
Abstract
Proteins encoded by extraordinarily polymorphic major histocompatibility complex (MHC) genes are involved in the adaptive immune response. Balancing selection is believed to maintain MHC polymorphism in the long term, although neutral processes also play a role in shaping MHC diversity. However, the relative contribution of these processes is poorly understood. Here we characterized MHC class II variation of a low-dispersal, pond-breeding newt (Triturus carnifex) over a restricted, geographically structured area. We aimed to (1) evaluate the contribution of selection and neutral processes to shaping MHC diversity at two geographic scales, and (2) test for signatures of divergent allele advantage (DAA), which is a potentially important mechanism of balancing selection. The dominant role of selection in shaping MHC variation was suggested by the lack of correlation between MHC and neutral (microsatellite) variation. Although most variation occurred within populations for both types of markers, they differed in the extent of structuring at the two spatial scales. MHC structuring was more pronounced at local scales, suggesting the role of local selection, while structuring was not detectable at a larger scale, possibly due to the effect of balancing selection. Microsatellites showed the opposite pattern. As expected under DAA, the observed genotypes combined more sequence diversity than expected under a random association of alleles. Thus, DAA may contribute to maintaining MHC polymorphism, which is ancient, as supported by signatures of historical positive selection and trans-species polymorphism. Our results point to the importance of a multi-scale approach in studying MHC variation, especially in low-dispersal taxa, which are genetically structured at fine spatial scales.
Similar content being viewed by others

Introduction
The major histocompatibility complex (MHC) is a family of extraordinarily polymorphic genes involved in the adaptive immune response of jawed vertebrates (Kelley et al. 2005). Proteins encoded by classical MHC class II genes present antigens from extra-cellular pathogens and parasites to T-cells, activating the adaptive immune cascade (Neefjes et al. 2011). Each MHC protein can recognize and bind a limited range of antigens, as determined by the amino-acid composition of the antigen-binding sites (ABS). These sites are scattered along exon 2 of MHC class II genes (Brown et al. 1993), which usually exhibits high levels of sequence variability (Kelley et al. 2005).
Positive selection for amino-acid replacement in ABS favours novel alleles, which are maintained for a long time by various forms of balancing selection, the detailed mechanisms of which remain incompletely understood (Spurgin and Richardson 2010). As a consequence, numerous alleles and substantial sequence variation are commonly found in MHC loci (Bernatchez and Landry 2003). Gene duplication (Edwards and Hedrick 1998) and micro-recombination (Richman et al. 2003) may additionally increase the overall MHC diversity. This diversity has adaptive significance, ultimately conferring resistance to a broad array of pathogens (Hughes and Yeager 1998). MHC polymorphism is often preserved for an extraordinarily long time, leading to trans-species polymorphism (TSP) (Klein 1987), which results from the retention of allelic lineages through multiple speciation events. Pathogen-driven balancing selection has been proposed as the dominant factor promoting and maintaining MHC polymorphism, although some forms of sexual selection may contribute as well (Piertney and Oliver 2005). The two main mechanisms of pathogen-driven balancing selection are negative frequency dependence and heterozygote advantage, also referred to as overdominance (Hughes and Yeager 1998; Spurgin and Richardson 2010). The first model, which received support in experimental and theoretical studies (Ejsmond and Radwan 2015; Phillips et al. 2018), postulates that novel alleles are favoured when rare, but are selected against when they become common, consistent with the host–parasite co-evolutionary Red Queen dynamics. The second model predicts that heterozygotes would be favoured over homozygotes because they possess different types of MHC molecules and thus can recognize more pathogens. Although evidence of heterozygote advantage from experimental studies has been frequently reported, excess of heterozygotes in natural populations has been detected only in some studies, probably because the tests lacked sufficient power (Garrigan and Hedrick 2003; Spurgin and Richardson 2010). Importantly, as rare alleles are present mostly in heterozygotes, these mechanisms are entangled, thus making it difficult to distinguish between them (Spurgin and Richardson 2010). Recent studies are increasingly focusing on an extended interpretation of heterozygote advantage, the divergent allele advantage concept (DAA) (Wakeland et al. 1990). According to this hypothesis, heterozygous genotypes composed of more divergent alleles are supposed to recognize a broader range of antigens, ultimately entailing higher fitness at the individual level. Therefore, MHC genotypes combining high sequence divergence, especially in ABS, are expected to be found more frequently than predicted under a random association of alleles. A correlation of sequence divergence between pairs of alleles with the ability of their products to recognize more antigens has been detected for the human MHC using computational antigen-binding prediction algorithms (Lenz 2011; Pierini and Lenz 2018). Pierini and Lenz (2018) found also a significant positive correlation between the mean divergence of an allele from other alleles and its frequency in humans. The DAA has been only rarely tested in other species, and results are not consistent between studies. For example, Lenz and collaborators (2009, 2013) demonstrated that high MHC IIB allele divergence was a predictor of fitness proxies in the long-tailed giant rat and sea lions. On the other hand, Gaigher et al. (2018) failed to detect shifts towards MHC haplotypes combining high diversity in a natural population of barn owl. Thus, the evidence for DAA in natural populations remains limited and further research is needed.
Various patterns of spatial MHC structuring have been observed, with structuring higher, lower or comparable to that in putatively neutral markers, such as microsatellites (Bernatchez and Landry 2003). A stronger MHC population structure is predicted in the case of spatially and temporally fluctuating (directional) selection (Spurgin and Richardson 2010), leading to substantial differentiation among populations, consistent with a local adaptation model (e.g. Miller et al. 2001, Ekblom et al. 2007, Li et al. 2016). Conversely, if selection promoting novel, rare alleles (i.e. negative frequency-dependent selection) increases the effective rate of MHC gene flow among populations (Schierup et al. 2000), a weaker geographic structuring of MHC would be observed compared to that of neutral variation (e.g. van Oosterhout et al. 2006; Rico et al. 2015). The same is expected even under isolation if balancing selection prevents loss of alleles and maintains similar allele frequencies. Demographic processes interact with selection in shaping MHC diversity, and their relative role may differ at various temporal and spatial scales (Radwan et al. 2010; Eimes et al. 2011). For example, Herdegen et al. (2014) found a strong MHC population structure within regions, while between regions MHC was weakly structured compared to microsatellites. The authors ascribe these differences to the dominant role of diversifying/fluctuating selection at the smaller scale and an interplay between (weak) drift and balancing selection at the larger scale. Therefore, it is essential to investigate MHC variation at adequate spatial scales reflecting environmental variation, the dispersal abilities of species and demographic processes (reviewed in Richardson et al. 2014). A hierarchical multi-scale approach is highly informative in this respect, and it may help mitigate possible bias, as, for example, sampling over too large a scale may mask the effects of selection due to differences in environment and limited gene flow (Lenormand 2002).
