
Abstract
Purpose
Next-generation sequencing has revealed the major impact of de novo variants (DNVs) in developmental disorders (DD) such as intellectual disability, autism, and epilepsy. However, a substantial fraction of these predicted pathogenic DNVs remains challenging to distinguish from background DNVs, notably the missense variants acting via nonhaploinsufficient mechanisms on specific amino acid residues. We hypothesized that the detection of the same missense variation in at least two unrelated individuals presenting with a similar phenotype could be a powerful approach to reveal novel pathogenic variants.
Methods
We looked for variations independently present in both our database of >1200 solo exomes and in denovo-db, a large, publicly available collection of de novo variants identified in patients with DD.
Results
This approach identified 30 variants with strong evidence of pathogenicity, including variants already classified as pathogenic or probably pathogenic by our team, and also several new variants of interest in known OMIM genes or in novel genes. We identified FEM1B and GNAI2 as good candidate genes for syndromic intellectual disability and confirmed the implication of ACTL6B in a neurodevelopmental disorder.
Conclusion
Annotation of local variants with denovo-db can highlight missense variants with high potential for pathogenicity, both facilitating the time-consuming reanalysis process and allowing novel DD gene discoveries.
Similar content being viewed by others


INTRODUCTION
The Deciphering Developmental Disorders project has estimated that up to 41.8% of the patients in their cohort of more than 4000 families harbor a pathogenic de novo variant (DNV).1 A substantial proportion of these predicted pathogenic DNVs remains challenging to distinguish from background DNVs, emphasizing the need for new and subtle approaches in data analysis. DNV pathogenicity is underlain by two distinct mechanisms that depend on the variant’s molecular effect. The first is the loss-of-function (LOF) mechanism, mediated notably by truncating variants in which the transcript and/or the protein levels are expected to be lowered. The second is the altered function mechanism, or nonhaploinsufficient mechanism, which results from a protein that is produced but with impaired function (e.g., a dominant negative effect or a toxic effect). It is becoming increasingly clear that most developmental diseases caused by LOF DNMs have already been identified. Despite some exceptions, the effects of truncating variants in a specific gene are roughly homogeneous, which denotes the binary nature of LOF variants. This binary aspect allows statistical models2 to be applied efficiently, either for the identification of genes depleted for LOF variations in general population (ExAC probability of loss-of-function intolerance [pLI]3), or, on the contrary, for the identification of genes enriched in truncating DNVs in affected populations.1,4 Conversely, missense variants with a nonhaploinsufficient mechanism are much more challenging to work with. The effects are highly heterogeneous, ranging from neutral polymorphisms to deleterious variants, greatly complicating the statistical modeling of these variants.5 Several diseases might be linked to a small number of possible pathogenic missense variants affecting the protein in a specific way, implicating highly unlikely and therefore rare mutational events. Identifying recurrence and/or variant clustering is essential for determining the genes and diseases associated with these variations.
Over the past decade, next-generation sequencing (NGS) and pangenomic analyses have enabled researchers to identify the genetic bases for several developmental disorders. The initial strategies used small cohorts with a homogeneous phenotype of unknown molecular basis to identify recurrently mutated genes.6,7 This phenotype-first approach has yielded many successful results but has reached its limitations in nonsyndromic diseases or atypical presentations. With the decrease in the cost of sequencing and maturation of the technology, NGS has been added to routine medical care, and pangenomic data is now being collected from large heterogeneous cohorts.
