
Abstract
Purpose
Phenylketonuria (PKU) is a rare metabolic disorder that requires life-long management to reduce phenylalanine (Phe) concentrations within the recommended range. The availability of pegvaliase (PALYNZIQ™, an enzyme that can metabolize Phe) as a new therapy necessitates the provision of guidance for its use.
Methods
A Steering Committee comprising 17 health-care professionals with experience in using pegvaliase through the clinical development program drafted guidance statements during a series of face-to-face meetings. A modified Delphi methodology was used to demonstrate consensus among a wider group of health-care professionals with experience in using pegvaliase.
Results
Guidance statements were developed for four categories: (1) treatment goals and considerations prior to initiating therapy, (2) dosing considerations, (3) considerations for dietary management, and (4) best approaches to optimize medical management. A total of 34 guidance statements were included in the modified Delphi voting and consensus was reached on all after two rounds of voting.
Conclusion
Here we describe evidence- and consensus-based recommendations for the use of pegvaliase in adults with PKU. The manuscript was evaluated against the Appraisal of Guidelines for Research and Evaluation (AGREE II) instrument and is intended for use by health-care professionals who will prescribe pegvaliase and those who will treat patients receiving pegvaliase.
Similar content being viewed by others

INTRODUCTION
Phenylketonuria (PKU, OMIM 261600) is a rare genetic disorder caused by pathogenic variants in the phenylalanine hydroxylase (PAH) gene, which results in elevated phenylalanine (Phe) concentrations in the blood and brain.1,2,3 The concentration of blood Phe in healthy individuals is ~60 (±30) µmol/L, but in individuals with PKU who have not restricted their Phe intake, can be ≥1200 µmol/L.4 Elevated blood Phe concentrations are associated with impairment of executive function, behavioral and psychiatric problems, including depression and anxiety, and have a negative impact on patient quality of life (QoL), mood, and attention span.2,5,6,7,8,9 Even on treatment, deficits in executive functioning, motor ability, and behavior can occur.7,10,11,12 Conversely, lowering of Phe levels is associated with improved neurological performance.13 Neurocognitive and psychiatric symptoms can develop later in life when it is more difficult to maintain Phe control, and a correlation has been reported between improved cognitive performance and control of blood Phe throughout the lifespan.14,15 Blood Phe concentration is therefore the primary biomarker for optimizing treatment. The American College of Medical Genetics and Genomics (ACMG) guidelines state that the goal of treatment is to maintain plasma Phe concentration in the range of 120–360 μmol/L.4 European guidelines state that the primary goal of treatment is normal neurocognitive and psychosocial functioning through maintaining plasma Phe concentration between 120 and 360 μmol/L up to the age of 12 years and up to 600 μmol/L thereafter.16
A Phe-restricted diet supplemented with medical food has been the standard of care for 50 years,17 but adherence is poor, particularly postadolescence. Tetrahydrobiopterin (BH4) supplementation is an adjunct therapy and until recently, sapropterin dihydrochloride (marketed in the United States as KUVAN®) was the only available pharmacologic treatment for patients with PKU. However, not all patients respond to BH4 and few can eliminate the need for medical foods. In a recent survey commissioned by the National PKU Alliance (NPKUA), of 625 patients, over half (51.7%) reported having difficulty in managing their PKU, including maintaining a Phe-restricted diet. Individuals with PKU desire new treatments that would allow them to increase their intake of natural protein, discontinue or reduce use of medical foods, improve mental health, and reduce blood Phe concentration.18 In a study conducted to assess change in QoL in adults returning to a PKU diet, although many individuals experienced positive well-being, the majority of those with symptoms of depression and anxiety experienced improvements in subjective well-being upon return to a controlled diet.19 In another study, more than half of patients reported improved QoL with a Phe-restricted diet.8 The ACMG guidelines state that current and future therapies should be evaluated not only for ability to lower Phe, but also for potential to enhance QoL.4
Pegvaliase (PEGylated recombinant [Anabaena variabilis] phenylalanine ammonia lyase [PAL]; marketed in the United States as PALYNZIQTM) is a novel enzyme substitution therapy administered via subcutaneous injection20 that lowers blood Phe independently of PAH and its BH4 cofactor.21 Two phase 3 studies, PRISM-1 (Study 301) and PRISM-2 (Study 302), evaluated the efficacy and safety of pegvaliase treatment in adults with PKU.21 In PRISM-1, pegvaliase-naïve participants with blood Phe >600 μmol/L were randomized 1:1 to a maintenance dose of 20 mg/day or 40 mg/day of pegvaliase. Participants in PRISM-1 continued treatment in PRISM-2, a four-part clinical trial that includes an ongoing, open-label, long-term extension (LTE) study of doses between 5 and 60 mg/day.22,23 Many patients were on unrestricted diets when entering these trials. Within 24 months, 68.4%, 60.7%, and 51.2% of participants achieved blood Phe concentrations ≤600 μmol/L, ≤360 μmol/L, and ≤120 μmol/L, respectively. Reduction in blood Phe was associated with improvements in neuropsychiatric outcomes that were sustained with long-term treatment. In the phase 3 study of 261 patients, the most common adverse events (AEs) were arthralgia (70.5%), injection site reaction (62.1%), injection site erythema (47.9%), and headache (47.1%). Acute systemic hypersensitivity events, per external adjudication according to the National Institute of Allergy and Infectious Diseases/Food Allergy and Anaphylaxis Network (NIAID/FAAN) criteria, were observed in 4.6% (12/261) of participants.22 Across all clinical trials of pegvaliase with induction/titration/maintenance dosing (n = 285), 26 (9%) patients experienced 37 anaphylaxis episodes. Based on these results, pegvaliase was recently licensed in the United States for use in adult patients with PKU with a blood Phe concentration >600 μmol/L on existing management (defined as prior or current restriction of dietary Phe and protein intake, and/or prior treatment with sapropterin dihydrochloride).21 The aim is for patients receiving pegvaliase to achieve life-long, reliable control of blood Phe concentration, thereby experiencing improvement in cognitive and psychosocial symptoms while consuming diets that are unrestricted in protein.
An expert Steering Committee (SC), comprising health-care professionals (HCPs) with experience with pegvaliase through its clinical development program, was convened to provide guidance on the optimal use of pegvaliase to improve the long-term outcomes for adults with PKU. This paper summarizes these evidence- and consensus-based recommendations and has been evaluated against the Appraisal of Guidelines for Research and Evaluation (AGREE II) Instrument.24,25 It is intended for use by all HCPs who will prescribe pegvaliase or manage patients receiving pegvaliase. It is expected that these recommendations will inform updates to existing guidelines.4,16,26
MATERIALS AND METHODS
Development of guidance statements
At the time of development of this guidance, pegvaliase was not approved; therefore, the SC comprised 17 HCPs who were principal investigators or investigators in pegvaliase clinical trials. The group was from across the United States and included metabolic physicians, geneticists, psychiatrists, and dietitians. They met through a series of face-to-face meetings and identified a list of 21 clinical questions (see Appendix Table 1) grouped into four areas of clinical focus (Table 1). Diagnosis and the broader management of PKU and a direct comparison of pegvaliase and sapropterin dihydrochloride were beyond the scope of the program and are not discussed. It is important to note that the guidance statements in this paper are based on clinical trial experience. However, with the recent availability of the prescribing information,21 the SC deemed it necessary in some instances to provide additional clarity in the supporting text and footnotes to supplement the statements.