Exploring how MHC variation is structured at local scales is particularly interesting in taxa that have low dispersal ability and show high genetic differentiation (detectable using neutral markers) even at micro-geographical scales. However, studies investigating MHC variation at fine scales are rather scarce (e.g. Gonzalez-Quevedo et al. 2016; Pearson et al. 2018). In this context, pond-breeding amphibians provide a good model, as they likely experience heterogeneous selective pressures (e.g. parasites), which may substantially differ among nearby sites (e.g. different biotopes), possibly leading to local adaptation. Their limited vagility, exacerbated by habitat fragmentation, often results in high structuring of populations even at small scales (e.g. Lampert et al. 2003, Beebee 2005, Smith and Green 2005, Cushman 2006).
In the present study, MHC class II variation of the Italian crested newt, Triturus carnifex (Laurenti, 1768), was analysed over a restricted (<200 km2), but geographically structured area. Our sampling covered three hierarchical scales: the overall region, two well-defined geographic subregions, and nine populations. Strong site fidelity has been reported for T. carnifex (Mori et al. 2017; but see also Denoël et al. 2018), and neutral genetic structuring in Triturus is detectable at fine spatial scales (i.e. <30 km2, Jehle et al. 2005). Thus, the adopted sampling scale is relevant for hierarchical analysis of the genetic structure and diversity of T. carnifex. We compared putative adaptive (MHC) and neutral variation (microsatellites) to assess the contribution of evolutionary forces to the pattern of MHC diversity observed across spatial scales. Additionally, taking advantage of a relatively simple organization of MHC II in Triturus, we tested for signatures of the DAA. Indeed, in the closely related T. cristatus a single expressed class II locus (some evidence for a second, possibly functional locus was detected in only 2% of individuals) and several pseudogene loci were described (Babik et al. 2009). Transcribed and pseudogene alleles could be distinguished on the basis of their sequence. Although MHC has not previously been characterized in T. carnifex, we expected a similar organization as in T. cristatus. Basically, we aimed to (1) characterize MHC class II genes in T. carnifex; (2) investigate the relative role of selection and neutral processes in shaping MHC diversity at a small geographic scale; and (3) test for balancing selection in the form of the divergent allele advantage.
Materials and methods
Study area, sample collection and DNA extraction
Triturus carnifex samples (tail clippings) were collected from nine sampling sites in the area of the Lepini Mountains, central Italy, during the 2018 breeding season (Table 1). The Italian Ministry of Environment approved all sampling procedures (authorization no. 36230/T-A31). The study area, about 200 km2, encompasses two distinct geographic subregions, separated by the Lepini mountain range. Due to the karst nature of the massif, natural ponds are scarce, so T. carnifex inhabits man-made biotopes (e.g. Roman wells, semi-natural ponds and watering places), which occur patchily at <1000 m above sea level (Corsetti 2006, Mattoccia 2015). Therefore, the mountain range likely acts as a physical barrier to dispersal. Four (COR, NOR, BAS, SEZ) and five (SEG1, SEG2, GOR, MON, CAR) populations fall into the Western and the Eastern subregion, respectively (Fig. 1). The average distance between sampling sites was 9.96 km, ranging from 1.40 to 19.46 km. Tissues were stored in ethanol at −20 °C until genomic DNA extraction, which was carried out using the GenElute™ Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich – Saint Louis, MO, USA), following the standard protocol for the mammalian tissue.

Frequencies of 13 MHC-DAB alleles in nine T. carnifex populations from the Lepini Mountains (Italy). The geographic location of the study area (red triangle) is shown on the top right of the map. Population abbreviations, coloured according to geographic subregion (red = Western; black = Eastern), refer to Table 1
MHC exon 2 PCR amplification, next-generation amplicon sequencing and post-processing
We used 144 newts (16 individuals per population) for MHC analyses. An ~200-bp fragment of MHC class II exon 2 was amplified using the primer pair TrMHCII10F and TrMHCII5R (Babik et al. 2009), with partial Illumina Nextera XT adaptors added to allow library generation, and along with a 4-bp tag (four forward and four reverse-tagged primers were combined to obtain 16 unique barcode combinations; SuppInfo 1). Each PCR reaction, in a total volume of 20 μl, contained 20–100 ng DNA, 0.375 μM each primer, 1× Phusion Buffer HF, 200 μM each dNTP, and 0.4U of Phusion Hot Start II High-Fidelity DNA polymerase (Thermo Scientific, Waltham, MA, USA). Amplification was carried out in a 2720 Thermal cycler (Applied Biosystems, Foster City, CA, USA) under the following conditions: initial denaturation at 98 °C for 2′, followed by 30 cycles at 98 °C for 10″, 58 °C annealing for 20″, 72 °C for 20″, and a final elongation step at 72 °C for 5′. We assembled overall nine batches of 16 PCR products each, individually labelled by a barcode combination and pooled approximately equimolarly, as assessed by visual examination of band intensities on 1% agarose gels. A commercial company (Bio-Fab Research, Rome, Italy) accomplished the library preparation (i.e. adding the remaining part of the Illumina adapters and indexes in a second PCR step) and performed a 300-bp paired-end sequencing on Illumina MiSeq (Illumina, San Diego, CA, USA). The Illumina technology enables efficient genotyping of complex MHC systems (e.g. Lighten et al. 2014, Gaigher et al. 2016, Biedrzycka et al. 2017).
The PRINSEQ online tool (Schmieder and Edwards 2011) was used to trim read ends (>250 bp) and to discard those with a low base-call accuracy (i.e. average quality < 25 Phred). Paired-end reads were merged in FLASH (Magoč and Salzberg 2011) with a minimum overlap of 20 bp. Finally, AmpliCHECK (Sebastian et al. 2016) was used for removal of primer sequences, de-multiplexing and counting variants for each amplicon.
MHC genotyping pipeline
The adopted genotyping procedure consisted of three main steps: identification of maximum per-amplicon frequencies (MPAF) for true alleles and putative artefacts (1), and actual genotyping, which included a clustering step (2) followed by a filtering step (3). AmpliCHECK was used for preliminary assessment of the results to check whether putative true variants can be distinguished from putative artefacts based on MPAF. After sorting all variants by descending MPAF a gap between 7.6 and 2.8% was detected. We confirmed that variants showing MPAF > 7.6% were putative true alleles. Conversely, variants with MPAF < 2.8% could be mostly identified as low-frequency artefacts (chimaeras or 1–2 base mismatch errors) derived from putative true variants with higher per-amplicon frequency (PAF) within the same amplicon (step 1). Hence we assumed the 2.8% PAF threshold (i.e. the maximum PAF observed for the putative artefacts) for the sequence similarity-based clustering procedure (step 2) performed by AmpliSAS (Sebastian et al. 2016). This method allowed to increase the depth of true variants and thus to simplify their identification in the filtering step. In this way, putative parental variants (in this case > 2.8% PAF) included depths of artefacts derived from these variants (i.e. putative descendant sequences showing 1–2 base mismatch errors and < 2.8% PAF) occurring within the same amplicon. Finally (step 3), to discard residual artefacts due to contaminations or tag-switching (Carlsen et al. 2012), we adopted the Degree Of Change (DOC) genotyping method (described in Lighten et al. 2014), implemented as a filtering step in AmpliSAS, which is reliable in case of ultra-deep sequencing (i.e. thousands of reads per amplicon). Briefly, under the assumption that true variants occur in higher PAF than artefacts, the break point separating true variants form artefacts is set within each amplicon separately by ordering variants by their PAF and finding the highest drop in cumulative frequency of consecutive variants (i.e. the maximum DOC). Additionally, we set up specific filters to discard chimaeras and variants differing by ± 30 bp from the expected length. All other settings were as default.