For the most part, the identification of the genetic bases of unrecognized developmental disorders has shifted to genotype-first approaches. Here, data are produced and then mined to identify new genetic bases using various methods, including the evaluation of “phenotypical recurrence” in patients with similar genotype, and an increasing number of statistical approaches. The power of these strategies suggests that the most frequent causes of genetic disease have been uncovered and that the remaining genotype–phenotype correlations are for ultrarare diseases only. Large-scale sharing of both clinical and genetic data will be needed to delineate these rare clinicobiological entities, and there have already been attempts to facilitate this process. GeneMatcher is one such solution in which users can share variants of interest online to find other individuals with similar genotypes and phenotypes.8 Several teams, including ours, have used GeneMatcher successfully for disease identification. On a wider scale, sharing large sets of unsorted variants can also yield positive results. For example, denovo-db is a database that gathers thousands of de novo variations identified in various cohort studies of mainly trio exome sequencing (ES) and trio genome sequencing (GS).9 In a recent study, the authors applied a statistical algorithm to denovo-db data to look for clusters of de novo missense variants, which resulted in the identification of three new genes, including ACTL6B.10
Recurrence of the same de novo variant in at least two unrelated individuals, what we call variant recurrence, is not exceptional in developmental disorders,11,12 but rare are the studies that take advantage of this recurrence for the identification of variants of interest.13 We hypothesized that patients from our local cohort with developmental disorder and negative solo exome sequencing could harbor variants identified as de novo in large trio cohort studies of developmental disorders, and that identifying a recurrence would weigh in favor of these variants’ pathogenicity. Our approach attempted to identify missense pathogenic variants in our database of >1200 solo exomes based on data obtained from denovo-db.
MATERIALS AND METHODS
Selection of variants of interest from denovo-db
We first created a set of missense variants of interest, identified at the de novo state in at least one individual with a developmental disorder. We downloaded a file containing the 283,888 variants of denovo-db version 1.5 from the publicly available website (http://denovo-db.gs.washington.edu/denovo-db/Download.jsp). For each variant, several data were available, including standard annotations such as the predicted functional effect of the variant and the frequency of the allele in various databases. The cohort study in which the variant was detected was also specified, providing basic clinical information about the patient harboring the de novo variant. Only variants of interest that matched the clinical presentations investigated in our institution were considered. From the 18 clinical cohorts available in denovo-db, we selected the variants from 6 subcohorts linked to pediatric developmental disorders: autism, intellectual disability, developmental disorder, epilepsy, neural tube defects, and acromelic frontonasal dysostosis (Fig. 1). We also excluded variants that were observed at least once in the ExAC database, because (1) ExAC is thought to be free of individuals with severe pediatric disorders, and (2) most single-nucleotide variants (SNVs) implicated in severe developmental disorders with a de novo mechanism appear fully penetrant, with very few resilient individuals in control databases.14,15 This filtration process resulted in a list of 7335 de novo events from denovo-db, corresponding to 7205 unique missense variants of interest.

Study flowchart. Denovo-db subcohorts used are highlighted in bold. DD developmental disorder, NGS next-generation sequencing.
Local ES and GS cohort
At the time of this study, our database contained 1271 entries from ES of probands with a developmental disorder. All individuals had signed written consent and the local ethical committee approved this study (Comité de Protection des Personnes, CPP, number DC2011–1332). The majority were sequenced with a solo strategy (n = 1036), but duos, trios, and trio+ were also present (n = 235). Raw data from exomes were analyzed using a standard in-house pipeline as previously described,16 leading to the identification of approximately 400 high quality rare sequence variants with a coding effect (or intronic variants located close to exon–intron junctions) in each patient. Variant interpretation was performed independently by two biologists following American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) recommendations with a focus on genes associated with a human disease in OMIM. Most of the negative exomes underwent serial reanalysis.17 Prior to this study, of the 1271 entries, 376 analyses (29.6%) were annotated as positive, 137 (10.8%) as inconclusive (indicating the presence of a variation of unknown significance), and 758 (59.6%) as negative.
