
Abstract
The contribution of de novo variants as a cause of intellectual disability (ID) is well established in several cohorts reported from the developed world. However, the genetic landscape as well as the appropriate testing strategies for identification of de novo variants of these disorders remain largely unknown in low-and middle-income countries like India. In this study, we delineate the clinical and genotypic spectrum of 54 families (55 individuals) with syndromic ID harboring rare de novo variants. We also emphasize on the effectiveness of singleton exome sequencing as a valuable tool for diagnosing these disorders in resource limited settings. Overall, 46 distinct disorders were identified encompassing 46 genes with 51 single-nucleotide variants and/or indels and two copy-number variants. Pathogenic variants were identified in CREBBP, TSC2, KMT2D, MECP2, IDS, NIPBL, NSD1, RIT1, SOX10, BRWD3, FOXG1, BCL11A, KDM6B, KDM5C, SETD5, QRICH1, DCX, SMARCD1, ASXL1, ASXL3, AKT3, FBN2, TCF12, WASF1, BRAF, SMARCA4, SMARCA2, TUBG1, KMT2A, CTNNB1, DLG4, MEIS2, GATAD2B, FBXW7, ANKRD11, ARID1B, DYNC1H1, HIVEP2, NEXMIF, ZBTB18, SETD1B, DYRK1A, SRCAP, CASK, L1CAM, and KRAS. Twenty-four of these monogenic disorders have not been previously reported in the Indian population. Notably, 39 out of 53 (74%) disease-causing variants are novel. These variants were identified in the genes mainly encoding transcriptional and chromatin regulators, serine threonine kinases, lysosomal enzymes, molecular motors, synaptic proteins, neuronal migration machinery, adhesion molecules, structural proteins and signaling molecules.
Similar content being viewed by others


Introduction
Intellectual disability (ID) is defined as a defect in cognitive functioning and adaptive behavior that originates before the age of 18 years and has a worldwide prevalence of ~2–3% [1]. The etiology of ID includes both acquired as well as genetic causes [2, 3]. The genetic etiology of disorders of ID is highly heterogeneous, encompassing a wide spectrum of genetic variations, including structural variants (SVs), copy number variants (CNVs), small insertions/deletions, and single-nucleotide variants (SNVs), identified across more than 1000 genes [4,5,6,7]. Monogenic disorders contribute to 30–50% of cases of ID, around 20% are due to disease-causing large or small CNVs and the cause of ~50% cases remains unknown till date [4, 5, 8, 9].
Disorders which have a component of ID and are associated with other systemic or behavioral abnormalities are referred to as syndromic ID (sID). These disorders follow all inheritance patterns and show extreme genetic heterogeneity. With the availability of rapidly advancing next generation sequencing (NGS) based platforms, mainly exome and genome sequencing (ES/GS) has led to the rapid and efficient diagnosis as well as discovery of several ID syndromes in the last two decades [10].
De novo variants are now a well-recognized cause of severe early-onset genetic diseases, including sIDs. Though the genetic basis of inherited sIDs could be identified through studies involving large family pedigrees, the sporadic ones remained largely unidentified until recently. NGS approaches, particularly proband-parents trio ES/GS is an effective method of understanding the distribution of variations and determining all types of de novo events throughout the genome, from SNVs, indels, CNVs to large SVs in addition to determining the parental origin and whether they occurred in the germline or post zygotically [4, 8, 9, 11].
The challenges faced by the low- and middle-income countries (LMIC) in terms of rare disease diagnosis using the evolving genomic technologies include the lack of coherent national policies, limited trained professionals, lagging research infrastructure, and lastly economic and cultural challenges [12, 13]. The extent of the burden posed by de novo variants associated with ID syndromes remains incompletely understood within several LMICs, including India. With the aim to address this challenge, we herein represent the clinical and genetic spectrum of 54 families with de novo variants underlying sID. We also highlight the utility of proband-only ES followed by segregation analysis as a first-tier testing in identification of de novo variants in resource-limited settings.