Guidance statements were developed based on results of a systematic literature and data review, supplemented with expert opinion where appropriate. To maintain editorial independence from the sponsor, all SC meetings were facilitated by an independent medical communications agency, who fulfilled the role of methodology experts and secretariat throughout the process. Due to the necessity of sharing data for this novel agent, BioMarin Pharmaceutical Inc. attended SC meetings and provided clinical data as requested; however, the final decision on the scope of the program and wording of the guidance statements was made by the SC. To ensure that the guidance represents the patient voice, the statements were reviewed at each round by the Executive Director of the US National PKU Alliance, and feedback incorporated.
Literature search methodology
The literature search was conducted in accordance with Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA)27 by four bibliographic fellows who were nominated by the SC. Given the limited availability of published pegvaliase data at the time of the program, the evaluation included a systematic review of published literature and a focused review of pegvaliase clinical trial data. This was directed by the SC, who identified the key data required to address the clinical questions. For details of the literature search methodology, please refer to the Appendix (Fig. 1, Tables 2 and 3). Briefly, search strings were defined based on the PICO methodology,28 review of gray literature included information provided by a hair loss specialist from the Boston Children’s Hospital, and evidence levels were assessed using the Oxford Centre for Evidence-Based Medicine criteria.29 (see Appendix Table 4a). Evidence summaries prepared for each clinical question were reviewed by the SC and used to develop guidance statements.
Consensus process
A modified Delphi (see Appendix for details) was employed to demonstrate consensus on the guidance statements among a wider group. It was sent to principal investigators from the 29 pegvaliase clinical trial sites across the United States for completion by their multidisciplinary teams (see Appendix Tables 5a and 5c for details). As per the Delphi methodology, individual results were anonymized to eliminate bias. Consensus was deemed to have been reached when >75% of respondents agreed with a given statement. Statements for which consensus was not reached were revised by the SC, for a maximum of three rounds of voting before a decision of “no agreement.” Based on comments received during the first round of voting, the SC made the decision to update and revote on statements that reached 75–85% consensus to improve their clarity. The manuscript was assessed according to AGREE II by three independent reviewers with expertise in guideline development.24,25
RESULTS
A total of 34 guidance statements were included in the modified Delphi voting. The final statements are listed in Tables 2–5 and a quick reference guide to all statements can be found in Appendix Table 7. Per the GRADE approach,30 language within the statements, e.g., the use of “should,” “consider,” and “may,” has been used to indicate strength of the recommendation. In the first voting round, 26 responses were received from 24 hospitals/institutions (11 included SC members). Consensus was reached on 33/34 statements (Appendix Table 5b). Seven statements achieved 75–85% agreement and were revised by the SC. Thus, eight statements were sent for a second round of voting, in which 22 responses were received from 18 hospitals/institutions (13 included SC members). Consensus was achieved on all eight statements (Appendix Table 5d); thus, a third round was not required. Please refer to Appendix Table 4b for evidence levels assigned for the publications used to develop each of the guidance statements.
Goal of pegvaliase treatment
Adults treated with pegvaliase can achieve blood Phe concentrations below the lower limit of the recommended range (<120 μmol/L) while maintaining consistent dietary protein intake,22 thereby avoiding severe restriction of dietary Phe intake that can be required during treatment with dietary management and/or sapropterin dihydrochloride. The goal of pegvaliase treatment, therefore, is to provide life-long maintenance of blood Phe concentration as low as possible (concentrations of 31–120 μmol/L should not be regarded as too low) while normalizing diet (defined as not requiring PKU medical food and containing at least the Dietary Reference Intake [DRI] for protein [0.8 g/kg/day]31).
Patient-/system-related considerations prior to initiating therapy
The recommendations for the use of pegvaliase are based on clinical trial data showing a significant reduction in blood Phe concentration in adults with PKU.22,23,32,33 In the 24-week, phase 2 multicenter, open-label, dose-finding study, pegvaliase resulted in a reduction in blood Phe, which appeared to be related to dose, treatment duration, and individual immune response.32 Analysis of participants who achieved maintenance dose during the first 24 weeks of the study versus those who did not revealed that participants who responded later (after additional treatment and dose titration) obtained a benefit similar to that of early responders, with a similar safety profile. In the phase 3 PRISM-1 study, patients randomized to a maintenance pegvaliase dose of 20 or 40 mg/day22 achieved mean decreases from baseline in blood Phe of 51% and 69% at 12 and 24 months, respectively, and 68%, 61%, and 51% of participants respectively achieved blood Phe concentrations of≤600 µmol/L, ≤360 µmol/L, and ≤120 µmol/L. The study included an 8-week discontinuation period to evaluate outcomes in participants who had achieved a ≥20% blood Phe reduction from pretreatment baseline (i.e., while receiving pegvaliase). Participants were randomized 2:1 to either continue pegvaliase (20 or 40 mg/day) or switch to matching placebo.23 The primary endpoint (change in blood Phe concentration from entry of this study period to week 8) was met with statistically significant differences between the pegvaliase and placebo groups. Across all pegvaliase clinical trials with induction/titration/maintenance dosing (n = 285), 26 patients (9%) experienced 37 anaphylaxis episodes, most of which (84%; 28/37) occurred within 1 hour of administration.21 Consideration should therefore be given to having a trained observer who can recognize and manage anaphylaxis present for at least 1 hour after administration of pegvaliase. Anaphylaxis episodes can occur at any time during treatment, but are more frequent during the first 6 months.21,22 Consequently, although not mandated in the pegvaliase prescribing information,21 the SC recommends that the presence of the trained observer would be most appropriate during induction/titration of pegvaliase and that consideration should be given to the patient’s ability to adhere to pegvaliase treatment, including requirements for the monitoring of adverse events prior to initiation, and that caution should be exercised if the necessary support is not in place. Both the patient and their observer should receive specific, face-to-face education (in person or via telemedicine, i.e., the provision of health care remotely by means of telecommunications technology) about appropriate use of the epinephrine autoinjector (including the need for the patient to always carry the autoinjector; when and how to administer the autoinjector; and when to contact emergency services/seek medical advice). If this is not possible, physicians may, after a thorough risk/benefit assessment, still prescribe pegvaliase and, if feasible, consider observation by other HCPs (e.g., home health services or local office visits).
There are no data describing concomitant use of pegvaliase and sapropterin dihydrochloride. Guidance on treatment with sapropterin dihydrochloride can be found in the ACMG4 and European16 guidelines.