Identifying recurrence within our local cohort
Using a custom Python script, we looked for the subset of 7205 variants extracted from denovo-db in our local data. With the hypothesis that variant recurrence could help to highlight critical amino acid residues, we extended our analysis not only to strictly similar missense variants, but also to missense variants resulting from a distinct substitution affecting the same nucleotide, and even from a substitution of a distinct nucleotide within the same codon. Following this process, the recurrent variants thus identified were reannotated using SNPEff and SNPSift, providing updated annotations from standard databases used for variant interpretation such as gnomAD, OMIM, ClinVar, and some missense pathogenicity predictors. Variants present in at least one individual in the gnomAD cohort were ruled out. We applied this strategy to the entire local cohort without considering patient phenotypes or the previously detected genetic alterations. The rare variants identified within our exome cohort with this strategy were manually assessed according to ACMG guidelines.18 We considered the concordance between the proband’s phenotype and the phenotype of the denovo-db subcohort, the functional relevance of the gene, its expression and its implication in human disease as reported in OMIM and in the literature. We also looked at the relevance of the variant according to the affected transcripts, the pathogenicity prediction algorithms, and the potential presence of the variant in databases such as ClinVar or HGMD. Free web-based resources for genome analysis, such as the University of California–Santa Cruz (UCSC) Genome Browser19 and VarSome (https://varsome.com/) were used for manual assessment. Confirmation and parental segregation of the variants of interest were performed by Sanger sequencing according to standard procedure. Polymerase chain reaction (PCR) primers are available upon request. Candidate research variants were further investigated through data sharing and collaboration, either with investigators of denovo-db cohort studies or with the GeneMatcher platform.8 Figure 1 summarizes the overall study procedure.
RESULTS
Characteristics of recurrent variants
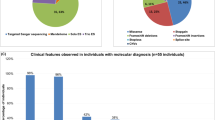
Our strategy identified 67 ultrarare good quality missense variants that were either strictly the same variant (n = 32), a distinct variant affecting the same nucleotide (n = 12), or a distinct nucleotide change affecting the same codon (n = 23) as a de novo missense variant identified in a patient with developmental disorder in the denovo-db database (Fig. 2a, Supplementary Figure 1).

Characteristics of the variants identified by variant recurrence. a Recurrence subtype. b Pathogenicity assessment. c Type of substitutions. DD developmental disorder, DNV de novo variant.
According to the OMIM database, a significant proportion of the 67 identified genes were known to be involved in a developmental disorder (intellectual disability, epileptic encephalopathy, or autism) caused by de novo pathogenic variants (29/67, 43.3%) (Fig. 2b). Moreover, 21 variants (31.3%) were annotated as pathogenic or likely pathogenic by one or more users in the ClinVar database.
We had already recognized 21 (31.3%) of the 67 variants as responsible (or strong candidates) for disease in the 23 corresponding affected patients (Supplementary table 1). These findings indicate that our approach is an efficient way to detect pathogenic variants via variant recurrence.
Identification of new variants of interest
We then sought to analyze the 46 remaining missense variants harbored by probands with developmental disorder whose previously solo ES analysis was negative (Supplementary Figure 1). As we did not only consider variants in known developmental disorder genes, we used gene expression data from GTEx to manually exclude genes with no brain expression, considered as poor candidate genes for intellectual disability and neurodevelopmental disorders. Twenty-five of these 46 variants were not retained for further analysis, because (1) they were affecting a nonrelevant gene (no brain expression), (2) they were also harbored by controls or unaffected parents either in denovo-db or our local database, or (3) they were identified in a patient with another pathogenic variant interpreted as fully contributing to the phenotype (Supplementary table 2).
The last 21 variants (31.3%) were considered as new candidate variants of interest and were subsequently validated by Sanger sequencing in the probands and their parents (and affected brother in the case of a dominant transmission of an ACTL6B variant). Eleven of these 21 candidate variants were ruled out because they were inherited from healthy parents (Supplementary table 2). The other ten variants were either de novo (n = 8, GNB1, DHDDS, GABRB2, CLTC, CACNA1A, FEM1B, PCGF2, and GNAI2) or displayed good segregation with the affected status (n = 2, ACTL6B and ZFX). Six de novo variants were affecting developmental disorder genes compatible with the patient’s phenotype and therefore considered as pathogenic (GNB1, DHDDS, GABRB2, CLTC, CACNA1A, and PCGF2). To assess why these variants were not selected after the first analysis of the exomes, we checked the release date of these OMIM entries, assuming they had been recently added. Indeed, 3 entries had been added less than three months prior to this study, two were between one and two years old, and one entry has not been added to OMIM yet (PCGF2 gene). Therefore, these novel diagnosis were related to clinico-biological entities of recent description compared to the recurrent variants already known as pathogenic described in Supplementary table 1. Four of the ten variants remained candidates: the de novo p.(Arg126Gln) variant in FEM1B, p.(Gly343Arg) in ACTL6B, p.(Arg179His) in GNAI2, and p.(Arg764Trp) variant in ZFX.