Material and methods
We evaluated and recruited 530 families with heterogeneous neurodevelopmental disorders (NDDs) in an ongoing mono centric study from October 2019 to December 2022. The clinical characteristics of the affected individuals were recorded through detailed clinical examination using human phenotype ontology (HPO terms). Informed consents for genetic testing, publication of data and clinical photographs were obtained from the families. The informed consents were approved by the Institutional ethics committee, Kasturba Medical College and Kasturba Hospital, India as per the declaration of Helsinki.
Genomic DNA was extracted from the peripheral blood sample of the proband, parents and siblings (as required) using the QIAamp DNA Blood Mini Kit (QIAGEN, Valencia, CA; cat # 51106). The testing strategy included either an exome first or a sequential testing approach in which a targeted test or chromosomal microarray (CMA) was followed by ES for the affected individuals based on the clinical phenotype. The NGS data processing, quality assessment, variant calling, annotation and analysis was performed as described earlier [14]. The Sanger validation and segregation analysis was carried out in all families with a singleton ES or Mendeliome while Sanger validation was carried out in families who achieved a diagnosis using a trio ES. CNV analysis from exome data was performed for individuals with no clinically relevant SNVs/indels detected on ES. The detailed description of Mendeliome, CNV and ES analysis is provided in the supplementary material.
Results
Of the 530 families recruited, 211 affected individuals from 196 families presented with a syndromic presentation characterized by major or minor morphologic anomalies and neurologic, cognitive, behavioral or sensory impairments. A molecular diagnosis could be achieved in 104 of the total 196 families (53%). Of these, 59 families (57%) carried de novo variants, six families (4%) had inherited variants underlying an autosomal dominant or an X-linked disorder, 16 families (16%) had biallelic variants underlying an autosomal recessive disorder, and 23 families (23%) were diagnosed with a chromosomal aberration. Consanguinity was noted in 25 families (24%). Within the 25 consanguineous families, 14 families (56%) had biallelic variants underlying an autosomal recessive disorder, two families (8%) had inherited variants underlying X-linked recessive disorders, six (24%) carried de novo variants causing autosomal dominant and X-linked dominant disorders, and three families (12%) had CNVs.
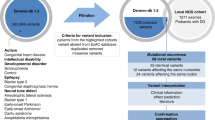
Of the 59 families with de novo variants, five families (five affected individuals) with novel disease-gene association, phenotypic expansion, and multiple genetic diagnoses have been published earlier [15,16,17]. The present cohort consists of 55 individuals from 54 families diagnosed using targeted Sanger sequencing, Mendeliome, singleton ES, and trio ES (Fig. 1A and Table 1). Thirty-one affected individuals were males (57%) and 24 were females (43%). Consanguinity was noted in six families (11%). The age ranged from newborn to 14 years. The clinical findings noted in 55 diagnosed individuals, in addition to ID, included global developmental delay, dysmorphism, malformations, seizures, autism spectrum disorder, hypotonia, and sensory dysfunction (Fig. 1C). A total of 53 disease-causing de novo variants underlying 46 distinct ID syndromes were identified in the current cohort (Table 1). Of which, 51 were SNVs and/or indels and two were CNVs (Fig. 1B). Notably, 39 (74%) of them were found to be novel. These SNVs and indels were classified according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) standards and guidelines for interpreting sequence variants [18]. Thirty variants were classified as pathogenic (58%) and 21 as likely pathogenic (40%). The two CNVs identified were classified as pathogenic according to the American College of Medical Genetics and Genomics and ClinGen standards and guidelines for CNVs (Table 1) [19]. The additional details pertaining to the genetic testing performed, disease-causing variants, and ClinVar submission IDs are provided in Supplementary Table S1.

A Types of genetic testing employed B Disease-causing de novo variants identified C Spectrum of the clinical findings observed in individuals with sID. GDD Global developmental delay, ASD Autism spectrum disorder.