MHC class II characterization
MHC class II alleles were numbered according to their frequency, separately for the putatively functional locus (Trca-DAB) and for pseudogenes (Trca-ps). Variants were aligned in MEGA7 (Kumar et al. 2016) using the MUSCLE algorithm and checked visually to assess the reliability of the alignment. The average pairwise sequence divergence (nucleotide distance = Kimura 2-Parameter model to correct for multiple hits; amino-acid distance = PAM distance accounting for amino-acid replacement probability, as described in Dayhoff et al. 1978), and the synonymous (dS) and non-synonymous (dN) divergence (modified Nei-Gojobori method with Jukes-Cantor correction) were computed in MEGA7 as well.
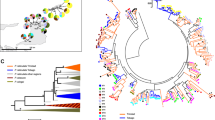
To examine the relationship among Triturus MHC class II sequence variants, a maximum likelihood tree (model: Jukes-Cantor with Gamma rate distribution and a nonzero proportion of invariant sites) was built in MEGA7 and its robustness was assessed with 1000 bootstrap replicates. Additionally, as MHC alleles often show complex genealogies departing from a bifurcating tree, a neighbour-net network (p-distance) was built in SplitsTree (Huson and Bryant 2006) to visualize conflicting signals due to gene duplication and putative recombination events. Analyses of relationships included 66 MHC II exon 2 sequences available for the genus Triturus, 42 deposited in GenBank (accessions FJ447986–FJ448028; Babik et al. 2009) and 24 available in Dryad (https://doi.org/10.5061/dryad.s9v06; Wielstra et al. 2015).
We tested for recombination using the following methods: SisScan and Bootscan (window = 30, step size = 3); RDP (window = 30); MaxChi and Chimaera (variable sites = 10); GENECONV (default settings). All methods were implemented in RDP v.4.95 (Martin et al. 2015), using the Bonferroni correction for multiple tests.
We looked for signatures of historical positive selection using two approaches. First, we tested in MEGA7 for ω (dN/dS ratio) > 1 by performing the one-tailed Z-test in three alignment partitions: (1) all sites; (2) 19 ABS inferred on the basis of human MHC (Brown et al. 1993); (3) non-ABS. Second, we compared three codon-based evolution models of putative functional alleles in PAML v4.8 (Yang 2007): (1) M0 assumes a single ω value for all codons; (2) M7 considers purifying selection and neutrality (ω ≤ 1); (3) M8 allows positive selection (a proportion of sites evolves with ω > 1). The Akaike information criterion (AIC) was used to choose the best model. Positively selected sites (PSS) were identified using several methods. We used the Bayes empirical Bayes (BEB) procedure in PAML, along with four methods implemented on the Datamonkey server (https://www.datamonkey.org/): the fast unconstrained Bayesian approximation (FUBAR), the fixed effects likelihood (FEL), the single-likelihood ancestor counting (SLAC) and the mixed-effects model of evolution (MEME) (Kosakovsky Pond and Frost 2005; Murrell et al. 2012, 2013). These analyses were carried out twice: namely, using only T. carnifex alleles, and, later, including both T. carnifex and T. cristatus putative functional variants (i.e. GenBank accession numbers FJ448004–FJ448028, and with 24 alleles in Dryad: https://doi.org/10.5061/dryad.s9v06) to improve the accuracy of PSS detection.
MHC supertyping
All Triturus putative functional variants were collapsed into supertypes. Supertypes are classes of functionally similar sequences as defined by the physico-chemical properties of amino acids in positions that determine the specificity of antigen binding. Amino-acid sites that are likely involved in peptide binding (i.e. all PSS detected by at least one method) were extracted and each site was characterized according to five physio-chemical descriptors as explained in Sandberg et al. (1998). Supertypes were defined in the adegenet R-package (Jombart 2008) using the K-means clustering algorithm. For each K the Bayesian information criterion (BIC) value was recorded, and we used the ΔBIC ≥ 2 criterion to choose the optimal number of clusters. Eventually, all variants within each cluster were collapsed into a single MHC supertype.
MHC variation
Standard indices of genetic variability, i.e. the number of alleles (MHCAN) and private alleles (MHCPA), and the observed (MHCHO) and expected (MHCHE) heterozygosity, were calculated in GenAlEx 6.5 (Peakall and Smouse 2012). In GenePop on the Web (Rousset 2008) we tested for significant departures from Hardy–Weinberg expectations (HWE) within each population, and p-values were adjusted for multiple testing (Holm–Bonferroni correction). Pie charts displaying allele and supertype frequencies were mapped and edited in QGIS 2.18 (Quantum GIS Development Team, http://qgis.osgeo.org).
STR amplification, genotyping, and variation
To assess neutral variation we analysed 10 microsatellite loci (hereafter STR): TCRI13, TCRI29, TCRI35, TCRI36 and TCRI46 (Krupa et al. 2002), A7, A8, A126, D5 and D127 (Dufresnes et al. 2016). PCR reactions were performed in a total volume of 12 μL containing 20–100 ng of genomic DNA, 2X PCR Master Mix (Thermo Scientific) and 0.83 μM of each primer. Amplifications were carried out in a 2720 thermal cycler (Applied Biosystems) as follows: initial denaturation at 94 °C for 5', followed by 35 cycles at 94 °C for 30'', a locus-specific annealing temperature (i.e. 56 °C for A7, D5, A126 and TCRI46; 58 °C for the others) for 45'', 72 °C for 30'', and a final elongation step at 72 °C for 5'. Amplicons were electrophoresed on a 1.5% agarose gel with ethidium bromide and visualized under UV light to assess successful reactions. Fragment analysis was conducted on an ABI 3730xl genetic analyzer (Applied Biosystems) by the DNA Analysis facility on Science Hill (Yale University, USA), and genotypes were determined using Peak Scanner v2 (Applied Biosystems).
We tested for departures from HWE and for linkage disequilibrium (LD) in GenePop (Rousset 2008). The frequency of null alleles was estimated with the EM algorithm in FreeNA (Chapuis and Estoup 2007). STR intra-population diversity (STRPA, STRHE, STRHO) was assessed as described for the MHC. Sample sizes for STR were unequal and larger than for MHC (see Table 1), because the former were obtained from another ongoing study (Pietrocini et al. in prep.). To precisely estimate the neutral diversity, instead of subsampling STR data to the MHC sample size, we retained all STR samples. For comparison of the number of alleles, which required equal MHC and STR sample sizes, we calculated mean allelic richness (STRAR) within the PopGenReport R-package (Adamack and Gruber 2014) by setting rarefaction to 32 alleles (i.e. 16 individuals) to allow comparison with MHCAN.