Pegvaliase has not been studied in pregnancy and women should be advised of the unknown potential risks of pegvaliase to the fetus.21 Consideration should be given to the known risks of poorly controlled Phe concentrations during pregnancy, which include birth defects (e.g., microcephaly and major cardiac malformations), intrauterine fetal growth retardation, and intellectual disability. Continued control of blood Phe concentrations and rigorous diet management during pregnancy are essential to reduce the teratogenic effects of hyperphenylalaninemia.34 Treatment of female mice with pegvaliase during pregnancy resulted in offspring that survived until adulthood, as compared with complete lethality of offspring of untreated mice, or limited survival of those from mice on a PKU diet.35 More studies are required to assess the safety profile of pegvaliase during pregnancy, and the use of pegvaliase in women who are planning pregnancy cannot be recommended at this time. Clinical data on the presence of pegvaliase in human milk or its effects on milk production or the breastfed infant are currently lacking. Therefore, the developmental and health benefits of breastfeeding should be considered alongside the mother’s need for pegvaliase and the associated potential adverse effects of PKU and/or pegvaliase on the breastfed child.21 Although no study data are available, there is no evidence to suggest that pegvaliase is contraindicated in men anticipating fatherhood.
Patient education required prior to initiation
Patients should be made aware that hypersensitivity reactions may occur following administration of pegvaliase. The most common are injection site reactions and arthralgia;21 with most events subsiding over time. Anaphylaxis reactions occur rarely, and although they can occur at any time during treatment, they are more frequent during the first 6 months (specific advice is detailed above).21,22 Other adverse events observed after administration of pegvaliase include headache, generalized skin reactions lasting at least 14 days, pruritus, nausea, abdominal pain, oropharyngeal pain, vomiting, cough, diarrhea, and fatigue.21 Patients should be encouraged to report all adverse events to their clinician.
The decision to prescribe pegvaliase should be made at the discretion of the clinician while considering patient preference, treatment goals, and potential barriers to receiving injections. A systematic review of diabetes therapies showed that barriers to initiation of injectable therapy originate from both patients and practitioners and include fear of injection-associated pain or needle phobia, concerns about using injectable therapy, side effects, and impact on QoL.36 Most barriers can be overcome with education;37,38 it is therefore important to educate patients on the potential adverse events associated with pegvaliase and provide counseling to ensure that psychological barriers do not affect adherence. Patients should also be provided with clear instructions on how to correctly dose and inject pegvaliase.
Response to pegvaliase appears to depend on individual immune response rather than dose; therefore, a variation in time to response has been observed.21 Patients should be educated that it may take longer than 1 year to respond.22
Definition of treatment efficacy
Blood Phe concentration fluctuates over time; therefore, the definition of “clinically meaningful efficacy” should be determined by the treating clinician. In pegvaliase clinical trials, a reduction from baseline in blood Phe concentration of ≥20% at any point within 52 weeks was considered the first sign of effect, suggesting that further blood Phe lowering may be achieved with continued treatment.21,22,23 Progression toward normalization of diet is defined as an ability to consume more intact (natural) protein and reduced requirement for medical foods. Additional monitoring to assess the impact of pegvaliase on cognitive function and/or QoL could be considered.
Introduction and titration of pegvaliase
The frequency of blood Phe and Tyr monitoring should be determined by the treating clinician on a patient-by-patient basis. More frequent monitoring can be considered during dose titration, diet/dose adjustment, and episodes of hypophenylalaninemia (Phe concentration of <30 µmol/L). When a patient is receiving a stable dose of pegvaliase, blood Phe and Tyr concentrations may be monitored less frequently. The pegvaliase prescribing information provides a recommended dosing regimen and timeline for induction, titration, and maintenance, which advises a minimum of 9 weeks for titrating to 20 mg/day, but confirms that the pharmacokinetics appear to be dictated by individual immune response.21 Modifications from the phase 3 dosing regimen were informed by data/experience generated throughout the pegvaliase clinical development and based on the totality of collected efficacy and safety data (outlined in the prescribing information).21 Patients who experience adverse events may require slower titration to safely achieve their target maintenance dose (defined as the lowest effective dose; 20 or 40 mg/day). While it may be prudent to discontinue pegvaliase in patients who do not show response within 52 weeks, consideration should be given to extending the duration of treatment in patients in whom dose escalation took longer, or in those who had their dose reduced or discontinued due to adverse events. In clinical trials, some patients have been successfully maintained on a dose >40 mg/day and are still being monitored. The US FDA prescribing information states that pegvaliase should be discontinued in patients who have not achieved ≥20% reduction in blood Phe concentration from pretreatment baseline or a blood Phe concentration ≤600 µmol/L after 16 weeks of continuous treatment with the maximum dosage of 40 mg once daily.21
Adjusting diet/dose
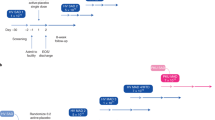
The SC recommends that as a precaution, extended periods of hypophenylalaninemia (<30 μmol/L for >3 months) should be avoided by Phe monitoring and adjusting diet and pegvaliase dose (see Fig. 1). In the clinical trial program, 16% and 42% of patients experienced hypophenylalaninemia in two or more consecutive measurements during induction/titration and maintenance phase treatments, respectively. However, this likely reflects the use of fixed dosing, and it is expected that incidence would decrease with flexibility in dosing.21 Some patients developed hair loss and/or skin abnormalities, but there was disagreement among the SC about whether these were related to hypophenylalaninemia. A systematic review of the literature identified case reports of children with low Phe concentrations with similar symptoms,39,40,41,42,43 but it is not clear whether these occurred due to low Phe concentrations, general malnourishment, or other causes, such as nutrient/protein deficiency or compromised immune status, as shown in other studies.44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60 The SC agreed that the clinical implications of hypophenylalaninemia are not clear and therefore recommended that extended periods of hypophenylalaninemia should be avoided. Although intake of intact protein should be increased as tolerated until ≥DRI is reached, the SC suggests that this alone may be insufficient to increase blood Phe concentrations in patients with hypophenylalaninemia and pegvaliase dose reduction may also be required. In patients on a normal diet, simple dose changes, for example removing one dose of pegvaliase per week, should be considered. Patients should be counseled on the importance of adhering to all other doses.

Diet and dose-adjustment algorithm. DRI dietary reference intake, Phe phenylalanine, Tyr tyrosine.
Resuming pegvaliase treatment following dose interruption
The recommendations for restarting pegvaliase following treatment interruption are based on findings from the pegvaliase discontinuation trial in which patients achieving >20% reduction from baseline in blood Phe concentration with pegvaliase were randomized (2:1) to either continue their randomized dose (20 or 40 mg/day) or receive matching placebo for 8 weeks. After 8 weeks, patients who received placebo were switched back to pegvaliase at their previous dose (20 mg/day [n = 11] or 40 mg/day [n = 8]) (ref. 23). The most common AEs for pooled pegvaliase and placebo groups were arthralgia (13.6% and 10.3%), headache (12.1% and 24.1%), anxiety (10.6% and 6.9%), fatigue (10.6% and 10.3%), and upper respiratory tract infection (1.5% and 17.2%) (ref. 23).
The statements are based on the small number of patients who completed this study.23 The SC commented that per the trial protocol, premedications were reintroduced following ≥4 days of missed doses. More data are required to understand the need for premedications in patients with shorter treatment interruptions.