Discussion
Several cohorts of individuals harboring disease-causing de novo variants and underlying sIDs have been reported in the last two decades. However, most of these cohorts have originated from Caucasian outbred populations of the developed world [20,21,22]. The spectrum as well as the burden of the variants contributing to sID in LMICs remains largely uninvestigated. The current study elucidates the clinical and genotypic spectrum of 54 families with de novo variants underlying sID in an Indian cohort of neurodevelopmental disorders. Though several sIDs are clinically recognizable and less challenging to diagnose than isolated ID, they are often associated with large variability in the phenotypes especially among different populations. This variability could be because of a difference in genetic background, environmental factors or a combination of both. In addition to ID, global developmental delay, dysmorphism, malformations, seizures, autism spectrum disorder, hypotonia and sensory dysfunction defects were the other more commonly observed comorbidities. Of the 46 disorders observed in this cohort, 24 disorders are being reported through our cohort for the first time in the Indian population, to the best of our knowledge.
Genetics of monogenic sID is extremely heterogeneous and follows all inheritance patterns. Rare de novo variants are known to contribute to causative variants in 40–70% individuals with ID [9, 21, 22]. Despite the recently published large cohort studies, the precise burden of de novo variants remains largely unknown. In the present study, we noted that 57% (54 families) of the 104 molecularly diagnosed families with sID carried de novo variants for autosomal dominant or X-linked disorders. Thirty-nine of the 53 (74%) disease-causing variants identified were novel, thus expanding the list of the disease-causing variants in sID causative genes. All these disease-causing variants were submitted to ClinVar to make them available to the medical genetics’ community worldwide.
Until recently, CMA was recommended as a first-tier test for investigating undiagnosed disorders of ID and congenital anomalies with diagnostic yields ranging from 16 to 28% [23, 24]. However, currently it is recommended that ES/GS which can identify the genetic etiology in 28–68% individuals be strongly considered as a first- or second-tier test [25, 26]. Previous studies have mainly utilized trio ES as a first-tier test to identify de novo variants in sIDs, resulting in a high diagnostic yield of 50–70% [9, 21, 22, 27]. In the current study, it was observed that 72% of the de novo variants could be identified using a singleton ES/Mendeliome. This can be attributed to additional phenotypic clues in sID aiding the interpretation of the singleton ES data and reaffirms the findings previously observed in Indian studies highlighting the effectiveness of deep phenotyping and singleton ES in diagnosis of clinically heterogeneous NDDs [13, 28, 29]. However, in families with isolated ID, variant prioritization often warrants familial testing through trio exome sequencing and/or testing of additional affected and unaffected family members. Moreover, the overall diagnostic yield of identifying monogenic de novo variants has increased from 55% (SNVs/indels) to 57% using a combinatorial approach of detecting SNVs/indels and CNVs from the exome data. These results are in line with previous studies highlighting the significance of incorporating exome based CNV analysis algorithms to increase the diagnostic yield of NDDs [30, 31].
Previous studies on syndromic/nonsyndromic ID cohort of de novo variations showed that genes encoding transcriptional and chromatin regulators were the most commonly mutated genes as compared to other neuronal regulators (synaptic maintenance and signaling) or fundamental cellular processes (translation, cell cycle control and energy metabolism) regulating genes [27, 32, 33]. In this study, we classified the genes carrying the disease-causing de novo variants in the current cohort based on its function, and our results were similar to those observed by Taskiran et al (2021). We observed that the transcriptional and chromatin regulators represented the largest class of ID-associated genes, followed by signaling molecules, serine threonine kinases, neuronal migration machinery, structural proteins, molecular motors, synaptic proteins, adhesion molecules, enzymes, microtubule formation, and cell proliferation (Supplementary Fig. 1).
It is known that approximately 80% of all de novo germline single nucleotide variants arise on the paternal allele [11, 34]. Thus, advanced paternal age at conception has been established as the major factor linked to the increase in the number of de novo variants, a subset of which might underlie developmental disorders [8, 11, 35]. However, we noted that in our cohort, the median paternal age at the time of conception was 39 years in families with autosomal recessive disorders, 29 years for inherited autosomal dominant/X-linked disorders, and 35 years in families with de novo variants.