Comparison between MHC and STR variation and genetic structuring
We assessed the correlation between putative adaptive and neutral variation by testing the relationship between MHC (response variable) and STR variation (predictive variable) across the examined populations. Linear models (lm function in R; R Development Core Team, 2017) were used to evaluate the association between MHCAN–STRAR and MHCHE–STRHE.
We followed five approaches to analyse and compare the genetic structures of MHC and STR. First, we tested for the overall signature of isolation by distance (IBD). A Mantel test (9999 permutations, adegenet package in R) was performed to assess the significance and magnitude of the relationship between population pairwise genetic and geographic distances for MHC and STR independently. Genetic distances (FST) were computed in FreeNA: MHC alleles were treated as equally different from each other; pairwise FST at STR were adjusted for the presence of null alleles according to the ENA correction (5000 replicates).
Second, we tested whether the genetic structure was stronger or weaker in MHC compared to STR loci using the FST outlier method in BayeScan 2.1 (Foll and Gaggiotti 2008). We first ran an analysis involving only STR data, which identified STR outliers. These STR outliers were removed and a second analysis was performed to test whether MHC is an outlier with respect to the remaining STR loci. The program was run by setting the default prior odds (PO = 10, meaning that the model with selection is 10 times more unlikely than the neutral one) and allowing a 5% threshold of false discovery rate (i.e. q-value < 0.05). Third, we tested for a correlation between the genetic differentiation (pairwise FST) among populations at MHC and STR using the Mantel test (9999 permutations, adegenet R-package). Fourth, the non-metric multidimensional scaling (NMDS), as performed in PAST (Hammer et al. 2001) was used to visualize and compare matrices of pairwise FST based on MHC and STR.
Finally, a hierarchical analysis of molecular variance (AMOVA, 10000 permutations) was performed in Arlequin v3.5 (Excoffier and Lischer 2010) to assess the partitioning of genetic variation among and within the two geographic subregions (i.e. the Eastern and Western one), as well as within populations. The analysis was carried out separately for MHC alleles, MHC supertypes and STR. Note that the consistency between geographical and genetic structure was assessed by an STR-based Bayesian clustering analysis in STRUCTURE v.2.3.4 (Pritchard et al. 2000) (details in SuppInfo 2).
Tests for MHC divergent allele advantage
To test for an evidence of the divergent allele advantage we compared the observed divergence between alleles within individuals to theoretical expectations obtained via simulations. Specifically, we used simulations to evaluate DAA for (1) each population separately, (2) each subregion and (3) all samples pooled. We tested if observed genotypes combine on average more sequence diversity than expected under a random association of alleles (i.e. random mating, no selection). We computed the mean genetic distance between functional alleles within individuals (DOBS) and compared it to the null distribution expected under a random assortment of alleles in individuals (DEXP, 9999 randomizations). The null distribution was obtained by random shuffling (i.e. without replacement) of alleles to form a simulated dataset of the same size as the observed dataset—during this procedure, all possible genotypes can be generated according to the alleles available in the pool. For tests at population and subregion levels, p-values were adjusted for multiple testing (Holm–Bonferroni). We performed the one-way Wilcoxon signed rank test to check whether DOBS was greater than DEXP across all populations. Furthermore, to separate the possible effect of a simple excess of heterozygotes from the actual DAA, we performed a modified version of the test where both DOBS and DEXP were computed only for heterozygous individuals (“heterozygotes only”). Tests were carried out using a custom R-function (code and input data examples are available in Dryad, https://doi.org/10.5061/dryad.ct40nm7). DAA was investigated at two partitions separately (all sites and PSS as identified using all Triturus alleles), each testing for three measures of genetic distance to take into account divergence among MHC alleles at multiple levels (i.e. nucleotide, amino acid and functional). Specifically, in MEGA we computed nucleotide p-distance (correction for multiple hits was computationally not possible when considering only PSS, so this distance was probably an underestimate) and PAM distance (to take into account different probabilities of various amino-acid substitutions). A script of Pierini and Lenz (2018) (available at https://sourceforge.net/projects/granthamdist/) was used to calculate the Grantham distance (Grantham 1974), which takes into consideration the physiochemical properties of amino acids, and has been found to more adequately reflect functional distances between (human) MHC alleles compared to other measures (Pierini and Lenz 2018).
Results
MHC characterization
We sequenced a fragment of MHC class II exon 2 in 144 newts from nine sampling sites. The individual amplicon coverage depth was high (mean 7363.8; range: 1,673–33,678) (SuppInfo 3). We found 2–6 variants per individual (mean = 4.25 ± 0.80), which indicated co-amplification of at least three loci (i.e. DAB locus and several pseudogenes).
Overall, 19 unique sequences of 189, 197 or 200 bp were detected, none of which were previously reported for the closely related T. cristatus. Six variants fell into a cluster (bootstrap support 89%, Fig. 2) that was previously identified in T. cristatus as grouping non-expressed, putative pseudogene sequences (Babik et al. 2009). The pseudogene status of T. carnifex variants in this cluster was supported by the presence of an internal stop codon (Trca-ps*06) or an 11-bp frameshift deletion (Trca-ps*01, *05, *06); it was not possible to assign pseudogene variants to loci based on sequence similarity or a co-occurrence pattern, and hence they were not analysed further (their frequencies are shown in SuppInfo 4). The remaining 13 alleles (197–200bp), each translating into a unique protein sequence (65 or 66 amino acids with no stop codons or frameshift mutations; SuppInfo 5), were intermixed on the tree with T. cristatus alleles from the putative functional locus (Fig. 2), a signature of TSP. Babik et al. (2009) suggested that a divergent allele, named Trcr-DBB*01, represents a separate locus of unknown expression status in T. cristatus. Here we found two alleles (Trca-DAB*04 and Trca-DAB*11) with similarity to Trcr-DBB*01, but it is unlikely that these come from a separate locus, for the following two reasons. First, no individual had more than two alleles, and alleles Trca-DAB*04 and Trca-DAB*11 occurred in combination with various other alleles (SuppInfo 3)—both alleles were rare, with the highest per-population frequency of 0.16, which implies that hypothetical haplotypes carrying the second locus would occur mostly in heterozygotes. Thus, three alleles per individual are expected unless the two-locus haplotype shares an allele with the single-locus haplotype present in the heterozygous individual. In such a case, however, identical genotypes with two alleles would be expected for all 13 and 2 individuals carrying alleles Trca-DAB*04 and Trca-DAB*11, respectively, which was not observed. Second, none of the populations showed strong departures from HWE (no p < 0.01 at the nominal level and none significant after Holm–Bonferroni correction, SuppInfo 6). Thus, the available evidence suggests that all 13 alleles come from a single putative functional locus, DAB. Nevertheless, as we are not able to rule out completely the possibility that Trca-DAB*04 and Trca-DAB*11 represent a separate locus, here we present the results of DAA also without these two alleles.