Patient education, monitoring, and supplementation requirements
Unlike PAH, which converts Phe to Tyr, pegvaliase converts Phe to trans-cinnamic acid and ammonia and does not therefore increase Tyr concentrations. The effect of Tyr supplementation on neurological outcomes is well studied;61,62,63,64,65,66,67 although there is good evidence that Tyr supplementation increases blood Tyr levels in patients with PKU, it is unclear whether this translates into improved cognitive processing and neurological outcomes.68,69 In the pegvaliase phase 3 trial, patients were instructed to take 500 mg of Tyr three times per day with meals and plasma Tyr concentrations were noted to be in the normal range.22 Although no recommendations are provided on Tyr supplementation, maintenance of physiologic Tyr blood concentrations in patients with PKU is recommended and therefore Tyr supplementation should be considered when postprandial blood concentrations are consistently below the lower limit of normal.70
Individuals with PKU are often advised to avoid all high-protein foods for life, and therefore may not know how to transition to a normal diet. Nutrient intake should be evaluated by a metabolic dietitian to determine if additional supplementation is necessary. If so, patients should be counseled on how to introduce high-protein foods into their diet, including advice on portion size, food safety, and cooking methods. Eating disorders are common among individuals with early-treated PKU, as many experience guilt and shame surrounding the consumption of high-protein foods. For these individuals, changing dietary restrictions may also be accompanied by a need to adjust their emotional response to foods that were previously forbidden. If individuals with PKU decide to follow a vegetarian diet, they should be encouraged to incorporate high-quality protein (eggs, soy) into their diet. If they choose to be vegan, they should be educated to consume 10–15% more protein than the DRI, due to the reduced bioavailability of plant proteins.71,72,73 Individuals who obtain most of their protein from plant sources are particularly at risk of micronutrient deficiencies, either because the micronutrients are found primarily in animal proteins or because of poor bioavailability of plant proteins,74 and should be advised to consider use of the above supplements.
Monitoring adverse events
The frequency of adverse events in patients receiving pegvaliase does not appear to correlate with dose or biochemical markers, but largely depends on individual immune response.21 As a result, it is difficult to predict which patients are likely to experience a hypersensitivity reaction. The risk of hypersensitivity reaction to other PEGylated or biologic products was assessed in the literature review; no clear evidence was found.75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97 Health-care providers should therefore assess all patients for hypersensitivity reactions, including local and generalized skin reactions, injection site reactions, arthralgia, and lymphadenopathy (inguinal, axillary, occipital regions). Telemedicine has been used successfully in other settings98,99,100,101,102,103,104,105 and should be considered for monitoring of adverse events if local infrastructure and training is in place.
Prevention of adverse events
In May 2014, risk mitigation strategies were implemented in pegvaliase clinical trials to avoid hypersensitivity reactions. These included mandatory premedication with antihistamines (H1/H2 receptor antagonists, e.g., ranitidine, cetirizine, fexofenadine) with or without antipyretics (e.g., acetaminophen or ibuprofen) during introduction and titration of pegvaliase. Prior to implementation, 15.4% of patients discontinued pegvaliase due to an adverse event in the first 6 months of treatment. Following the mandate, this figure was reduced to 5.9%, however, slowing the titration was also permitted and is a confounding factor.22 Overall, these data suggest that premedication is successful in reducing the rate and intensity of hypersensitivity reactions (including life-threatening anaphylaxis) and concurs with the broad evidence base from other therapy areas uncovered in the literature review.106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124 Based on patient preference and safety considerations associated with chronic use, the expert panel recommends the use of nondrowsy antihistamines such as cetirizine or fexofenadine.
Management of anaphylaxis
Management of anaphylaxis in the pegvaliase clinical trials included administration of autoinjectable epinephrine, corticosteroids, antihistamines, and/or oxygen.21 In the event of suspected acute systemic hypersensitivity reaction, the clinician should assess the risks and benefits of readministering pegvaliase and make the decision to interrupt/discontinue treatment using appropriate assessment criteria and clinical judgment. The NIAID/FAAN and Brown’s criteria can be used to assess the severity of suspected anaphylaxis; however, it is important to use clinical judgment to identify true anaphylaxis events.125,126 In the phase 3 trial, anaphylaxis assessed by external expert allergists/immunologists using NIAID/FAAN criteria and clinical judgment was reported in 4.6% (12/261) of patients.22 According to FDA adjudication, across all pegvaliase clinical trials with induction/titration/maintenance dosing, 9% (26/285) of patients experienced a total of 37 anaphylaxis-type episodes.21 Eighteen (69%) of the 26 patients were rechallenged; 5 had recurrence of symptoms, but all anaphylaxis episodes resolved without sequelae. The predominant mechanism of hypersensitivity was considered to be type III non-IgE-mediated, therefore rechallenge can be considered in a controlled medical setting.21 When antibiotic, anticancer, and biological treatments are administered to patients with a history of severe immediate infusion reactions, desensitization protocols are routinely used; this approach may reduce the rate and/or severity of breakthrough reactions with pegvaliase.76,127,128,129,130,131,132,133,134,135,136,137
Management of arthralgia
Episodes consistent with arthralgia were reported in 83% of the 285 patients who received induction/titration/maintenance dosing in the pegvaliase clinical trial program; events were most frequent during the induction/titration phase.21 Most (>91%) events resolved without requiring a change in pegvaliase dose, and were managed with nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and/or acetaminophen.21 The rate in the phase 3 study (in 261 patients) was 70.5% (ref. 22). The SC suggests that dose reduction to the last tolerated dose for days or weeks could be an alternative approach.
Management of injection site reactions
During induction/titration of pegvaliase, injection site reactions occurred in 88% of patients and generalized skin reactions (not limited to the injection site) were seen in 21% of patients.21 Injection site reactions were more frequent during the induction/titration phase; incidence then decreased and stabilized over time.21 There is insufficient evidence to recommend use of cool compresses; some patients reported relief with topical antipruritics, including steroid creams. Pegvaliase should be stored in a refrigerator at 36 °F to 46 °F (2 °C to 8 °C) and should be kept refrigerated between use, although if necessary, it can be stored in the original carton at room temperature for up to 30 days.21 Recommended injection sites include the thighs and the lower abdomen, excepting the 2-inch (5-centimeter) area directly around the navel. If a caregiver is giving the injection, the top of the buttocks and the back of the upper arms can also be used. It is useful to separate injection sites by an inch or more. Patients should be advised to not inject into moles, scars, birthmarks, bruises, rashes, or areas where the skin is hard, tender, red, damaged, burned, inflamed, or tattooed, and to maintain adequate cleansing of the skin prior to injection. Use of telemedicine—conducted either by store-and-forward technology, or as a live remote review via photo/audio or video link—can be considered for remote diagnosis/review and to support the management of adverse events. This approach has been used successfully in dermatology.98,99,100,101,102,103,104,105,138
Discussion
The robust methodology including use of a systematic literature review and use of a modified Delphi process to gain consensus are strengths. As consensus does not equate to 100% agreement among the expert group (Tables 5a and 5b), although ≥85% consensus was reached across all statements, physician discretion should be used when applying this guidance in clinical practice according to individual patient needs. The manuscript was appraised by three independent reviewers using the AGREE II tool (www.agreetrust.org). All three reviewers gave an initial overall quality score of 5/7 and assigned a “recommended for use with modifications” classification (see Appendix Table 6). Two domains, Scope and Purpose and Clarity of Presentation, were strong, both achieving a score of 83%. The Stakeholder Involvement and Rigor of Development domains received scores of 67% and 74%, respectively, with the lowest scoring domains being Applicability (47%) and Editorial Independence (33%). Where possible, suggested amendments were addressed, and two of the AGREE II reviewers conducted a second review (the third was unable to do so due to time commitments). The amended average domain scores were Scope and Purpose (86%), Stakeholder Involvement (75%), Rigor of Development (79%), Clarity of Presentation (86%), Applicability (54%), and Editorial Independence (50%). Another strength of the process was that the manuscript, and the statements themselves, were reviewed by the Executive Director of the National PKU Alliance, to ensure that the patient view was represented.
