 116 parsimoniously informative sites out of 200 aligned base pairs. The TN93 (Tamura and Nei 1993) genetic distances between populations of L. trilineata and L. viridis are provided in SI Appendix, Table S10. MHC class I divergence estimated with TN93 for the whole data (or hybrid in parenthesis) between the two species ranged from 11.1% (11.0%) to 15.2% (15.2%) for exon 2 and from 16.8% (16.8%) to 19.4% (19.4%) for exon 3. This high sequence divergence was further reflected between populations within species [9.4% (9.2%) to 15.4% (15.4%) for exon 2 and from 15.8% (15.7%) to 19.1% (19.5) for exon 3 for L. trilineata, and 12.9% (13.1%) to 14.3% (14.4%) for exon 2 and from 17.8% (17.9%) to 18.4% (18.5%) for exon 3 for L. viridis)]. Additional details on MHC diversity are available in SI Appendix, SI Results, Section SI.2.
Signatures of selection and recombination
The overall nucleotide diversity for exon 2 and exon 3 was 2.99 and 1.96 times higher at antigen-binding sites than at non-antigen-binding sites, respectively. Although nonsynonymous substitutions showed significant excess at the antigen-binding sites of exon 2 (Z = 1.84, P = 0.034), this was not the case for exon 3 (Z = 0.76, P = 0.452). We found no evidence for positive selection using whole sequences for either exon (SI Appendix, Table S11). However, the likelihood ratio test in comparing models of codon evolution indicated that models integrating positive selection (M2a and M8) fitted significantly better to the observed allelic variation of our data than those that did not (M1 and M7; Table 5). CODEML found evidence of positive selection at 18 sites, of which 13 occur in the antigen-binding region. Almost 90% of sites identified by CODEML were also identified by MEME, but MEME suggested 28 total sites (i.e. 10 additional) that had experienced episodic events of positive selection (Table 5). Although defining ABS and non-ABS based on crystallography data from distantly related species brings several limitations, since MHC genes and especially ABS are known to vary greatly between species, the findings from CODEML and MEME that the pre-defined ABS overlap with the detected sites under selection.
Historical recombination events seem to be a significant source of genetic variation at the MHC class I loci in Lacerta species as we detected one to three recombination events in almost half of the 30 data subsets for both exons examined.
Introgression, trans-species polymorphism, and convergent evolution
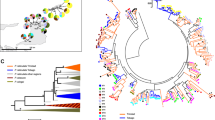
The high degree of MHC class I genetic diversity lead to poorly resolved branches in all phylogenetic trees (ML and BI) for both exons. We did not find a clear pattern of paralog clustering or species/lineages clustering for exon 2 and exon 3 (Fig. 3; only BI results are shown since also ML showed similar results). Interestingly, sympatric populations of the two species (central region: ~44%; Sterea Ellada, and Thessaly; northern region: ~53%; Epirus, Macedonia, Thrace, and Serbia) shared more MHC alleles than allopatric (southern region: 31%; Peloponnese; islands: 21%) ones (chi-square test, for all pairwise comparisons P < 0.05) (Figs. 1, 2). Therefore, the lower average pairwise FST between northern L. trilineata and L. viridis (0.204 ± 0.07) compared to allopatric L. trilineata and L. viridis (0.275 ± 0.10) comes as no surprise (t = 3.54, df = 88, P < 0.001). Furthermore, AMOVA (Table 4) revealed that MHC variation for populations in sympatry (4.67%, P = 0.08) was not significant, but it was highly significant for allopatric L. trilineata and L. viridis (7.13%, P < 0.001). Furthermore, population hierarchical level (i.e. geography) explained almost 3.5 times more variance than region did for all groups.

Bayesian inference trees illustrating the phylogenetic relationships of a exon 2 variants and b exon 3 variants using all sites. Asterisks indicate posterior probabilities higher than 0.75. Black dots denote outgroup sequences. Black lines indicate alleles that were only found in Lacerta trilineata, blue lines for L. viridis, whilst red lines identical alleles that are shared among species
Structure analyses for L. trilineata and L. viridis suggested the presence of five MHC clusters often from both L. trilineata and L. viridis populations (Fig. 2), though island populations had greater structure than mainland conspecifics (Fig. 2 and SI Appendix, Table S12 and Figure S2B). Genetic admixture was inferred in sympatric populations with L. trilineata and L. viridis from the northern and central parts of the mainland distribution. Remarkably, L. trilineata populations from Macedonia, Thrace, and Serbia exhibited extremely high admixture from genetic clusters found in L. viridis and vice versa. Furthermore, MHC allele pools composition differed significantly among populations (ANOSIM; r = 0.22, P < 0.001). Mantel’s test and Mantel correlogram (Table 3) between MHC allele pool compositions revealed isolation by distance, where geographically closer mainland populations had more similar allele pools (r = 0.61, P < 0.001).