Consanguinity and inbreeding are widely prevalent in specific communities and geographic regions of India [14, 36]. Though, the high rate of parental consanguinity and inbreeding is expected to precipitate rare autosomal recessive disorders including those causative of sIDs, we observed de novo variants in 24% of our consanguineous families with sID [37]. Previously Kahrizi et al. and Mercan et al. have reported de novo variants in approximately 17% and 28% of the individuals with ID born to consanguineous parents, respectively, thus highlighting the significant occurrence of de novo events within highly inbred populations [38, 39].
There are few limitations of our study. Though we report a high rate of identifying de novo disease-causing variants underlying sIDs using a singleton ES in the known disease-causing genes, only one proband with new disease gene association could be ascertained [15]. This could be explained by an inability to perform trio ES or GS in the undiagnosed families due to resource limitations. Also, further investigation may be needed for individuals with undiagnosed phenotypes, such as exploring variants beyond the exonic regions, somatic alterations, digenic as well as oligogenic etiologies.
The knowledge of rare genetic disorders, their diagnoses using the rapidly emerging genomic testing techniques, genetic counseling, prenatal testing, early intervention, and management in LMICs is improving owing to the availability of better infrastructure, cost effective genomic testing, manpower and trained professionals [40, 41]. We herein consolidate the phenotypic and genotypic spectrum of de novo variants underlying monogenic sIDs highlighting the utility of singleton ES as an excellent diagnostic tool for the diagnosis of heterogeneous sIDs in LMIC like India. Our study reiterates de novo variants are likely to contribute significantly as the most frequent cause of sIDs even in populations with consanguinity and endogamy.
Data availability
Additional data are available from the corresponding author on reasonable request.
References
Russell PSS, Nagaraj S, Vengadavaradan A, Russell S, Mammen PM, Shankar SR, et al. Prevalence of intellectual disability in India: a meta-analysis. World J Clin Pediatr. 2022;11:206–14.
Winnepenninckx B, Rooms L, Kooy RF. Mental retardation: a review of the genetic causes. Br J Dev Disabilities. 2003;49:29–44.
Milani D, Ronzoni L, Esposito S. Genetic advances in intellectual disability. J Pediatr Genet. 2015;4:125–7.
Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–12.
Topper S, Ober C, Das S. Exome sequencing and the genetics of intellectual disability. Clin Genet. 2011;80:117–26.
Buckley N, Glasson EJ, Chen W, Epstein A, Leonard H, Skoss R, et al. Prevalence estimates of mental health problems in children and adolescents with intellectual disability: a systematic review and meta-analysis. Aust N Z J Psychiatry. 2020;54:970–84.
Kochinke K, Zweier C, Nijhof B, Fenckova M, Cizek P, Honti F, et al. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet. 2016;98:149–64.
Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016;17:241.
Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7.
Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385:1305–14.
Goldmann JM, Wong WS, Pinelli M, Farrah T, Bodian D, Stittrich AB, et al. Parent-of-origin-specific signatures of de novo mutations. Nat Genet. 2016;48:935–9.
Tekola-Ayele F, Rotimi CN. Translational genomics in low- and middle-income countries: opportunities and challenges. Public Health Genomics. 2015;18:242–7.
Pranav Chand R, Vinit W, Vaidya V, Iyer AS, Shelke M, Aggarwal S, et al. Proband only exome sequencing in 403 Indian children with neurodevelopmental disorders: Diagnostic yield, utility and challenges in a resource-limited setting. Eur J Med Genet. 2023;66:104730.
Kausthubham N, Shukla A, Gupta N, Bhavani GS, Kulshrestha S, Das Bhowmik A, et al. A data set of variants derived from 1455 clinical and research exomes is efficient in variant prioritization for early-onset monogenic disorders in Indians. Hum Mutat. 2021;42:e15–e61.
Narayanan DL, Rivera Alvarez J, Tilly P, do Rosario MC, Bhat V, Godin JD, et al. Further delineation of KIF21B-related neurodevelopmental disorders. J Hum Genet. 2022;67:729–33.
Narayanan DL, Udyawar D, Kaur P, Sharma S, Suresh N, Nampoothiri S, et al. Multilocus disease-causing genomic variations for Mendelian disorders: role of systematic phenotyping and implications on genetic counselling. Eur J Hum Genet. 2021;29:1774–80.