A limitation of the process was the lack of high-quality evidence available to support the recommendations, which was a result of the lack of patients and randomized controlled studies in rare diseases. Despite this, the recommendations in this consensus are based on data from the largest randomized controlled interventional study carried out to date in patients with PKU. Another limitation of this program is that the SC and wider Delphi responders, by necessity, comprised individuals who had experience of using pegvaliase through participation in the clinical development program. Therefore, absolute elimination of potential bias is impossible to avoid. However, the timeliness of this guidance was deemed important enough to proceed with this small group of experts. Although the initiative was funded by the sponsor, which is not uncommon in rare diseases,139 the process was facilitated and managed by an independent medical communications secretariat to ensure that the views of the sponsor did not influence the direction of the program. All content, including the development of guidance statements, was led by the SC with editorial support provided by the secretariat. The sponsor attended meetings to present supporting clinical trial data and contribute to discussions; however, the SC had the final decision on the wording of the statements. Furthermore, the anonymous modified Delphi process, involving all institutions with experience of using pegvaliase in the clinical trial setting, further minimized sponsor influence.
The SC discussed potential barriers to the use of pegvaliase, which included lack of understanding of the burden of PKU; physicians' lack of confidence and concerns about allergic reaction; the length of time it may take to see a clinical response; a shrinking pool of metabolic physicians, which may result in care falling to more inexperienced health-care providers; and lack of resources to provide adequate time/commitment to patients. Potential solutions to these barriers were discussed and included creating centers of excellence (which would also help with reimbursement); using mentors to train HCPs; and using telemedicine as an educational tool, noting that costs should be kept as low as possible. A HCP website is under development by BioMarin, which will hold materials including the prescribing information, a summary brochure, a dosing and administration guide, a HCP enrollment tool, and risk evaluation and mitigation strategies. Similarly, a patient website will be developed. Patient materials will include a patient educational brochure, a patient summary leaflet, a dosing and administration guide, a patient enrollment tool and a welcome kit (to include items such as an injection placemat and injection site rotation calendar).
As pegvaliase was not commercially available during the development of this guidance, cost-effectiveness was not assessed. As for all therapies, value is important and consideration should be given to the unique challenges of developing therapies for patients with rare diseases.140 For patients with PKU, this includes the lack of therapeutic alternatives, particularly for those who do not respond to sapropterin dihydrochloride, and the impact of normalization of Phe concentrations and diet on QoL. Observational and safety follow-up studies are planned.
This guidance has been developed to coincide with the FDA approval of pegvaliase for adults with PKU in the United States, based on clinical trial experience limited to the United States. There is a need to translate this clinical experience to other countries to coincide with the potential granting of marketing authorization of pegvaliase in other countries/regions.
As clinical experience with pegvaliase grows beyond the clinical trial program, understanding and guidance for the general management of PKU will evolve. In particular, additional evidence-based recommendations with regard to dosing, timing and comedication management, and management in specific populations would be of value. To facilitate this, there is a need for further research to be conducted and published, both by the pharmaceutical industry and independent investigators. In addition to referring to the guidance presented here, clinicians without experience of pegvaliase are advised to seek support and advice from colleagues in the medical community who have already used the drug to ensure appropriate use of pegvaliase. It is also anticipated that professional groups including the ACMG, European Society for Phenylketonuria and Allied Disorders Treated as Phenylketonuria (ESPKU), and Genetic Metabolic Dietitians International (GMDI) will update their respective guidelines4,16,26 with regard to pegvaliase per their published timeframes for updating.
References
Williams RA, Mamotte CD, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29:31–41.
ten Hoedt AE, de Sonneville LM, Francois B, et al. High phenylalanine levels directly affect mood and sustained attention in adults with phenylketonuria: a randomised, double-blind, placebo-controlled, crossover trial. J Inherit Metab Dis. 2011;34:165–171.
Burton B, Grant M, Feigenbaum A, et al. A randomized, placebo-controlled, double-blind study of sapropterin to treat ADHD symptoms and executive function impairment in children and adults with sapropterin-responsive phenylketonuria. Mol Genet Metab. 2015;114:415–424.
Vockley J, Andersson HC, Antshel KM, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med. 2014;16:188–200.
Moyle JJ, Fox AM, Arthur M, Bynevelt M, Burnett JR. Meta-analysis of neuropsychological symptoms of adolescents and adults with PKU. Neuropsychol Rev. 2007;17:91–101.
Pietz J, Fatkenheuer B, Burgard P, Armbruster M, Esser G, Schmidt H. Psychiatric disorders in adult patients with early-treated phenylketonuria. Pediatrics. 1997;99:345–350.
Smith I, Knowles J. Behaviour in early treated phenylketonuria: a systematic review. Eur J Pediatr. 2000;159 suppl 2:S89–93.
Gassio R, Campistol J, Vilaseca MA, Lambruschini N, Cambra FJ, Fuste E. Do adult patients with phenylketonuria improve their quality of life after introduction/resumption of a phenylalanine-restricted diet? Acta Paediatr. 2003;92:1474–1478.
Weglage J, Fromm J, van Teeffelen-Heithoff A, et al. Neurocognitive functioning in adults with phenylketonuria: results of a long term study. Mol Genet Metab. 2013;110 suppl:S44–48.
Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376:1417–1427.
Jahja R, van Spronsen FJ, de Sonneville LMJ, et al. Social-cognitive functioning and social skills in patients with early treated phenylketonuria: a PKU-COBESO study. J Inherit Metab Dis. 2016;39:355–362.
DeRoche K, Welsh M. Twenty-five years of research on neurocognitive outcomes in early-treated phenylketonuria: intelligence and executive function. Dev Neuropsychol. 2008;33:474–504.
Bilder DA, Noel JK, Baker ER, et al. Systematic review and meta-analysis of neuropsychiatric symptoms and executive functioning in adults with phenylketonuria. Dev Neuropsychol. 2016;41:245–260.
Palermo L, Geberhiwot T, MacDonald A, Limback E, Hall SK, Romani C. Cognitive outcomes in early-treated adults with phenylketonuria (PKU): a comprehensive picture across domains. Neuropsychology. 2017;31:255–267.
Romani C, Palermo L, MacDonald A, Limback E, Hall SK, Geberhiwot T. The impact of phenylalanine levels on cognitive outcomes in adults with phenylketonuria: effects across tasks and developmental stages. Neuropsychology. 2017;31:242–254.
van Wegberg AMJ, MacDonald A, Ahring K, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis. 2017;12:162.
van Spronsen FJ, de Groot MJ, Hoeksma M, Reijngoud DJ, van Rijn M. Large neutral amino acids in the treatment of PKU: from theory to practice. J Inherit Metab Dis. 2010;33:671–676.