We did not find significant evidence of convergent evolution at the antigen-binding region between sympatric populations of L. trilineata and L. viridis (Thessaly, Epirus, Macedonia, Thrace, and Serbia). For exon 2, we found that 45 132 out of 82 134 amino acids in the antigen-binding region analyzed were identical between species, 42 657 of which were coded by the same codon. These numbers were higher than the expected codon similarity under a scenario of a common ancestor (i.e. trans-species polymorphism; 39 104 ± 70; P < 0.001) and much higher than the expected codon similarity under convergent evolution (25 487.04 ± 99; P < 0.001). Similar results were obtained for exon 3: 31 802 out of the 33 460 pairs of identical amino acids were coded by identical codon, which was closer to the common ancestor scenario (30 362.97 ± 51; P < 0.001) than the convergent evolution scenario (21 934.64 ± 71; P < 0.001). The comparison between pairs of allopatric populations revealed similar results; observed codon similarity in the antigen-binding region fits better with a common ancestor scenario (for exon 2: out of 224 640 amino acids, 120 856 were identical between species and 114 889 coded by the same codon and for exon 3 we found 100 708 identical amino acids of which 95 273 were coded by the same codon).
Effects of insularity on MHC class I diversity
The comparison between mainland and island populations of L. trilineata revealed significant differences in MHC class I allele diversity (Fig. 4). In particular, MHCAr (t = −5.51, df = 33.1, P < 0.001) and NaI (t = −4.51, df = 33.99, P < 0.001) were higher for mainland than island populations, whereas estimates of π (t = −0.20, df = 33.49, P = 0.841) and D (t = −0.61, df = 33.44, P = 0.543) did not differ between groups. Interestingly, the re-sampling analysis showed that the observed nucleotide diversity (t = 3.06, df = 397.45, P = 0.002) and amino-acid divergence (t = 4.27, df = 396.63, P < 0.001) on islands were higher than expected by chance.

MHC diversity indices between mainland and island populations of Lacerta trilineata. MHCAR MHC allele richness, NaI the average number of alleles per individual, π the average nucleotide diversity, D the mean intra-individual evolutionary distance between amino-acid variants. We also provide the correlation between MHCAr and size of the island and distance from the mainland for insular L. trilineata populations. Green line refers to the best-fit line
MHCAr (a measure of population MHC genetic diversity) was significantly positively correlated with the size of the island, and negatively with the distance from the mainland (Table 2 and Fig. 4), whereas nucleotide diversity per site (π) and allele amino-acid distance (D) were not affected by island size (all P-values > 0.05) or island isolation (all P-values > 0.05). We found no relationship between the number of MHC variants per individual (NaI; a measure of individual MHC genetic diversity) and island size (F1,6 = 0.05, P = 0.825) or distance from the continent (F1,6 = 0.44, P = 0.533) (Table 2). Mantel’s test between MHC similarity and pairwise FST showed no significant correlation (r = 0.23, P = 0.125).
As expected under a drift scenario, the structure analysis showed that island L. trilineata populations had greater MHC structure and were more differentiated from mainland populations than mainland populations were from each other (Fig. 2). In line with this, the MHC allele divergence was higher between island–mainland pairwise comparisons compared to mainland–mainland pairs (t-test; t = 3.96, df = 151, P < 0.001) (SI Appendix, Table S4C).
Discussion
We found that positive selection, introgressive hybridization, and genetic drift have jointly molded MHC class I diversity in these two species of green lizards. In particular, the high MHC polymorphism, the high nucleotide diversity on ABR, and the pattern of trans-species polymorphism suggest that positive selection has been a significant force shaping MHC diversity across island and mainland populations. Introgressive hybridization, on the other hand, has contributed significantly to the increased sharing of allelic lineages and polymorphism in sympatric populations. In island populations, we observed a reduction in neutral genetic diversity and MHC allele richness and polymorphism but found no effect on MHC amino-acid and nucleotide diversity. This is consistent with a scenario where positive selection has counteracted the effects of drift on islands. We also detected strong support for recombination as a source of MHC diversity in this study species complex.