The variant recurrence of the FEM1B missense variant was highlighted in a patient with a syndromic global developmental delay from our cohort and a patient from the DDD study (individual DDD4K.00478). Segregation analysis showed that this variant had also occurred de novo in our patient. International collaboration through GeneMatcher identified a third individual with a developmental disorder of unknown cause and the same de novo missense variant. FEM1B is a small ubiquitous gene that has never been linked to a developmental disorder. These three patients share a strikingly similar phenotype that will be the object of a future study.
We identified the p.(Gly343Arg) variant in the ACTL6B gene in two siblings presenting with severe global developmental delay and hypotonia. The variant was not inherited from the mother, and the father, who presented an intellectual disability, was not available for testing. This variant, though possibly inherited, was therefore of clinical relevance within the scope of a dominant mode of inheritance. Insights about the pathogenicity of de novo ACTL6B missense variants came from a recent study that identified a significant cluster of de novo variants in patients with developmental disorders.10 This cluster had the same missense variant found de novo in three unrelated individuals. The phenotype of these patients has not yet been described. Our approach identified a distinct sequence variant, but one that led to the same missense effect p.(Gly343Arg). Several patients with ACTL6B pathogenic variants have recently been brought together to characterize the associated clinical phenotype (manuscript in process by collaborators).
De novo p.(Arg179His) in the GNAI2 gene was identified at in a proband with a syndromic developmental disorder. Denovo-db includes a distinct substitution of the same residue in a patient with a developmental disorder: p. (Arg179Cys). Intriguingly, both amino acid changes p.(Arg179His) and p.(Arg179Cys) demonstrate an old-established oncogenic potential,20 and have been observed at a somatic state in several endocrine tumors, with a documented activating effect. We hypothesize that these activating GNAI2 variants could, at a constitutive state, lead to a developmental disorder. In accordance with this hypothesis, a collaborating team has gathered a series of patients and will expose the associated phenotype in the near future.
The ZFX variant p.(Arg764Trp) was identified in a hemizygous state in a proband with a syndromic disorder of unknown cause characterized by global developmental delay, dysmorphic facial features, and several other abnormalities such as angiomatosis, hyperoxaluria, and deficit of corticotropic hormones. The variant was inherited from an asymptomatic mother and no further segregation analysis was possible. The X-linked ZFX gene has not yet been associated to developmental disorders. The same missense variant was identified de novo in the denovo-db database in a male proband with a developmental disorder. Altogether, these results are promising but will require further investigation.
Mechanisms of variant recurrence
We identified two potential mechanisms that might explain the observation of the unlikely events of strict variant recurrence. First, we observed a higher rate of CpG transitions within recurrent variants (19/32, 59.4%) than in the initial set of denovo-db variants used in this study (17.7%, p = 1.9 × 10−7, Fisher’s exact test), which was similar to the CpG transition rate in DNV from the general population21 (17.33%, p = 0.58) (Fig. 2d). This finding suggests a strong and rational association between variant recurrence and the likely variant event of CpG transitions,22 which has previously been observed by another group.13 The second possible mechanism is the model of “selfish spermatogonial selection”.23 One variant in the PPP2R5D gene displayed extreme variant recurrence since it was found in ten independent individuals in denovo-db in addition to one patient from our cohort. Indeed, the gain-of-function mechanism of this variant and the overgrowth aspect of the associated syndrome are in line with this hypothesis.24
Mechanisms of pathogenicity
Missense variants can lead to disease either by a loss-of-function mechanism in haploinsufficient genes, or by more specific alterations of the protein, grouped under the term nonhaploinsufficient (NHI) mechanism. We hypothesized that the active selection of very specific residues by variant recurrence could specifically highlight variants acting via NHI mechanism. We used the DD gene2phenotype database (https://www.ebi.ac.uk/gene2phenotype/) to annotate the mechanism of pathogenicity of the definite pathogenic variants identified by variant recurrence (Table 1 and Supplementary Table 1). This analysis showed a NHI mechanism for 72% of the variants (n = 18/25, either “activating”, “dominant negative”, or “all missense/in frame”), and a loss-of-function mechanism for seven variants.