Brown CS, Lichter-Konecki U. Phenylketonuria (PKU): a problem solved? Mol Genet Metab Rep. 2016;6:8–12.
Bik-Multanowski M, Didycz B, Mozrzymas R, et al. Quality of life in noncompliant adults with phenylketonuria after resumption of the diet. J Inherit Metab Dis. 2008;31 suppl 2:S415–418.
Longo N, Harding CO, Burton BK, et al. Single-dose, subcutaneous recombinant phenylalanine ammonia lyase conjugated with polyethylene glycol in adult patients with phenylketonuria: an open-label, multicentre, phase 1 dose-escalation trial. Lancet. 2014;384:37–44.
Palynziq. [prescribing information]. Novato, CA: BioMarin Pharmaceutical Inc.; 2018.
Thomas J, Levy H, Amato S, et al. Pegvaliase for the treatment of phenylketonuria: results of a long-term phase 3 clinical trial program (PRISM). Mol Genet Metab. 2018;124:27–38.
Harding CO, Amato RS, Stuy M, et al. Pegvaliase for the treatment of phenylketonuria: a pivotal, double-blind randomized discontinuation Phase 3 clinical trial. Mol Genet Metab. 2018;124:20–26.
Brouwers MC, Kho ME, Browman GP, et al. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182:E839–842.
Brouwers MC, Kho ME, Browman GP, et al. Development of the AGREE II, part 1: performance, usefulness and areas for improvement. CMAJ. 2010;182:1045–1052.
Singh RH, Cunningham AC, Mofidi S, et al. Updated, web-based nutrition management guideline for PKU: An evidence and consensus based approach. Mol Genet Metab. 2016;118:72–83.
Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:e1000097.
Schardt C, Adams MB, Owens T, Keitz S, Fontelo P. Utilization of the PICO framework to improve searching PubMed for clinical questions. BMC Med Inform Decis Mak. 2007;7:16.
Oxford Centre for Evidence-Based Medicine. Levels of evidence (March 2009). https://www.cebm.net/2009/06/oxford-centre-evidence-based-medicine-levels-evidence-march-2009/. Accessed March 2018.
Woolf S, Schunemann HJ, Eccles MP, Grimshaw JM, Shekelle P. Developing clinical practice guidelines: types of evidence and outcomes; values and economics, synthesis, grading, and presentation and deriving recommendations. Implement Sci. 2012;7:61.
National Institutes of Health Office of Dietary Supplements. Nutrient recommendations: Dietary Reference Intakes (DRI). https://ods.od.nih.gov/Health_Information/Dietary_Reference_Intakes.aspx. Accessed April 2018.
Zori R, Thomas J, Shur N, et al. Induction, titration, and maintenance dosing regimen in a Phase 2 study of pegvaliase for control of blood phenylalanine in adults with phenylketonuria. Mol Genet Metab. 2018 Nov;125:217-227. https://doi.org/10.1016/j.ymgme.2018.06.010.
Longo N, Zori R, Wasserstein MP, et al. Long-term safety and efficacy of pegvaliase for the treatment of phenylketonuria in adults: combined Phase 2 outcomes through PAL-003 extension study. Orphanet J Rare Dis. 2018 Jul 4;13:108. https://doi.org/10.1186/s13023-018-0858-7.
Committee opinion no. 636: management of women with phenylketonuria. Obstet Gynecol. 2015;125:1548–1550.
Zeile WL, McCune HC, Musson DG, et al. Maternal phenylketonuria syndrome: studies in mice suggest a potential approach to a continuing problem. Pediatr Res. 2018;83:889–896.
Kruger DF, LaRue S, Estepa P. Recognition of and steps to mitigate anxiety and fear of pain in injectable diabetes treatment. Diabetes Metab Syndr Obes. 2015;8:49–56.
Costello K, Kennedy P, Scanzillo J. Recognizing nonadherence in patients with multiple sclerosis and maintaining treatment adherence in the long term. Medscape J Med. 2008;10:225.
Cox D, Mohr DC, L E. Treating self-injection phobia in patients prescribed injectable medications: a case example illustrating a six-session treatment model. Cogn Behav Pract. 2006;11:278–283.
Hanley WB, Linsao L, Davidson W, Moes CA. Malnutrition with early treatment of phenylketonuria. Pediatr Res. 1970;4:318–327.
Pode-Shakked B, Shemer-Meiri L, Harmelin A, et al. Man made disease: clinical manifestations of low phenylalanine levels in an inadequately treated phenylketonuria patient and mouse study. Mol Genet Metab. 2013;110 suppl:S66–70.
Donati A, Vincenzi C, Tosti A. Acute hair loss in phenylketonuria. J Eur Acad Dermatol Venereol. 2009;23:613–615.
Cohn RM, Yudkoff M, Yost B, Segal S. Phenylalanine-tyrosine deficiency syndrome as a complication of the management of hereditary tyrosinemia. Am J Clin Nutr. 1977;30:209–214.
Samycia M, McCourt C, Shojania K, Au S. Experiences from a combined dermatology and rheumatology clinic: a retrospective review. J Cutan Med Surg. 2016;20:486–489.
Guo EL, Katta R. Diet and hair loss: effects of nutrient deficiency and supplement use. Dermatol Pract Concept. 2017;7:1–10.
Park H, Kim CW, Kim SS, Park CW. The therapeutic effect and the changed serum zinc level after zinc supplementation in alopecia areata patients who had a low serum zinc level. Ann Dermatol. 2009;21:142–146.
Abdel Fattah NS, Atef MM, Al-Qaradaghi SM. Evaluation of serum zinc level in patients with newly diagnosed and resistant alopecia areata. Int J Dermatol. 2016;55:24–29.
Jin W, Zheng H, Shan B, Wu Y. Changes of serum trace elements level in patients with alopecia areata: a meta-analysis. J Dermatol. 2017;44:588–591.
Karashima T, Tsuruta D, Hamada T, et al. Oral zinc therapy for zinc deficiency-related telogen effluvium. Dermatol Ther. 2012;25:210–213.
Slonim AE, Sadick N, Pugliese M, Meyers-Seifer CH. Clinical response of alopecia, trichorrhexis nodosa, and dry, scaly skin to zinc supplementation. J Pediatr. 1992;121:890–895.
Moeinvaziri M, Mansoori P, Holakooee K, Safaee Naraghi Z, Abbasi A. Iron status in diffuse telogen hair loss among women. Acta Dermatovenerol Croat. 2009;17:279–284.
Park SY, Na SY, Kim JH, Cho S, Lee JH. Iron plays a certain role in patterned hair loss. J Korean Med Sci. 2013;28:934–938.
Amor KT, Rashid RM, Mirmirani P. Does D matter? The role of vitamin D in hair disorders and hair follicle cycling. Dermatol Online J. 2010;16:3.
Rasheed H, Mahgoub D, Hegazy R, et al. Serum ferritin and vitamin D in female hair loss: do they play a role? Skin Pharmacol Physiol. 2013;26:101–107.
Ghafoor R, Anwar MI. Vitamin D deficiency in alopecia areata. J Coll Physicians Surg Pak. 2017;27:200–202.