Pande S, Mascarenhas S, Venkatraman A, Bhat V, Narayanan DL, Siddiqui S, et al. Further validation of craniosynostosis as a part of phenotypic spectrum of BCL11B-related BAFopathy. Am J Med Genet A. 2023;191A:2175–2180.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22:245–57.
Limwongse C. Medical genetic services in a developing country: lesson from Thailand. Curr Opin Pediatr. 2017;29:634–9.
Brunet T, Jech R, Brugger M, Kovacs R, Alhaddad B, Leszinski G, et al. De novo variants in neurodevelopmental disorders—experiences from a tertiary care center. Clin Genet. 2021;100:14–28.
Järvelä I, Määttä T, Acharya A, Leppälä J, Jhangiani SN, Arvio M, et al. Exome sequencing reveals predominantly de novo variants in disorders with intellectual disability (ID) in the founder population of Finland. Hum Genet. 2021;140:1011–29.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64.
Narayanan DL, Girisha KM. Genomic testing for diagnosis of genetic disorders in children: chromosomal microarray and next-generation sequencing. Indian Pediatr. 2020;57:549–54.
Srivastava LK, Ju Z, Ghagre A, Ehrlicher AJ. Spatial distribution of lamin A/C determines nuclear stiffness and stress-mediated deformation. J Cell Sci. 2021;134jcs248559.
Malinowski J, Miller DT, Demmer L, Gannon J, Pereira EM, Schroeder MC, et al. Systematic evidence-based review: outcomes from exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability. Genet Med. 2020;22:986–1004.
Hamdan FF, Srour M, Capo-Chichi JM, Daoud H, Nassif C, Patry L, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014;10:e1004772.
Kaur P, do Rosario MC, Hebbar M, Sharma S, Kausthubham N, Nair K, et al. Clinical and genetic spectrum of 104 Indian families with central nervous system white matter abnormalities. Clin Genet. 2021;100:542–50.
Sheth H, Pancholi D, Bhavsar R, Mannan AU, Ganapathy A, Chowdhury M, et al. Assessing utility of clinical exome sequencing in diagnosis of rare idiopathic neurodevelopmental disorders in Indian population. Neurol India. 2021;69:1729–36.
Jiao Q, Sun H, Zhang H, Wang R, Li S, Sun D, et al. The combination of whole-exome sequencing and copy number variation sequencing enables the diagnosis of rare neurological disorders. Clin Genet. 2019;96:140–50.
Zhai Y, Zhang Z, Shi P, Martin DM, Kong X. Incorporation of exome-based CNV analysis makes trio-WES a more powerful tool for clinical diagnosis in neurodevelopmental disorders: a retrospective study. Hum Mutat. 2021;42:990–1004.
Taşkıran EZ, Karaosmanoğlu B, Koşukcu C, Ürel-Demir G, Akgün-Doğan Ö, Şimşek-Kiper PÖ, et al. Diagnostic yield of whole-exome sequencing in non-syndromic intellectual disability. J Intellect Disabil Res. 2021;65:577–88.
Brookes E. New insights into intellectual disability caused by mutations in a chromatin regulator. EBioMedicine. 2016;6:2–3.
Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–5.
Taylor JL, Debost J-CPG, Morton SU, Wigdor EM, Heyne HO, Lal D, et al. Paternal-age-related de novo mutations and risk for five disorders. Nat Commun. 2019;10:3043.
Dalal A, Bhavani GS, Togarrati PP, Bierhals T, Nandineni MR, Danda S, et al. Analysis of the WISP3 gene in Indian families with progressive pseudorheumatoid dysplasia. Am J Med Genet A. 2012;158a:2820–8.
Temaj G, Nuhii N, Sayer JA. The impact of consanguinity on human health and disease with an emphasis on rare diseases. J Rare Dis. 2022;1:2.
Mercan S, Akcakaya NH, Salman B, Yapici Z, Ozbek U, Ugur Iseri SA. Clinical and genetic analyses in syndromic intellectual disability with primary microcephaly reveal biallelic and de novo variants in patients with parental consanguinity. Genes Genomics. 2023;45:13–21.