DISCUSSION
The approach described here retrospectively identified a small set of rare missense variants of interest within our >1200 exomes database. This set was highly enriched with clinically relevant variants that were either known as the cause of our patient’s disease (21/67) or newly discovered (10/67).
More than one-third of this set of variants (26/67) was confirmed to have occurred de novo in our probands. Thus, the presence of a recurrence with denovo-db, and particularly a strict recurrence (Fig. 2a), appears to be a major predictor of the de novo status of rare variants. This characteristic is particularly valuable when trio sequencing is not available, as for the majority of our exomes. Variant recurrence can be coincidental (Supplementary table 2), due to the increasing size of the cohorts,3 but two factors allowed us to effectively overcome this background noise. First, we only considered variants not found in the general population (minor allele frequency [MAF] = 0 in the ExAC database), thus excluding many highly mutable genomic positions unrelated to the disease. Secondly, the phenotypic data we were able to access allowed us to compare the phenotype of our probands with the information from the clinical cohort of the denovo-db patients. This proved a powerful means to spot strong candidates.
Within our cohort, most of the diagnostic variants with known genotype–phenotype correlations identified by variant recurrence had already been found during the initial clinical exome analysis (23/29 patients). Six additional diagnoses were obtained thanks to articles published subsequent to the initial analyses (GNB1,25 DHDDS,12 GABRB2,12 CLTC,12 PCGF2,26 and CACNA1A27).
Our analysis has led to further consideration of the pathogenicity of known variants. A de novo missense variant (NM_001429.3:c.4783T>G, p.Phe1595Val) in EP300 was identified in a patient from the DDD study who presented with very mild features evocative of Rubinstein–Taybi syndrome (RTS).28 This variant was considered pathogenic and was identified as such in the ClinVar database. We observed the same variant in a proband with neurodevelopmental disorder but lacking the characteristic morphologic features of RTS. This variant was inherited from an asymptomatic father with no evidence of mosaicism, so it can either represent a false positive finding of trio sequencing in the DDD cohort or a variant with incomplete penetrance. We did not return this result, which we considered a variant of unknown significance.
Our approach highlighted variants of interest in genes with a neurodevelopmental phenotype not concordant with the literature. This includes variants in KCNMA1, KCNQ3, and SMARCA2 genes, described respectively in paroxysmal nonkinesigenic dyskinesia with or without generalized epilepsy (OMIM 609446), neonatal benign seizures (OMIM 121201), and Nicolaides–Baraitser syndrome (OMIM 601358). These patients and their clinical description, within wider cohorts assembled through data sharing, are currently the object of distinct work to delineate new genotype–phenotype associations.
Finally, variant recurrence also helped us to identify novel candidate genes. Two of them were already under consideration prior to this study, including one variant in PACS2 recently published29 in two unrelated individuals and one variant in TRAF7.30 OMIM disease references for both genes have recently been published (#618067 and #618164). New candidates in the FEM1B, ACTL6B, GNAI2, and ZFX genes were identified though this approach. These results highlight the strength of variant recurrence as a strategy to identify new disease-associated genes that harbor highly specific and clustered missense variants. The phenotype associated with FEM1B, ACTL6B, and GNAI2 genes will be the object of future publications. The ZFX variant requires further replication and segregation before a conclusion can be reached.