Relationships among Triturus MHC class II exon 2 sequence variants. a Neighbour-net network, p-distance method; the dashed line separates the group of putative functional alleles (DAB locus) from pseudogenes (at least two loci). b Maximum likelihood tree, Jukes–Cantor model with gamma-distributed rate variation and a non-zero proportion of invariant sites (JC+G+I); bootstrap support > 70 is shown; the outgroup is Ambystoma tigrinum (GenBank: DQ071906). Black squares and green circles indicate T. carnifex and T. cristatus variants, respectively. Putative functional alleles are labelled without the prefix (Trca-DAB* and Trcr-DAB*) for a better visualization, and labels are coloured according to supertypes
The putatively functional DAB locus showed high sequence diversity, particularly in the ABS codons, as well as clear signatures of historical positive selection: excess of nonsynonymous substitutions at ABS (Table 2) and strong support (ΔAIC = 7.4) for the PAML model M8, which allows for positive selection (SuppInfo 7). Only SisScan, out of the six methods used, identified a single recombination event (results not shown), so recombination was not substantial. Four positively selected sites (PSS) were detected in T. carnifex by at least one method (i.e. amino-acid sites 6, 38, 48 and 64), and the number increased to eight (additional sites: 8, 45, 49, 51) after including T. cristatus sequences (SuppInfo 5). These sites are likely involved in peptide binding since most of them (75%) matched the positions of human ABS (p < 0.01, Fisher’s Exact Test) (SuppInfo 5). Four supertypes were identified in Triturus on the basis of the eight PSS. All four occurred in T. carnifex, grouping from two to five alleles (Fig. 2, SuppInfo 8).
MHC-DAB vs STR: variation and genetic structuring
We detected three to eight DAB alleles (two to four supertypes) per population; some populations had a single private allele (Table 1), but no private supertypes were detected. The geographic distribution of alleles or supertypes did not show a clear pattern (Fig. 1 and SuppInfo 8, respectively).
All STR loci were polymorphic, showing between 4 and 29 alleles (SuppInfo 9). The standard indices of variation within populations are summarized in Table 1. LD was significant (Holm-Bonferroni adjusted p < 0.05) in 8 out of 405 pairwise comparisons, all in population SEG1, which indicates no physical linkage between any STR loci. Ten out of 90 tests showed significant departures from HWE (Holm-Bonferroni adjusted p < 0.05). Locus D5 showed departures in multiple populations (SuppInfo 6), probably due to the presence of null alleles (SuppInfo 10). Therefore, we removed this locus from AMOVA and outlier analyses, in which correction for null alleles was not possible. For the other analyses, allele frequencies were corrected for the presence of null alleles.
The comparisons between MHC-DAB and the neutral (STR) variation showed (1) no significant association between DAB and STR variation (MHCAN and STRAR: R2 = 0.01, p = 0.82; MHCHE and STRHE: R2 = 0.05, p = 0.57; further details in SuppInfo 11); (2) the IBD model explained a fraction of the neutral differentiation (Mantel: r = 0.38, p = 0.03), but not DAB differentiation (Mantel: r = −0.01, p = 0.50) (Fig. 3); (3) a similar level of geographic structuring for DAB and STR loci was suggested by the FST outlier analysis (results not shown); and (4) an appreciable correlation between population differentiation at DAB and STR (Mantel test: r = 0.52, p = 0.02; SuppInfo 12).

Relationship between pairwise genetic (FST) and geographic distance (m) for nine T. carnifex populations. Results based on MHC-DAB (light grey; Mantel r = −0.01ns) and STR (dark grey; Mantel r = 0.38*) are given
The analysis of the population genetic structure at STR loci (i.e. NMDS and STRUCTURE) supported the geographic structuring that we assumed a priori based on the presence of the mountain barrier (SuppInfo 2). The Western group was more heterogeneous (Fig. 4a), which probably led to a secondary ΔK peak in STRUCTURE analysis supporting five clusters (SuppInfo 2). In contrast to STR, DAB did not show significant differentiation between subregions, while it did show substantial differentiation between populations (Table 3, Fig. 4b). As expected for highly polymorphic markers, the vast majority of variation was within populations (Table 3).

Non-metric two-dimensional scaling of the pairwise FST matrix computed for a 10 STR loci (stress = 0.23); b the MHC-DAB (stress = 0.05). Population abbreviations refer to Table 1; dots are coloured according to the geographic subregion (light grey = Western; dark grey = Eastern)
Tests of divergent allele advantage in MHC-DAB
No clear evidence for DAA was detected within populations (SuppInfo 13): DOBS was significantly greater than DEXP in BAS only, possibly due to a slight excess of heterozygotes in this population (the DOBS excess was no longer significant when considering heterozygotes only; SuppInfo 13). On the other hand, there was an excess of populations with values of DOBS > DEXP (Wilcoxon signed-rank test, p < 0.05) for three combinations of distance/data partition/test version (all with the PAM distance, data not shown), suggesting a tendency in the direction expected under DAA.
When tests were performed at the subregion level, an unambiguous DAA signature was observed in the Western subregion (p < 0.05 in all but one tests; Table 4), while no signal was found in the Eastern subregion (Table 4).
Support for DAA was found also in the total sample (Table 4). We detected evidence for DAA considering PSS at both amino-acid (PAM p < 0.01, Grantham p < 0.1) and nucleotide (p < 0.1) levels. Only a weak DAA signature was observed using the amino-acid PAM distance when all sites were taken into account. Interestingly, strong support for DAA (p < 0.05) was found in all “heterozygotes only” tests at all examined partitions and measures of distance (Table 4). This indicates that the detected effect is not due to an excess of heterozygotes.
Finally, when tests were conducted without the highly divergent DAB alleles, the signature of DAA was observed just in some “heterozygotes only” tests for the pooled samples, and in all “heterozygotes only” tests for the Western subregion (SuppInfo 13).
Discussion
Characterization of MHC class II genes
Here we provide the first characterization of MHC class II in Triturus carnifex. At least three loci were simultaneously amplified, but just one (i.e. the DAB locus) appeared functional. Unequivocal signatures of historical positive selection on putative peptide binding sites were detected for this locus. A single functional locus is consistent with the MHC II organization detected in some other urodele amphibians, such as Ambystoma tigrinum (Bos and DeWoody 2005) or the congeneric T. cristatus (Babik et al. 2009, Wielstra et al. 2015). Otherwise, functional MHC II loci appear at least duplicated in other European salamanders (Babik et al. 2008; Nadachowska-Brzyska et al. 2012). Despite the lack of shared variants, we found extensive trans-species polymorphism between T. cristatus and T. carnifex, evident from exon-based genealogies. This indicates the retention of MHC allelic lineages for a long time, as the two species diverged ca. 5.4 Mya (Wielstra et al. 2019). The DAB sequence polymorphism is similar to that in T. cristatus (data in SuppInfo 14). Interestingly, the allelic richness (MHCAR = 4.4) is roughly comparable to that observed in populations of T. cristatus within the glacial refugia (mean MHCAR = 5.3 ± 1.1) rather than within post-glacial expansion areas where DAB variability was substantially depleted (mean MHCAR = 1.8 ± 0.3) (Babik et al. 2009). It is currently unclear whether this relatively high diversity results from the generally higher MHC variability in T. carnifex or from the proximity of refugial areas. In fact, multiple lowland refugia have been suggested for T. carnifex in the peninsular Italy (Canestrelli et al. 2012).