Adaptive genetic diversity in mainland populations
The comparison of MHC diversity between sympatric populations of the two species showed that L. viridis populations had higher MHC allelic richness and MHC allelic polymorphism compared to L. trilineata (SI Appendix, Supplementary Results, Section SI.2). MHC nucleotide diversity and amino-acid distance between alleles, however, showed no differences (SI Appendix, Supplementary Results). The higher number of MHC alleles in L. viridis populations could be potentially related to different evolutionary histories of the two species in southern Balkans (c.f. Nadachowska-Brzyska et al. 2012). Phylogenetic and phylogeographic studies on L. viridis have showed that its current distribution in the southern Balkans is the result of several postglacial migration events of distinct isolated lineages that were restricted to several microrefugia during the last glaciations within Europe (Godinho et al. 2005; Böhme et al. 2007). In contrast, L. trilineata rapidly dispersed into the Balkans from Anatolia (Sagonas et al. 2014). It is possible thus that the past population structure of L. viridis has lead to the relatively high MHC allele richness we observe among contemporary L. viridis populations. An alternative explanation is provided by the results from hybridization experiments (Rykena 1996; Rykena 2001), as well as sequence and allozyme data (Amann et al. 1997; Joger et al. 2001; Keller and Vassilakaki 2002; Godinho et al. 2005), which suggest hybridization and gene flow between L. viridis and L. bilineata (another green lizard species) in southern Balkans (Joger et al. 2001; Mayer and Beyerlein 2002). Hence, it is possible that historical population structure, and/or inter-specific gene flow from a third lizard species, is responsible for the comparatively high level of MHC diversity in L. viridis.
We found inconsistent MHC allelic genealogies and extensive allele sharing between L. viridis and L. trilineata. In addition, the highly isolated insular population of Crete showed high levels of allele sharing with L. viridis. Positive selection can maintain such trans-species polymorphism (Klein 1987; Těšický and Vinkler 2015; Gillingham et al. 2016). However, the degree of MHC allele sharing between populations of the two species was differentiated along a north–south gradient, where northern (Serbia, Thrace, Macedonia, and Epirus) and central (Thessaly and Sterea Ellada) populations of L. trilineata shared more MHC alleles with L. viridis than southern (Peloponnese) and insular populations, where L. viridis is absent (Figs. 1 and 2). Convergent evolution to a similar habitat/parasite community in sympatry could explain the north–south gradient pattern. But our codon-usage analysis showed that the similarity of motifs at the functionally relevant polymorphic sites at the MHC across sympatric populations were less likely due to convergent evolution but compatible with a scenario of common ancestor.
Most likely introgressive hybridization in sympatric regions has contributed (together with positive selection causing trans-species polymorphism) to the extensive MHC allele sharing that we observed. Introgression as a source of adaptive genetic diversity at multigene families has attracted recent interest (Nadachowska-Brzyska et al. 2012; Hedrick 2013; Grossen et al. 2014; Phillips et al. 2018). MHC introgression can provide novel alleles that confer an advantage for disease susceptibility and pathogen resistance (Wegner and Eizaguirre 2012; Phillips et al. 2018). Nettman (2001) suggested that these two Lacerta species can hybridize when in sympatry. Here ongoing hybridization and gene flow in sympatric populations of the two species is suggested by (i) the weak microsatellite structure, (ii) the significantly lower pairwise FST of sympatric populations, (iii) the presence of hybrid individuals, and (iv) the findings from Migrate-N. To add on, AMOVA indicated a nonsignificant between-group component of MHC variation in regions where the two species occur in sympatry (Table 4). Yet, it could be argued, given the evidence of admixture for microsatellites, that this MHC introgression is the result of incomplete isolation. Thus, whether adaptive hybridization contributes on MHC diversity needs further assessment. To do so, we tested the hypothesis that if hybridization has a selective advantage then MHC genes should introgress easier and more quickly than neutral markers (Schierup et al. 2000; Barton 2001). Our findings from the AMOVA with regions as higher levels of hierarchical clustering showed different patterns of deviation for microsatellite and MHC data in the north. As such, MHC genes but not microsatellite markers denoted lower differentiation among regions compared to among populations. The latter supports the scenario of preferential introgression (Nadachowska-Brzyska et al. 2012; Herdegen et al. 2014). Interestingly, this admixture signal that we observed in northern populations (Serbia, Thrace, and Macedonia; Fig. 2), together with the high number of F1 hybrids of L. trilineata backcrossed with L. viridis (SI Appendix, Table S6) indicate asymmetric gene flow, with a predominant direction from L. viridis to L. trilineata. In contrast, southern and insular populations of L. trilineata (i.e. populations further away from the distribution range of L. viridis) had more diverged MHC allele pools, suggesting lower levels of introgression, compared to northern populations, probably due to different parasitic communities and longer period of isolation.