Kahrizi K, Hu H, Hosseini M, Kalscheuer VM, Fattahi Z, Beheshtian M, et al. Effect of inbreeding on intellectual disability revisited by trio sequencing. Clin Genet. 2019;95:151–9.
Kingsmore SF. 2022: a pivotal year for diagnosis and treatment of rare genetic diseases. Cold Spring Harb Mol Case Stud. 2022;8:a006204.
Maltese PE, Poplavskaia E, Malyutkina I, Sirocco F, Bonizzato A, Capodicasa N, et al. Genetic tests for low- and middle-income countries: a literature review. Genet Mol Res. 2017;16:gmr16019466.
Acknowledgements
We would like to sincerely acknowledge and thank the affected individuals and their families for their consent and participation in the study. We are grateful to all the referring physicians who made this work possible. We would like to acknowledge and thank the National Institutes of Health, United States, for funding the study, “Genetic Diagnosis of Neurodevelopmental Disorders in India” (1R01HD093570-01A1). This grant is awarded to AS and KMG. We would also like to acknowledge and thank DBT/Wellcome Trust India Alliance for immense support by the grant [Grant number: IA/CRC/20/1/600002] awarded to KMG. We also thank the Indian council of Medical Research for awarding the Nurturing Clinical Scientist Fellowship (HRD/NCS-2019-03) to SP.
“We thank the “SG10K_Pilot Investigators” for providing the SG10K_Pilot data (EGAD00001005337). The data from the “SG10K_Pilot Study” reported here were obtained from EGA. This manuscript was not prepared in collaboration with the “SG10K_Pilot Study” and does not necessarily reflect the opinions or views of the “SG10K_Pilot Study”.
Funding
1. National Institutes of Health, United States, for funding the study, “Genetic Diagnosis of Neurodevelopmental Disorders in India” (1R01HD093570-01A1). 2. DBT/Wellcome Trust India Alliance for funding the study, “Centre for Rare Disease Diagnosis, Research and Training” (IA/CRC/20/1/600002). 3. Indian Council of Medical Research, New Delhi, India for supporting the “Nurturing Clinical Scientist Fellowship” (HRD/Head-NCS-2019-03). Open access funding provided by Manipal Academy of Higher Education, Manipal.
Author information
Authors and Affiliations
Contributions
SP and PM contributed to data collection, analysis of the exome sequencing data, Sanger validation, reporting the variant and drafting the manuscript, preparing figures and tables. NK, KN, NJ, AC, BH contributed to operating and modulating the in-house exome sequencing and copy number variant analysis pipeline and analysis of the data. LPR, SM, NK, MDR, JK has contributed to analyzing the exome sequencing data and validation of the results. SX has contributed to systematic maintenance of the recruited families’ demographic, phenotypic and genotypic data. GSB, S Nayak, S Bielas has a contribution to exome data analysis and planning and executing wet laboratory experiments. VB, DN has contributed to clinical assessment, analysis, interpretation of genomic data and genetic counseling of the recruited families. YBL, SJP, S Nampoothiri, NK, SA, RBY, LEL, S Sharma, SB, S Bajaj, NS have referred families and contributed to interpretation of genetic testing results and genetic counseling. S Siddiqui has contributed to radiodiagnosis. KMG, AS, and S Bielas have contributed to funding acquisition, conceptualization, patient evaluation, study recruitment, analysis, interpretation of the genomic testing, genetic counseling to the families, planning and conceptualizing the manuscript and overall supervision. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
We obtained the informed consent and consent for medical photography from the families for genetic testing, publication of data and clinical photographs. This was approved by our institutional ethics committee of Kasturba Medical College and Kasturba Hospital, India.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pande, S., Majethia, P., Nair, K. et al. De novo variants underlying monogenic syndromes with intellectual disability in a neurodevelopmental cohort from India. Eur J Hum Genet (2023). https://doi.org/10.1038/s41431-023-01513-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41431-023-01513-7