Evolutionary forces driving the spatial pattern of MHC variation
Two lines of evidence suggest the role of selection as the dominant mechanism driving MHC evolution and shaping MHC-DAB variation at a fine geographic scale. First, MHC variation did not correlate with neutral variation (STR) across populations (SuppInfo 11). Second, no IBD was detected for MHC, while IBD was significant for STR (Fig. 3). Thus, selection likely outweighs the effects of drift and distance-dependent gene flow, which are expected to shape diversity at neutrally evolving markers, especially in low-dispersal species. The FST outlier analysis suggested similar extents of genetic structuring at MHC and STR, and the Mantel test detected a positive correlation between FST matrices for the two types of markers. However, both the hierarchical analysis of genetic structure (AMOVA) and NMDSs revealed a more complex picture, indicating a stronger MHC differentiation at a local scale and stronger STR differentiation at a regional scale. MHC allele frequencies can differ substantially between neighbouring sampling sites, with no evidence of a larger-scale geographic structure, i.e. non-significant differentiation between subregions. In contrast, STRs are differentiated (although weakly) between subregions, while differentiation within subregions appears lower compared to MHC (Table 3). A similar scale-dependent partitioning of MHC variation was previously reported for guppies (Herdegen et al. 2014). Reduced differentiation at higher levels of geographic structuring is expected under overdominance and negative frequency dependence, as the effect of balancing selection compared to drift is more pronounced at lower migration rates (Schierup et al. 2000; Currat et al. 2015), e.g. where barriers to migration occur between geographic regions (the spatial structuring of neutral variation supports such a scenario in the present case; Fig. 4a, SuppInfo 2). Therefore, if balancing selection acts on MHC, but not on STR loci, the pattern we observe in T. carnifex may emerge. On the other hand, little structuring would be expected at smaller scales (i.e. within subregions) for both kinds of markers if gene flow would be high enough to homogenize allele frequencies even at neutral loci (Currat et al. 2015). However, we observed MHC differentiation between populations within subregions (Table 3, Fig. 4b), suggesting a more intricate scenario. Such fine-scale differentiation may result from host–parasite coevolutionary dynamics (i.e. Red Queen dynamics), which likely occurs asynchronously across sites (Herdegen et al. 2014). As a consequence of these non-synchronized cyclic dynamics, MHC allele frequencies are expected to fluctuate independently within populations, ultimately generating spatial genetic structuring even without substantial differences of the parasite communities across populations. Moreover, we cannot rule out that the detected pattern results from directional selection varying in space and time (Spurgin and Richardson 2010). However, it may be difficult to distinguish between Red Queen dynamics and fluctuating directional selection, especially if fluctuating selection is weak or local adaptation is at an early stage, and the two processes are not mutually exclusive.
Evidence for the divergent allele advantage
Our study provides evidence for DAA in T. carnifex. While we found only a relatively weak tendency for DAA within populations (possibly due to the limited power because of small sample sizes), signatures of DAA were detected when individuals from populations within the Western region or from all populations were pooled. This result is remarkable, because pooling should result in an apparent deficit of heterozygotes (Wahlund effect), making detection of DAA more difficult. This test was therefore conservative and we consider the evidence from pooled samples suggestive. In addition, when considering heterozygotes only, the DAA signal appears even stronger. As expected, the DAA was better supported for PSS than for all other sites—the former should be the immunologically most relevant sites, because they determine the spectrum of antigens recognized by a MHC molecule (Brown et al. 1993; Lenz 2011).
An excess of genotypes combining highly divergent alleles may result from direct selection caused by pathogens, but also from mate choice with respect to MHC dissimilarity. This has been reported in some studies (e.g. Forsberg et al. 2007, Juola and Dearborn 2011, Evans et al. 2012), but involvement of MHC in mate choice remains controversial (reviewed in Bernatchez and Landry 2003, Piertney and Oliver 2005). Whatever the mechanism underpinning the DAA, balancing selection mediated by DAA can contribute to the long-term maintenance of MHC diversity (Wakeland et al. 1990), explaining, at least in part, the retention of the (ancient) MHC variation in T. carnifex.
The extent of DAA across taxa remains poorly known due to the paucity of studies. To the best of our knowledge, this is the first report of DAA in an amphibian. Previously, DAA has been demonstrated in humans (Lenz 2011, Pierini and Lenz 2018) and wild terrestrial mammals (Lenz et al. 2009, 2013), but not in a bird (Gaigher et al. 2018). Notably, DAA has recently been invoked to explain the occurrence of highly divergent alleles in MHC-depleted population(s) of the Maui’s dolphin and the Apennine yellow-bellied toad, which have experienced recent or historical bottlenecks, respectively (Heimeier et al. 2018, Talarico et al. 2019). Clearly, further investigations of DAA in wild populations are needed.
Conclusions
Studies investigating evolutionary processes driving the pattern of MHC variation at a fine spatial scale are infrequent, although they are potentially highly informative. Therefore, the mechanisms actually involved in such dynamics remain poorly understood. Our study revealed a subtle, scale-dependent structuring of MHC variation compared to that of neutral markers. We interpret these differences as resulting from the action of recent or ongoing selection on MHC in T. carnifex. These findings emphasize the importance of hierarchical sampling, still rarely adopted, for understanding the evolutionary mechanisms shaping MHC diversity. We also found evidence for balancing selection in the form of divergent allele advantage, adding to a small number of studies that have tested this mechanism in natural populations. In general, our study contributes to the understanding of the evolutionary dynamics of MHC genes in taxa with low dispersal abilities.
Data archiving
MHC and STR individual genotypes, paired-end Fastq files (along with individual tags), and the R-function for DAA tests (i.e. code, usage and input data examples) have been archived in Dryad (https://doi.org/10.5061/dryad.ct40nm7). We deposited MHC sequences in GenBank (accessions: MH880293–MH880311).