Mahamid M, Abu-Elhija O, Samamra M, Mahamid A, Nseir W. Association between vitamin D levels and alopecia areata. Isr Med Assoc J. 2014;16:367–370.
Sengupta A, Lichti UF, Carlson BA, et al. Selenoproteins are essential for proper keratinocyte function and skin development. PLoS ONE. 2010;5:e12249.
Wolf B. Biotinidase deficiency. In: Adam MP, Ardinger HH, Pagon RA, et al. GeneReviews. Seattle (WA): University of Washington, 2016. https://www.ncbi.nlm.nih.gov/books/NBK1322/ Accessed December 2018.
Fujimoto W, Inaoki M, Fukui T, Inoue Y, Kuhara T. Biotin deficiency in an infant fed with amino acid formula. J Dermatol. 2005;32:256–261.
Bradfield RB, Bailey MA, Margen S. Morphological changes in human scalp hair roots during deprivation of protein. Science. 1967;157:438–439.
McLaren DS. Skin in protein energy malnutrition. Arch Dermatol. 1987;123:1674–1676a.
Mazzocco MM, Yannicelli S, Nord AM, van Doorninck W, Davidson-Mundt AJ, Greene CL. Cognition and tyrosine supplementation among school-aged children with phenylketonuria. Am J Dis Child. 1992;146:1261–1264.
Pietz J, Landwehr R, Kutscha A, Schmidt H, de Sonneville L, Trefz FK. Effect of high-dose tyrosine supplementation on brain function in adults with phenylketonuria. J Pediatr. 1995;127:936–943.
Webster D, Wildgoose J Tyrosine supplementation for phenylketonuria. Cochrane Database Syst Rev. 2013;6:CD001507.
Smith ML, Hanley WB, Clarke JT, et al. Randomised controlled trial of tyrosine supplementation on neuropsychological performance in phenylketonuria. Arch Dis Child. 1998;78:116–121.
Hase A, Jung SE, aan het Rot M. Behavioral and cognitive effects of tyrosine intake in healthy human adults. Pharmacol Biochem Behav. 2015;133:1–6.
Lou HC, Lykkelund C, Gerdes AM, Udesen H, Bruhn P. Increased vigilance and dopamine synthesis by large doses of tyrosine or phenylalanine restriction in phenylketonuria. Acta Paediatr Scand. 1987;76:560–565.
Lykkelund C, Nielsen JB, Lou HC, et al. Increased neurotransmitter biosynthesis in phenylketonuria induced by phenylalanine restriction or by supplementation of unrestricted diet with large amounts of tyrosine. Eur J Pediatr. 1988;148:238–245.
Kalsner LR, Rohr FJ, Strauss KA, Korson MS, Levy HL. Tyrosine supplementation in phenylketonuria: diurnal blood tyrosine levels and presumptive brain influx of tyrosine and other large neutral amino acids. J Pediatr. 2001;139:421–427.
Yano S, Moseley K, Azen C. Melatonin and dopamine as biomarkers to optimize treatment in phenylketonuria: effects of tryptophan and tyrosine supplementation. J Pediatr. 2014;165:184–189 e181.
van Spronsen FJ, van Rijn M, Bekhof J, Koch R, Smit PG. Phenylketonuria: tyrosine supplementation in phenylalanine-restricted diets. Am J Clin Nutr. 2001;73:153–157.
Position of the American Dietetic Association, Dietitians of Canada, and the American College of Sports Medicine: nutrition and athletic performance. J Am Diet Assoc. 2000;100:1543–1556.
Kniskern MA, Johnston CS. Protein dietary reference intakes may be inadequate for vegetarians if low amounts of animal protein are consumed. Nutrition. 2011;27:727–730.
American Dietetic Association, Dietitians of Canada. Position of the American Dietetic Association and Dietitians of Canada: vegetarian diets. Can J Diet Pract Res. 2003;64:62–81.
Evans S, Daly A, MacDonald J, et al. The micronutrient status of patients with phenylketonuria on dietary treatment: an ongoing challenge. Ann Nutr Metab. 2014;65:42–48.
Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Drug allergy: an updated practice parameter. Ann Allergy Asthma Immunol. 2010;105:259–273.
Lichtenstein L, Ron Y, Kivity S, et al. Infliximab-related infusion reactions: systematic review. J Crohns Colitis. 2015;9:806–815.
Swierczewska M, Lee KC, Lee S. What is the future of PEGylated therapies? Expert Opin Emerg Drugs. 2015;20:531–536.
Stidl R, Fuchs S, Bossard M, Siekmann J, Turecek PL, Putz M. Safety of PEGylated recombinant human full-length coagulation factor VIII (BAX 855) in the overall context of PEG and PEG conjugates. Haemophilia. 2016;22:54–64.
Turecek PL, Bossard MJ, Schoetens F, Ivens IA. PEGylation of biopharmaceuticals: a review of chemistry and nonclinical safety information of approved drugs. J Pharm Sci. 2016;105:460–475.
Hasson F, Keeney S, McKenna H. Research guidelines for the Delphi survey technique. J Adv Nurs. 2000;32:1008–1015.
Lubich C, Allacher P, de la Rosa M, et al. The mystery of antibodies against polyethylene glycol (PEG)—what do we know? Pharm Res. 2016;33:2239–2249.
Ivens IA, Achanzar W, Baumann A, et al. PEGylated biopharmaceuticals: current experience and considerations for nonclinical development. Toxicol Pathol. 2015;43:959–983.
Webster R, Didier E, Harris P, et al. PEGylated proteins: evaluation of their safety in the absence of definitive metabolism studies. Drug Metab Dispos. 2007;35:9–16.
Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood. 2015;126:1078–1085.
Abbott LS, Zakova M, Shaikh F, Shewaramani N, Punnett A, Dupuis LL. Allergic reactions associated with intravenous versus intramuscular pegaspargase: a retrospective chart review. Paediatr Drugs. 2015;17:315–321.
Meller S, Gerber PA, Kislat A, et al. Allergic sensitization to pegylated interferon-alpha results in drug eruptions. Allergy. 2015;70:775–783.
Lubbe J, Kerl K, Negro F, Saurat JH. Clinical and immunological features of hepatitis C treatment-associated dermatitis in 36 prospective cases. Br J Dermatol. 2005;153:1088–1090.
Basaranoglu M, Celebi S, Karaaslan H, Demir A. Case study on drug-related adverse effects of hepatitis C therapy. Adv Ther. 2006;23:769–771.
Henriksen LT, Harila-Saari A, Ruud E, et al. PEG-asparaginase allergy in children with acute lymphoblastic leukemia in the NOPHO ALL2008 protocol. Pediatr Blood Cancer. 2015;62:427–433.
Wenande E, Garvey LH. Immediate-type hypersensitivity to polyethylene glycols: a review. Clin Exp Allergy. 2016;46:907–922.
Bernardeschi C, Valeyrie-Allanore L, Ortonne N, et al. Dermatological side-effects in hepatitis C infected patients under a triple regimen associating pegylated interferon, ribavirin and telaprevir. J Eur Acad Dermatol Venereol. 2016;30:143–146.
Degen A, Weichenthal M, Ugurel S, et al. Cutaneous side effects of combined therapy with sorafenib and pegylated interferon alpha-2b in metastatic melanoma (phase II DeCOG trial). J Dtsch Dermatol Ges. 2013;11:846–853.