Besides missense variants, we also investigated other types of variants (data not shown). The recurrence of truncating variants, either stop-gain and indels frameshift variants, highlighted several pathogenic de novo variants, all of which were previously identified variants. Overall, the information of recurrence in LOF variants was not as useful as in missense variants, since LOF variants are much easier to implicate. Conversely, the observation of recurrence in in-frame indels (potentially leading to a disease via a nonhaploinsufficient mechanism) could be very useful, but we did not observe any.
Our approach relied on the powerful insights brought by variant recurrence in ultrarare diseases. The originality of this work was to highlight candidate variants in an unbiased way, initially independent from any interpretation process and candidate variants or genes. This straightforward method used accessible and easy-to-manage data from denovo-db. We created a color-coded custom track for the UCSC Genome Browser including the whole denovo-db data set, which turned out to be a useful tool in routine exome interpretation. It provided an overview of gene mutability, variant clustering, and variant type in diverse clinical presentations found in denovo-db. In addition to occasional retrospective analysis, we believe that prospective variant annotation with denovo-db could help highlight variants of interest in an unbiased approach, with limited overlap from ClinVar and HGMD. As publicly available trio-based sequencing cohorts are growing, so will the observation of variant recurrence. We therefore believe that our approach has potential to gain power in the future.
Our team showed the benefits of prospective annual reanalysis of ES data, and a new diagnosis was possible in up to 15.4% of our undiagnosed probands17 thanks to recent breakthroughs in research in the field of developmental disorders. However, complete reanalysis of NGS data continues to be tedious and expensive, raising questions about medium- and long-term feasibility. A focus on variant recurrence limits the number of variants to consider and is compatible with large-scale occasional reanalysis.
We hypothesize that variant recurrence will be a key consideration for future identification of new ultrarare diseases, notably those associated with specific nonhaploinsufficient missense variants. Recurrence of de novo variants at the nucleotide scale could also be a powerful tool to highlight other types of variants in which functional predictions are still lacking, including intronic, intergenic variants, or variants harbored by noncoding RNAs. It may also provide a useful unbiased approach to help uncover new phenotype–genotype relationships in genes that are already known to cause a distinct disease, for which standard variant interpretation would fail due to presumed phenotype incompatibility. Systematic variant aggregation such as denovo-db is a particularly pertinent approach. Otherwise, the pooling of unsolved cases into large cohorts might result in an increase in the signal of new pathogenic variant recurrence. The approach used for the present study will certainly play a key role in the European Solve-RD initiative, which plans to aggregate data from over 18,000 unsolved cases of presumed monogenic disorders, hopefully increasing our understanding of the genetic origins of ultrarare diseases.
References
Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–438.
Samocha KE, Robinson EB, Sanders SJ, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944–950.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Lelieveld SH, Reijnders MRF, Pfundt R, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19:1194–1196.
Sivley RM, Dou X, Meiler J, Bush WS, Capra JA. Comprehensive analysis of constraint on the spatial distribution of missense variants in human protein structures. Am J Hum Genet. 2018;102:415–426.
Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793.
Clayton-Smith J, O’Sullivan J, Daly S, et al. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am J Hum Genet. 2011;89:675–681.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–930.
Turner TN, Yi Q, Krumm N, et al. denovo-db: a compendium of human de novo variants. Nucleic Acids Res. 2017;45(D1):D804–D811.
Lelieveld SH, Wiel L, Venselaar H, et al. Spatial clustering of de novo missense mutations identifies candidate neurodevelopmental disorder-associated genes. Am J Hum Genet. 2017;101:478–484.
Wilfert AB, Sulovari A, Turner TN, Coe BP, Eichler EE. Recurrent de novo mutations in neurodevelopmental disorders: properties and clinical implications. Genome Med. 2017;9:101.
Hamdan FF, Myers CT, Cossette P, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet. 2017;101:664–685.
Geisheker MR, Heymann G, Wang T, et al. Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nat Neurosci. 2017;20:1043–1051.
Chen R, Shi L, Hakenberg J, et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol. 2016;34:531–538.