References
Adamack AT, Gruber B (2014) PopGenReport: simplifying basic population genetic analyses in R. Methods Ecol Evol 5:384–387
Babik W, Pabijan M, Arntzen JW, Cogalniceanu D, Durka W, Radwan J (2009) Long-term survival of a urodele amphibian despite depleted major histocompatibility complex variation. Mol Ecol 18:769–781
Babik W, Pabijan M, Radwan J (2008) Contrasting patterns of variation in MHC loci in the Alpine newt. Mol Ecol 17:2339–2355
Beebee TJC (2005) Conservation genetics of amphibians. Heredity 95:423–427
Bernatchez L, Landry C (2003) MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol 16:363–377
Bos DH, DeWoody JA (2005) Molecular characterization of major histocompatibility complex class II alleles in wild tiger salamanders (Ambystoma tigrinum). Immunogenetics 57:775–781
Biedrzycka A, Sebastian A, Migalska M, Westerdahl H, Radwan J (2017) Testing genotyping strategies for ultra-deep sequencing of a co-amplifying gene family: MHC class I in a passerine bird. Mol Ecol Resour 17:642–655
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL et al. (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39
Canestrelli D, Salvi D, Maura M, Bologna MA, Nascetti G (2012) One species, three Pleistocene evolutionary histories: Phylogeography of the Italian crested newt, Triturus carnifex. PLoS ONE. 7:e41754
Carlsen T, Aas AB, Lindner D, Vrålstad T, Schumacher T, Kauserud H (2012) Don’t make a mista(g)ke: is tag switching an overlooked source of error in amplicon pyrosequencing studies? Fungal Ecol 5:747–749
Corsetti L (2006) Distribuzione e preferenze ambientali degli anfibi urodeli nel Lazio meridionale (Italia centrale). In: Zuffi MAL (ed) Atti V Congresso Nazionale Societas Herpetologica Italica. Firenze University Press, Calci (Pisa), p. 7–18
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Currat M, Gerbault P, Di D, Nunes JM, Sanchez-Mazas A (2015) Forward-in-time, spatially explicit modeling software to simulate genetic lineages under selection. Evol Bioinf 11:27–39
Cushman SA (2006) Effects of habitat loss and fragmentation on amphibians: a review and prospectus. Biol Conserv 128:231–240
Dayhoff MO, Schwartz RM, Orcutt BC (1978) A model for evolutionary change in proteins. In: Dayhoff MO (ed) Atlas of protein sequence and structure. National Biochemical Research Foundation, Washington DC, p. 345–352
Denoël M, Dalleur S, Langrand E, Besnard A, Cayuela H (2018) Dispersal and alternative breeding site fidelity strategies in an amphibian. Ecography 41:1543–1555
Dufresnes C, Pellet J, Bettinelli-Riccardi S, Thiébaud J, Perrin N, Fumagalli L (2016) Massive genetic introgression in threatened northern crested newts (Triturus cristatus) by an invasive congener (T. carnifex) in Western Switzerland. Conserv Genet 17:839–846
Edwards SV, Hedrick PW (1998) Evolution and ecology of MHC molecules: from genomes to sexual selection. Trends Ecol Evol 13:305–311
Eimes JA, Bollmer JL, Whittingham LA, Johnson JA, van Oosterhout C, Dunn PO (2011) Rapid loss of MHC class II variation in a bottlenecked population is explained by drift and loss of copy number variation. J Evol Biol 24:1847–1856
Ejsmond MJ, Radwan J (2015) Red Queen processes drive positive selection on major histocompatibility complex (MHC) genes. PLoS Comput Biol 11:e1004627
Ekblom R, Saether SA, Jacobsson P, Fiske P, Sahlman T, Grahn M et al. (2007) Spatial pattern of MHC class II variation in the great snipe (Gallinago media). Mol Ecol 16:1439–1451
Evans ML, Dionne M, Miller KM, Bernatchez L (2012) Mate choice for major histocompatibility complex genetic divergence as a bet-hedging strategy in the atlantic salmon (Salmo salar). Proc R Soc Lond B Biol Sci 279:379–386
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10:564–567
Foll M, Gaggiotti O (2008) A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993
Forsberg LA, Dannewitz J, Petersson E, Grahn M (2007) Influence of genetic dissimilarity in the reproductive success and mate choice of brown trout – females fishing for optimal MHC dissimilarity. J Evol Biol 20:1859–1869
Gaigher A, Burri R, Gharib WH, Taberlet P, Roulin A, Fumagalli L (2016) Family-assisted inference of the genetic architecture of major histocompatibility complex variation. Mol Ecol Resour 16:1353–1364
Gaigher A, Roulin A, Gharib WH, Taberlet P, Burri R, Fumagalli L (2018) Lack of evidence for selection favouring MHC haplotypes that combine high functional diversity. Heredity 120:396–406
Garrigan D, Hedrick PW (2003) Perspective: detecting adaptive molecular polymorphism: lessons from the MHC. Evolution 57:1707–1722
Gonzalez-Quevedo C, Davies RG, Phillips KP, Spurgin LG, Richardson DS (2016) Landscape-scale variation in an anthropogenic factor shapes immune gene variation within a wild population. Mol Ecol 25:4234–4246
Grantham R (1974) Amino acid difference formula to help explain protein evolution. Science 185:862–864
Hammer Ø, Harper DAT, Ryan PD(2001) PAST: Paleontological statistics software package for education and data analysis Palaeontol Electron 4:9
Heimeier D, Alexander A, Hamner RM, Pichler F, Baker CS (2018) The influence of selection on MHC DQA and DQB haplotypes in the endemic New Zealand Hector’s and Māui dolphins. J Hered 109:744–756
Herdegen M, Babik W, Radwan J (2014) Selective pressures on MHC class II genes in the guppy (Poecilia reticulata) as inferred by hierarchical analysis of population structure. J Evol Biol 27:2347–2359
Hughes AL, Yeager M (1998) Natural selection at major histocompatibility complex loci of vertebrates. Annu Rev Genet 32:415–435
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Jehle R, Burke T, Arntzen JW (2005) Delineating fine-scale genetic units in amphibians: probing the primacy of ponds. Conserv Genet 6:227–234
Juola FA, Dearborn DC (2011) Sequence-based evidence for major histocompatibility complex-disassortative mating in a colonial seabird. Proc R Soc Lond B Biol Sci 279:153–162
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Kelley J, Walter L, Trowsdale J (2005) Comparative genomics of major histocompatibility complexes. Immunogenetics 56:683–695
Klein J (1987) Origin of major histocompatibility complex polymorphism: the trans-species hypothesis. Hum Immunol 19:155–162
Kosakovsky Pond SL, Frost SDW (2005) Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222
Krupa AP, Jehle R, Dawson DA, Gentle LK, Gibbs M, Arntzen JW et al. (2002) Microsatellite loci in the crested newt (Triturus cristatus) and their utility in other newt taxa. Conserv Genet 3:87–89
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Lampert KP, Rand AS, Mueller UG, Ryan MJ (2003) Fine-scale genetic pattern and evidence for sex-biased dispersal in the túngara frog, Physalaemus pustulosus. Mol Ecol 12:3325–3334
Lenormand T (2002) Gene flow and the limits to natural selection. Trends Ecol Evol 17:183–189