Effects of genetic drift and selection
We found that microsatellite allelic richness and expected heterozygosity were considerably lower on islands, as expected due to genetic drift in isolated populations with small population size (Jordan and Snell 2008; Hurston et al. 2009). Moreover, island populations (especially the small ones) had lost a significant proportion of MHC alleles. However, despite this loss, the remaining MHC variants were highly diverged, and the average allele nucleotide diversity and amino-acid divergence were comparable to mainland conspecifics (Fig. 4), implying that the reduction of variants had not resulted in any loss of functional diversity. This suggests that despite strong genetic drift, selection for diverse functions maintains nucleotide and amino-acid divergence in insular L. trilineata populations (c.f. Alcaide 2010). As expected under a drift scenario, we also found inter-island variation in allele composition.
Pathogen-mediated selection is well known to counteract the effects of drift on MHC by maintaining diverse alleles that can recognize a broader spectrum of pathogens (Garrigan and Hedrick 2001; Hedrick 2002; Sutton et al. 2011). Several lines of evidence suggest that selection has shaped the high variation in MHC genes in L. trilineata. First, the rate of nonsynonymous substitutions exceeded significantly the synonymous rate in the antigen-binding region. Second, there was no correlation between microsatellite divergence (pairwise microsatellite FST) and the number of shared MHC alleles (pairwise Bray-Curtis similarity) across islands, and, third, we found no clear population MHC structure among island populations, as islands tended to form clusters based on their geographic area (SI Appendix, Figure S3). Mechanisms causing positive selection at the MHC include negative frequency-dependent selection (Takahata and Nei 1990), heterozygote advantage (Doherty and Zinkernagel 1975), and divergent allele advantage, a special case of heterozygote advantage (Wakeland et al. 1990; Lenz 2011). In our case, the small sample size within populations, and the lack of data on the parasite community, host fitness, and survival, do not allow for distinguishing between these explanations. However, the significantly high average intra-individual amino-acid and genetic divergence fits with a scenario where individuals with more diverged MHC alleles have an advantage over genotypes with more similar alleles (Wakeland et al. 1990), as it has been reported from other taxa (e.g. Aguilar et al. 2004; Alcaide et al. 2008; Oliver and Piertney 2012; Lenz et al. 2013a, 2013b; Vlček et al. 2016).
Variations in island size and isolation allowed us to examine the factors influencing MHC diversity. Neutral diversity was not correlated to island size and distance from the mainland, but the number of MHC alleles at the population level decreased significantly with island size and distance from the continent (Table 2). Interestingly, the allele loss from these populations affected neither the MHC amino-acid distance nor the individual polymorphism. In other words, MHC sequence and functional diversity of insular populations was maintained even when MHC allele richness declined. This pattern most likely complies with pathogen-mediated positive selection preserving diverse MHC functioning, which recognize more pathogens, in the face of genetic drift (Eimes et al. 2011; Sutton et al. 2011; Vlček et al. 2016). In addition, simulations by Ejsmond and Radwan (2011) suggest that selection at the MHC might also has adverse effects, with advantageous alleles becoming fixed while rare disadvantageous alleles getting removed, depleting the total number of MHC alleles at a given population (MHCAR herein). Alternatively, the loss of alleles from a population’s MHC pool could mainly involve rare alleles that usually contribute less to individuals polymorphism (Vlček et al. 2016).
In conclusion, we found that the MHC diversity of these two lizard species is shaped by selection, introgression, and drift. As expected, a signature of positive selection on MHC diversity was seen for both species. Moreover, introgression increased MHC diversity in sympatric mainland populations of the two species, probably a sign of adaptive introgression of alleles that confer an advantage for the host (Nadachowska-Brzyska et al. 2012; Hedrick 2013; Grossen et al. 2014). Finally, we found a significant reduction in number of MHC alleles, but maintained MHC sequence and functional diversity, in insular populations compared to mainland conspecifics, suggesting that positive selection maintains a diverse function even in the face of drift.
Data availability
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.d3kk74r.