Tarailo-Graovac M, Zhu JYA, Matthews A, van Karnebeek CDM, Wasserman WW. Assessment of the ExAC data set for the presence of individuals with pathogenic genotypes implicated in severe Mendelian pediatric disorders. Genet Med. 2017;19:1300–1308.
Thevenon J, Duffourd Y, Masurel-Paulet A, et al. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet. 2016;89:700–707.
Nambot S, Thevenon J, Kuentz P, et al. Clinical whole-exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genet Med. 2017.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Karolchik D, Hinrichs AS, Kent WJ. The UCSC Genome Browser. Curr Protoc Bioinformatics. 2012;40:1.4.1–1.4.33.
Lyons J, Landis CA, Harsh G, et al. Two G protein oncogenes in human endocrine tumors. Science. 1990;249:655–659.
Kong A, Frigge ML, Masson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475.
Francioli LC, Polak PP, Koren A, et al. Genome-wide patterns and properties of de novo mutations in humans. Nat Genet. 2015;47:822–826.
Goriely A, McGrath JJ, Hultman CM, Wilkie AOM, Malaspina D. “Selfish spermatogonial selection”: a novel mechanism for the association between advanced paternal age and neurodevelopmental disorders. Am J Psychiatry. 2013;170:599–608.
Shang L, Henderson LB, Cho MT, et al. De novo missense variants in PPP2R5D are associated with intellectual disability, macrocephaly, hypotonia, and autism. Neurogenetics. 2016;17:43–49.
Petrovski S, Küry S, Myers CT, et al. Germline de novo mutations in GNB1 cause severe neurodevelopmental disability, hypotonia, and seizures. Am J Hum Genet. 2016;98:1001–1010.
Turnpenny PD, Wright MJ, Sloman M, et al. Missense mutations of the Pro65 residue of PCGF2 cause a recognizable syndrome associated with craniofacial, neurological, cardiovascular, and skeletal features. Am J Hum Genet. 2018;103:786–793.
Riant F, Ducros A, Ploton C, Barbance C, Depienne C, Tournier-Lasserve E. De novo mutations in ATP1A2 and CACNA1A are frequent in early-onset sporadic hemiplegic migraine. Neurology. 2010;75:967–972.
Hamilton MJ, Newbury-Ecob R, Holder-Espinasse M, et al. Rubinstein-Taybi syndrome type 2: report of nine new cases that extend the phenotypic and genotypic spectrum. Clin Dysmorphol. 2016;25:135–145.
Olson HE, Jean-Marçais N, Yang E, et al. A recurrent de novo PACS2 heterozygous missense variant causes neonatal-onset developmental epileptic encephalopathy, facial dysmorphism, and cerebellar dysgenesis. Am J Hum Genet. 2018;102:995–1007.
Tokita MJ, Chen C-A, Chitayat D, et al. De novo missense variants in TRAF7 cause developmental delay, congenital anomalies, and dysmorphic features. Am J Hum Genet. 2018;103:154–162.
Acknowledgements
The authors thank Suzanne Rankin for proofreading the manuscript. François Lecoquierre received funding from Rouen University Hospital, Rouen, France.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lecoquierre, F., Duffourd, Y., Vitobello, A. et al. Variant recurrence in neurodevelopmental disorders: the use of publicly available genomic data identifies clinically relevant pathogenic missense variants. Genet Med 21, 2504–2511 (2019). https://doi.org/10.1038/s41436-019-0518-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0518-x
Keywords
This article is cited by
-
Systematic reanalysis of genomic data by diagnostic laboratories: a scoping review of ethical, economic, legal and (psycho)social implications
European Journal of Human Genetics (2024)
-
Structural insights into the ubiquitylation strategy of the oligomeric CRL2FEM1B E3 ubiquitin ligase
The EMBO Journal (2024)
-
Evaluating the use of paralogous protein domains to increase data availability for missense variant classification
Genome Medicine (2023)
-
Landscape of mSWI/SNF chromatin remodeling complex perturbations in neurodevelopmental disorders
Nature Genetics (2023)
-
Defining E3 ligase–substrate relationships through multiplex CRISPR screening
Nature Cell Biology (2023)