Lenz TL (2011) Computational prediction of MHC II-antigen binding supports divergent allele advantage and explains trans-species polymorphism. Evolution 65:2380–2390
Lenz TL, Mueller B, Trillmich F, Wolf JBW (2013) Divergent allele advantage at MHC-DRB through direct and. Proc R Soc Lond B Biol Sci 280:20130714
Lenz TL, Wells K, Pfeiffer M, Sommer S (2009) Diverse MHC IIB allele repertoire increases parasite resistance and body condition in the long-tailed giant rat (Leopoldamys sabanus). BMC Evol Biol 9:269
Li J, Shen H, Wang H, Zhao M, Luo Z, Wu H (2016) Diversifying selection is the dominant factor affecting the geographical variation of MHC class II genes in the Omei tree frog. J Zool 300:197–204
Lighten J, van Oosterhout C, Paterson IG, Mcmullan M, Bentzen P (2014) Ultra-deep Illumina sequencing accurately identifies MHC class IIb alleles and provides evidence for copy number variation in the guppy (Poecilia reticulata). Mol Ecol Res 14:753–767
Magoč T, Salzberg SL (2011) FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 1:1–5
Mattoccia M (2015) Erpetofauna dei Monti Lepini (Lazio). In: Corsetti L, Angelini C, Copiz R, Mattoccia M, Nardi G (eds) Biodiversità dei Monti Lepini. Atti del convegno, Carpineto Romano (RM), 17 maggio 2014. Edizioni Belvedere, Le Scienze, Latina, p. 247–252
Miller K, Kaukinen K, Beacham T, Withler RE (2001) Geographic heterogeneity in natural selection on an MHC locus in sockeye salmon. Genetica 111:237–257
Mori E, Menchetti M, Cantini M, Bruni G, Santini G, Bertolino S (2017) Twenty years’ monitoring of a population of Italian crested newts Triturus carnifex: strong site fidelity and shifting population structure in response to restoration. Ethol Ecol Evol 29:460–473
Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL et al. (2013) FUBAR: A fast, unconstrained Bayesian approximation for inferring selection. Mol Biol Evol 30:1196–1205
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL (2012) Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764
Nadachowska-Brzyska K, Zieliński P, Radwan J, Babik W (2012) Interspecific hybridization increases MHC class II diversity in two sister species of newts. Mol Ecol 21:887–906
van Oosterhout C, Joyce DA, Cummings SM, Blais J, Barson NJ, Ramnarine IW et al. (2006) Balancing selection, random genetic drift, and genetic variation at the major histocompatibility complex in two wild populations of guppies (Poecilia reticulata). Evolution 60:2562–2574
Neefjes J, Jongsma MLM, Paul P, Bakke O (2011) Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11:823–836
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539
Pearson SK, Bull CM, Gardner MG (2018) Selection outweighs drift at a fine scale: Lack of MHC differentiation within a family living lizard across geographically close but disconnected rocky outcrops. Mol Ecol 27:2204–2214
Phillips KP, Cable J, Mohammed RS, Herdegen-radwan M, Raubic J, Przesmyckaa KJ et al. (2018) Immunogenetic novelty confers a selective advantage in host-pathogen coevolution. Proc Natl Acad Sci USA. 115:1552–1557
Pierini F, Lenz TL (2018) Divergent allele advantage at human MHC genes: Signatures of past and ongoing selection. Mol Biol Evol 35:2145–2158
Piertney SB, Oliver MK (2005) The evolutionary ecology of the major histocompatibility complex. Heredity 96:7–21
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Radwan J, Biedrzycka A, Babik W (2010) Does reduced MHC diversity decrease viability of vertebrate populations? Biol Conserv 143:537–544
Richardson JL, Urban MC, Bolnick DI, Skelly DK (2014) Microgeographic adaptiation and the spatial scale of evolution. Trends Ecol Evol 29:165–176
Richman AD, Herrera LG, Nash D, Schierup MH (2003) Relative roles of mutation and recombination in generating allelic polymorphism at an MHC class II locus in Peromyscus maniculatus. Genet Res 82:89–99
Rico Y, Morris-Pocock J, Zigouris J, Nocera JJ, Kyle CJ (2015) Lack of spatial immunogenetic structure among wolverine (Gulo gulo) populations suggestive of broad scale balancing selection. PLoS ONE 10:e0140170
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Res 8:103–106
Sandberg M, Eriksson L, Jonsson J, Sjöström M, Wold S (1998) New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J Med Chem 41:2481–2491
Schierup MH, Vekemans X, Charlesworth D (2000) The effect of subdivision on variation at multi-allelic loci under balancing selection. Genet Res 76:51–62
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864
Sebastian A, Herdegen M, Migalska M, Radwan J (2016) AMPLISAS: A web server for multilocus genotyping using next-generation amplicon sequencing data. Mol Ecol Res 16:498–510
Smith MA, Green DM (2005) Dispersal and the metapopulation paradigm in amphibian ecology and conservation: are all amphibian populations metapopulations? Ecography 28:110–128
Spurgin LG, Richardson DS (2010) How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc R Soc Lond B Biol Sci 277:rspb.2009.2084
Talarico L, Babik W, Marta S, Mattoccia M (2019) Genetic drift shaped MHC IIB diversity of an endangered anuran species within the Italian glacial refugium. J Zool 371:61–70
Wakeland EK, Boehme S, She JX, Lu C-C, McIndoe RA, Cheng I et al. (1990) Ancestral polymorphisms of MHC class II genes: Divergent allele advantage. Immunol Res 9:115–122
Wielstra B, Babik W, Arntzen JW (2015) The crested newt Triturus cristatus recolonized temperate Eurasia from an extra-Mediterranean glacial refugium. Biol J Linn Soc 114:574–587
Wielstra B, Mccartney-Melstad E, Arntzen JW, Butlin RK, Shaffer HB (2019) Phylogenomics of the adaptive radiation of Triturus newts supports gradual ecological niche 1 expansion towards an incrementally aquatic lifestyle 2. Mol Phylogenet Evol 133:120–127
Yang Z (2007) PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Acknowledgements
We thank to Ilaria Grossi, Jacopo Lorusso and Adelaide Rakaj, for their kind help in fieldwork, and to Marcello Giorgi for technical support in lab activities. We are also grateful to Prof. Barbara Mable and the three anonymous reviewers, which carefully revised our manuscript providing valuable comments and advices. This work was part of the PhD thesis of Lorenzo Talarico.
Authors’ contributions:
LT, WB, SM and MM conceived and designed the study. VP, MM and LT collected DNA samples. LT and VP conducted MHC and microsatellites lab experiments, respectively. LT analysed the data and wrote the paper. WB revised the manuscript. MM and SM approved the final version.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Talarico, L., Babik, W., Marta, S. et al. MHC structuring and divergent allele advantage in a urodele amphibian: a hierarchical multi-scale approach. Heredity 123, 593–607 (2019). https://doi.org/10.1038/s41437-019-0221-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-019-0221-3
This article is cited by
-
Divergent and non-parallel evolution of MHC IIB in the Neotropical Midas cichlid species complex
BMC Ecology and Evolution (2022)