References
Aguilar A, Roemer G, Debenham S, Binns M, Garcelon D, Wayne RK (2004) High MHC diversity maintained by balancing selection in an otherwise genetically monomorphic mammal. Proc Natl Acad Sci USA 101:3490–3494
Alcaide M (2010) On the relative roles of selection and genetic drift in shaping MHC variation. Mol Ecol 19:3842–3844
Alcaide M, Edwards SV, Negro JJ, Serrano D, Tella JL (2008) Extensive polymorphism and geographicalvariation at a positively selected MHC class II B gene of the lesser kestrel (Falco naumanni). Mol Ecol 17:2652–2665
Amann T, Rykena S, Joger U, Nettmann HK, Veith M (1997) Zur Trennung von Lacerta bilineata Daudin, 1802 und L. viridis (Laurenti, 1768). Salamandra 33:255–268
Amos W, Hoffman JI, Frodsham A, Zhang L, Best S, Hill AVS (2007) Automated binning of microsatellite alleles: problems and solutions. Mol Ecol Notes 7:10–14
Anderson EC, Thompson EA (2002) A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160:1217–1229
Barton NH (2001) The role of hybridization in evolution. Mol Ecol 10:551–568
Bates D, Maechler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67:1–48
Beerli P, Felsenstein J (2001) Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci USA 98:4563–4568
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2000) Genetix, a WindowsTM based software for population genetic analyses. laboratoire génome, populations, interactions CNRS UMR 5000. Université de Montpellier II Place. Montpellier, France
Böhme MU, Fritz U, Kotenko T, Dzukic G, Ljubisavljevic K, Tzankov N et al. (2007) Phylogeography and cryptic variation within the Lacerta viridis complex (Lacertidae, Reptilia). Zool Scr 36:119–131
Borcard D, Legendre P (2012) Is the Mantel correlogram powerful enough to be useful in ecological analysis? A simulation study. Ecology 93:1473–1481
Boudjemadi K, Martin O, Simon JC, Estoup A (1999) Development and cross-species comparison of microsatellite markers in two lizard species, Lacerta vivipara and Podarcis muralis. Mol Ecol Notes 8:518–520
Cornwall DH, Kubinak JL, Zachary E, Stark DL, Seipel D, Potts WK (2018) Experimental manipulation of population‐level MHC diversity controls pathogen virulence evolution in Mus musculus. J Evol Biol 31:314–322
Diniz-Filho JAF, Soares TN, Lima JS, Dobrovolski R, Landeiro VL, de Campos Telles MP et al. (2013) Mantel test in population genetics. Genet Mol Biol 36:475–485
Doherty PC, Zinkernagel RM (1975) Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature 256:50–52
Eimes JA, Bollmer JL, Whittingham LA, Johnson JA, Van Oosterhout C, Dunn PO (2011) Rapid loss of MHC class II variation in a bottlenecked population is explained by drift and loss of copy number variation. J Evol Biol 24:1847–1856
Eizaguirre C, Baltazar-Soares M (2014) Evolutionary conservation—evaluating the adaptive potential of species. Evol Appl 7:963–967
Eizaguirre C, Lenz TL, Kalbe M, Milinski M (2012a) Divergent selection on locally adapted major histocompatibility complex immune genes experimentally proven in the field. Ecol Lett 15:723–731
Eizaguirre C, Lenz TL, Kalbe M, Milinski M (2012b) Rapid and adaptive evolution of MHC genes under parasite selection in experimental vertebrate populations. Nat Comm 3:621–626
Ejsmond MJ, Radwan J (2011) MHC diversity in bottlenecked populations: a simulation model. Conserv Genet 12:129–137
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Garrigan D, Hedrick PW (2001) Class I MHC polymorphism and evolution in endangered California Chinook and other Pacific salmon. Immunogenetics 53:483–489
Gillingham MAF, Courtiol A, Teixeira M, Galan M, Bechet A, Cezilly F (2016) Evidence of gene orthology and trans-species polymorphism, but not of parallel evolution, despite high levels of concerted evolution in the major histocompatibility complex of flamingo species. J Evol Biol 29:438–454
Godinho R, Crespo EG, Ferrand N, Harris DJ (2005) Phylogeny and evolution of the green lizards, Lacerta spp. (Squamata: Lacertidae) based on mitochondrial and nuclear DNA sequences. Amphib-Reptil 26:271–285
Goudet J (2005) HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol Ecol Resour 5:184–186
Grossen C, Keller L, Biebach I, Croll D, The International Goat Genome C (2014) Introgression from domestic goat generated variation at the major histocompatibility complex of Alpine Ibex. PLoS Genet 10:e1004438
Hedrick PW (2002) Pathogen resistance and genetic variation at MHC loci. Evolution 56:1902–1908
Hedrick PW (2013) Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol Ecol 22:4606–4618
Herdegen M, Babik W, Radwan J (2014) Selective pressures on MHC class II genes in the guppy (Poecilia reticulata) as inferred by hierarchical analysis of population structure. J Evol Biol 27:2347–2359
Hersch-Green EI, Turley NE, Johnson MTJ (2011) Community genetics: what have we accomplished and where should we be going? Philos Trans R Soc B 366:1453–1460
Hurston H, Voith L, Bonanno J, Foufopoulos J, Pafilis P, Valakos E et al. (2009) Effects of fragmentation on genetic diversity in island populations of the Aegean wall lizard Podarcis erhardii (Lacertidae, Reptilia). Mol Phylogenet Evol 52:395–405
Huyghe K, Breugelmans K, Small M, TadiĆ Z, Van Damme R, Vanhooydonck B et al. (2009) Characterization of polymorphic microsatellite markers in the Dalmatian wall lizard Podarcis melisellensis (Squamata: Lacertidae). Mol Ecol Resour 9:299–301
Joger U, Amann T, Veith M (2001) Phylogeographie und genetische differenzierung im Lacerta viridis/bilineata komplex. Mertensiella 13:60–68
Jordan MA, Snell HL (2008) Historical fragmentation of islands and genetic drift in populations of Galápagos lava lizards (Microlophus albemarlensis complex). Mol Ecol 17:1224–1237
Keller M, Vassilakaki M (2002) Differenzierungsmuster bei Smaragdeidechsen (Lacerta bilineata und L. viridis) aus Italien und Griechenland: erste Ergebnisse, multivarianter Analysen von Pholidosemerkmalen. Mertensiella 13:69–77
Kirk H, Freeland JR (2011) Applications and implications of neutral versus non-neutral markers in molecular ecology. Int J Mol Sci 12:3966–3988
Klein J (1986) Natural history of the major histocompatibility complex. John Wiley and Sons. New York, USA
Klein J (1987) Origin of major histocompatibility complex polymorphism: The trans-species hypothesis. Hum Immunol 19:155–162
Kriener K, O’HUigin C, Tichy H, Klein J (2000) Convergent evolution of major histocompatibility complex molecules in humans and New World monkeys. Immunogenetics 51:169–178
Lenz TL (2011) Computational prediction of MHC II-antigen binding supports divergent allele advantage and explains trans-species polymorphism. Evolution 65:2380–2390
Lenz TL, Eizaguirre C, Kalbe M, Milinski M (2013a) Evaluating patterns of convergent evolution and trans-species polymorphism at MHC immunogenes in two sympatric stickleback species. Evolution 67:2400–2412
Lenz TL, Mueller B, Trillmich F, Wolf JBW (2013b) Divergent allele advantage at MHC-DRB through direct and maternal genotypic effects and its consequences for allele pool composition and mating. Proc R Soc B Biol Sci 280:20130714
Librado P, Rozas J (2009) DnaSPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lohm J, Grahn M, Langefors Å, Andersen Ø, Storset A, Von Schantz T (2002) Experimental evidence for major histocompatibility complex-allele-specific resistance to a bacterial infection. Proc R Soc B Biol Sci 269:2029–2033
Martin D, Rybicki E (2000) RDP: detection of recombination amongst aligned sequences. Bioinformatics 16:562–563
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P (2010) RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:1–5
Mayer W, Beyerlein P (2002) Genetische Differenzierung des Lacerta viridis/bilineata Komplexes und von Lacerta trilineata in Griechenland: Mitochondriale DNA-Sequenzen. Mertensiella 13:52–59
Miller HC, Allendorf F, Daugherty CH (2010) Genetic diversity and differentiation at MHC genes in island populations of tuatara (Sphenodon spp.). Mol Ecol 19:3894–3908
Miller HC, Belov K, Daugherty CH (2006) MHC class I genes in the Tuatara (Sphenodon spp.): evolution of the MHC in an ancient reptilian order. Mol Biol Evol 23:949–956
Murphy K, Weaver C (2016) Janeway’s immunobiology, 9th edn. Garland Science, Taylor & Francis Group, LL. New York, USA
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL (2012) Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764
Nadachowska-Brzyska K, ZieliŃSki P, Radwan J, Babik W (2012) Interspecific hybridization increases MHC class II diversity in two sister species of newts. Mol Ecol 21:887–906
Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426
Nembrini M, Oppliger A (2003) Characterization of microsatellite loci in the wall lizard Podarcis muralis (Sauria: Lacertidae). Mol Ecol Notes 3:123–124
Nettman HK (2001) Die Smaragdeidechsen (Lacerta s. str.)—Eine Übersicht über Verwandtschaft und Formenvielfalt. Mertensiella 13:11–32
Nielsen EEG, Bach LA, Kotlicki P (2006) HYBRIDLAB (version 1.0): a program for generating simulated hybrids from population samples. Mol Ecol Notes 6:971–973
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB et al (2013) Vegan: community ecology package. R package version 2.0-3 http://CRAN.Rproject.org/package=vegan
Oliver MK, Piertney SB (2012) Selection maintains MHC diversity through a natural population bottleneck. Mol Biol Evol 29:1713–1720
Padidam M, Sawyer S, Fauquet CM (1999) Possible emergence of new geminiviruses by frequent recombination. Virology 265:218–225
Phillips KP, Cable J, Mohammed RS, Herdegen-Radwan M, Raubic J, Przesmycka KJ et al (2018) Immunogenetic novelty confers a selective advantage in host–pathogen coevolution. Proc Natl Acad Sci USA.115:201708597
Pinho C, Sequeira F, Godinho R, Harris DJ, Ferrand N (2004) Isolation and characterization of nine microsatellite loci in Podarcis bocagei (Squamata: Lacertidae). Mol Ecol Notes 4:286–288
Poulakakis N, Goulielmos G, Antoniou A, Zouros E, Mylonas M (2005) Isolation and characterization of polymorphic microsatellite markers in the wall lizard Podarcis erhardii (Squamata: Lacertidae). Mol Ecol Notes 5:549–551
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Development Core Team (2015) R: a language and environment for statistical computing. R Foundation for Statistical Computing (https://www.r-project.org). Vienna, Austria
Radwan J, Biedrzycka A, Babik W (2010) Does reduced MHC diversity decrease viability of vertebrate populations? Biol Conserv 143:537–544
Reusch T, Langefors Å (2005) Inter- and intralocus recombination drive MHC class IIB gene diversification in a teleost, the three-spined stickleback Gasterosteus aculeatus. J Mol Evol 61:531–541
Richard M, Stevens VM, Le Hénanff M, Coulon A, Lourdais O, Clobert J (2012) Fourteen new polymorphic microsatellite loci for the wall lizard Podarcis muralis (Sauria: Lacertidae). Mol Ecol Resour 12:374–376.
Richardson DS, Jury FL, Blaakmeer K, Komdeur J, Burke T (2001) Parentage assignment and extra-group paternity in a cooperative breeder: the Seychelles warbler (Acrocephalus sechellensis). Mol Ecol 10:2263–2273
Roca V, Foufopoulos J, Valakos E, Pafilis P (2009) Parasitic infracommunities of the Aegean wall lizard Podarcis erhardii (Lacertidae, Sauria): Isolation and impoverishment in small island populations. Amphib-Reptil 30:493–503
Runemark A, Hansson B, Ljungqvist M, Brydegaard M, Svensson EI (2013) Has the inbreeding load for a condition-dependent sexual signalling trait been purged in insular lizard populations? Mol Ecol 22:1310–1321
Rykena S (1996) Experimental interspecific hybridization in the genus Lacerta. Isr J Zool 42:171–184
Rykena S (2001) Experimental hybridization in green lizards (Lacerta s. str.), a tool to study species boundaries. Mertensiella 13:78–88
Rykena S, Nettman H-K, Mayer W (2002) Lacerta viridis guentherpetersi ssp. nov., eine neue unterart der smaragdeidechse aus Griechenland. Mertensiella 13:89–97
Sagonas K, Poulakakis N, Lymberakis P, Parmakelis A, Pafilis P, Valakos ED (2014) Molecular systematics and historical biogeography of the green lizards (Lacerta) in Greece: Insights from mitochondrial and nuclear DNA. Mol Phylogenet Evol 76:144–154
Schierup MH, Mikkelsen AM, Hein J (2001) Recombination, balancing selection and phylogenies in MHC and self-incompatibility genes. Genetics 159:1833–1844
Schierup MH, Vekemans X, Charlesworth D (2000) The effect of subdivision on variation at multi-allelic loci under balancing selection. Genet Res 76:51–62
Sebastian A, Herdegen M, Migalska M, Radwan J (2015) AMPLISAS: a web server for multilocus genotyping using next-generation amplicon sequencing data. Mol Ecol Resour 16:498–510
Smith JM (1992) Analyzing the mosaic structure of genes. J Mol Evol 34:126–129
Sommer S (2005) The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 2:16
Spurgin LG, Van Oosterhout C, Illera JC, Bridgett S, Gharbi K, Emerson BC et al. (2011) Gene conversion rapidly generates major histocompatibility complex diversity in recently founded bird populations. Mol Ecol 20:5213–5225
Srithayakumar V, Castillo S, Mainguy J, Kyle CJ (2012) Evidence for evolutionary convergence at MHC in two broadly distributed mesocarnivores. Immunogenetics 64:289–301
Strand TM, Segelbacher G, Quintela M, Xiao L, Axelsson T, Höglund J (2012) Can balancing selection on MHC loci counteract genetic drift in small fragmented populations of black grouse? Ecol Evol 2:341–353
Sutton JT, Nakagawa S, Robertson BC, Jamieson IG (2011) Disentangling the roles of natural selection and genetic drift in shaping variation at MHC immunity genes. Mol Ecol 20:4408–4420
Takahata N, Nei M (1990) Allelic genealogy under overdominant and frequency-dependent selection and polymorphism of major histocompatibility complex loci. Genetics 124:967–978
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Těšický M, Vinkler M (2015) Trans-species polymorphism in immune genes: general pattern or MHC-restricted phenomenon. J Immunol Res 2015:1–10
Toonen RJ, Hughes S (2001) Increased throughput for fragment analysis on ABI Prism 377 automated sequencer using a membrane comb and STRand software. Biotechniques 31:1320–1324
Vlček J, Hoeck PEA, Keller LF, Wayhart JP, Dolinová I, Štefka J (2016) Balancing selection and genetic drift create unusual patterns of MHCIIβ variation in Galápagos mockingbirds. Mol Ecol 25:4757–4772
Wakeland EK, Boehme S, She JX, Lu C-C, McIndoe RA, Cheng I et al. (1990) Ancestral polymorphisms of MHC class II genes: Divergent allele advantage. Immunol Res 9:115–122
Wallny H-J, Avila D, Hunt LG, Powell TJ, Riegert P, Salomonsen J et al. (2006) Peptide motifs of the single dominantly expressed class I molecule explain the striking MHC-determined response to Rous sarcoma virus in chickens. Proc Natl Acad Sci USA 103:1434–1439
Wegner KM, Eizaguirre C (2012) New(t)s and views from hybridizing MHC genes: introgression rather than trans-species polymorphism may shape allelic repertoires. Mol Ecol 21:779–781
Wikelski M, Foufopoulos J, Vargas H, Snell H (2004) Galápagos birds and diseases: invasive pathogens as threats for island species. Ecol Soc 9:5
Wong WSW, Yang Z, Goldman N, Nielsen R (2004) Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168:1041–1051
Wright S (1943) Isolation by distance. Genetics 28:114–138
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang Z, Nielsen R, Goldman N, Pedersen A-MK (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yang Z, Wong WSW, Nielsen R (2005) Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Zagalska-Neubauer M, Babik W, Stuglik M, Gustafsson L, Cichon M, Radwan J (2010) 454 sequencing reveals extreme complexity of the class II major histocompatibility complex in the collared flycatcher. BMC Evol Biol 10:395
Acknowledgements
We thank Dr. Lenz (Max Planck Institute for Evolutionary Biology, Plön, Germany) for providing us Perl scripts to test for convergent evolution, Dr. Crnobrnja-Isailovic (Department of Evolutionary Biology, University of Belgrade, Serbia) for sending us tissue samples of L. trilineata and L. viridis from Serbia, and Tomas Johansson (Ecology Building, Lund University, Sweden) for running the pyrosequencing. We also thank Dr. Radwan, Dr. Gillingham, and one more anonymous reviewer for valuable comments that significantly improved the manuscript. This study was funded by the Swedish Research Council (621-2014-5222 and 621-2016-689 to BH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Sagonas, K., Runemark, A., Antoniou, A. et al. Selection, drift, and introgression shape MHC polymorphism in lizards. Heredity 122, 468–484 (2019). https://doi.org/10.1038/s41437-018-0146-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0146-2
This article is cited by
-
Immunogenetic-pathogen networks shrink in Tome’s spiny rat, a generalist rodent inhabiting disturbed landscapes
Communications Biology (2024)
-
Balancing selection versus allele and supertype turnover in MHC class II genes in guppies
Heredity (2021)
-
Habitat fragmentation differentially shapes neutral and immune gene variation in a tropical bird species
Heredity (2021)
-
Surviving despite reduce MHC variation: selection patterns and genetic variation of the endangered Huillín (Lontra provocax)
Mammal Research (2021)