Kim RJ, Peterson G, Kulp B, Zanotti KM, Markman M. Skin toxicity associated with pegylated liposomal doxorubicin (40 mg/m2) in the treatment of gynecologic cancers. Gynecol Oncol. 2005;97:374–378.
Hayat AS, Shaikh N, Masood N. Study for frequency and aetiology of lymphadenopathy during combination therapy for chronic hepatitis C (pegylated interferon alpha plus ribavirin) at a tertiary care hospital in Hyderabad. J Pak Med Assoc. 2011;61:986–989.
Wilhelmi M, Gubler C, Renner EL, Mullhaupt B. Lymph node enlargement during combination therapy for chronic hepatitis C with pegylated interferon alpha and ribavirin: harmless reaction or harmful disease? Swiss Med Wkly. 2006;136:65–67.
Bielory L, Gascon P, Lawley TJ, Young NS, Frank MM. Human serum sickness: a prospective analysis of 35 patients treated with equine anti-thymocyte globulin for bone marrow failure. Medicine (Baltimore). 1988;67:40–57.
Lawley TJ, Bielory L, Gascon P, Yancey KB, Young NS, Frank MM. A study of human serum sickness. J Invest Dermatol. 1985;85 1 suppl:129s–132s.
Bashshur RL, Shannon GW, Tejasvi T, Kvedar JC, Gates M. The empirical foundations of teledermatology: a review of the research evidence. Telemed J E Health. 2015;21:953–979.
Ribas J, Cunha Mda G, Schettini AP, Ribas CB. Agreement between dermatological diagnoses made by live examination compared to analysis of digital images. An Bras Dermatol. 2010;85:441–447.
Lepe V, Moncada B, Castanedo-Cazares JP, Martinez-Rodriguez A, Mercado-Ceja SM, Gordillo-Moscoso A. [First study of teledermatology in Mexico. A new public health tool]. Gac Med Mex. 2004;140:23–26.
Hailey D, Roine R, Ohinmaa A. Systematic review of evidence for the benefits of telemedicine. J Telemed Telecare. 2002;8 suppl 1:1–30.
Vyas KS, Hambrick HR, Shakir A, et al. A systematic review of the use of telemedicine in plastic and reconstructive surgery and dermatology. Ann Plast Surg. 2017;78:736–768.
Simonaro CM, Haskins ME, Schuchman EH. Articular chondrocytes from animals with a dermatan sulfate storage disease undergo a high rate of apoptosis and release nitric oxide and inflammatory cytokines: a possible mechanism underlying degenerative joint disease in the mucopolysaccharidoses. Lab Invest. 2001;81:1319–1328.
Fruhauf J, Krock S, Quehenberger F, et al. Mobile teledermatology helping patients control high-need acne: a randomized controlled trial. J Eur Acad Dermatol Venereol. 2015;29:919–924.
Shapiro M, James WD, Kessler R, et al. Comparison of skin biopsy triage decisions in 49 patients with pigmented lesions and skin neoplasms: store-and-forward teledermatology vs. face-to-face dermatology. Arch Dermatol. 2004;140:525–528.
Choquette D, Bensen W, Nantel F. Reduced incidence of infusion reactions to infliximab in patients pre-medicated with acetaminophen. J Rheumatol. 2010;37:1324.
Zidan J, Hussein O, Abzah A, Tamam S, Farraj Z, Friedman E. Oral premedication for the prevention of hypersensitivity reactions to paclitaxel. Med Oncol. 2008;25:274–278.
Sasada S, Hirashima T, Nakamura Y, et al. Preliminary experience with a modified premedication protocol that included intravenous diphenhydramine and calcium bromide for the prophylaxis of paclitaxel-related hypersensitivity reactions. Int J Clin Oncol. 2007;12:274–278.
Raisch DW, Campbell W, Garg V, et al. Description of anaphylactic reactions to paclitaxel and docetaxel reported to the FDA, with a focus on the role of premedication. Expert Opin Drug Saf. 2011;10:521–528.
Quock J, Dea G, Tanaka M, Gandara D, Lara P, Lau D. Premedication strategy for weekly paclitaxel. Cancer Invest. 2002;20:666–672.
Peereboom DM, Donehower RC, Eisenhauer EA, et al. Successful re-treatment with taxol after major hypersensitivity reactions. J Clin Oncol. 1993;11:885–890.
Kidera Y, Satoh T, Ueda S, et al. High-dose dexamethasone plus antihistamine prevents colorectal cancer patients treated with modified FOLFOX6 from hypersensitivity reactions induced by oxaliplatin. Int J Clin Oncol. 2011;16:244–249.
Sudarshan MK, Kodandaram NS, Venkatesh GM, Mahendra BJ, Ashwath Narayana DH, Parasuramalu BG. Evaluation of a new premedication protocol for administration of equine rabies immunoglobulin in patients with hypersensitivity. Indian J Public Health. 2007;51:91–96.
Navo M, Kunthur A, Badell ML, et al. Evaluation of the incidence of carboplatin hypersensitivity reactions in cancer patients. Gynecol Oncol. 2006;103:608–613.
Kimura K, Adachi M, Kubo K. H1- and H2-receptor antagonists prevent histamine release in allergic patients after the administration of midazolam-ketamine. A randomized controlled study. Inflamm Res. 1999;48:128–132.
Brockow K, Kiehn M, Riethmuller C, Vieluf D, Berger J, Ring J. Efficacy of antihistamine pretreatment in the prevention of adverse reactions to Hymenoptera immunotherapy: a prospective, randomized, placebo-controlled trial. J Allergy Clin Immunol. 1997;100:458–463.
Mervak BM, Davenport MS, Ellis JH, Cohan RH. Rates of breakthrough reactions in inpatients at high risk receiving premedication before contrast-enhanced CT. AJR Am J Roentgenol. 2015;205:77–84.
Kim SH, Lee SH, Lee SM, et al. Outcomes of premedication for non-ionic radio-contrast media hypersensitivity reactions in Korea. Eur J Radiol. 2011;80:363–367.
Bae YJ, Hwang YW, Yoon SY, et al. The effectiveness of automatic recommending system for premedication in reducing recurrent radiocontrast media hypersensitivity reactions. PLoS ONE. 2013;8:e66014.
Trautmann A, Anders D, Stoevesandt J. H1-antihistamine premedication in NSAID-associated urticaria. J Allergy Clin Immunol Pract. 2016;4:1205–1212.
Mach CM, Lapp EA, Weddle KJ, et al. Adjunct histamine blockers as premedications to prevent carboplatin hypersensitivity reactions. Pharmacotherapy. 2016;36:482–487.
Bartoli F, Bruni C, Cometi L, et al. Premedication prevents infusion reactions and improves retention rate during infliximab treatment. Clin Rheumatol. 2016;35:2841–2845.
Aeschlimann FA, Hofer KD, Cannizzaro Schneider E, Schroeder S, Lauener R, Saurenmann RK. Infliximab in pediatric rheumatology patients: a retrospective analysis of infusion reactions and severe adverse events during 2246 infusions over 12 years. J Rheumatol. 2014;41:1409–1415.
Jacobstein DA, Markowitz JE, Kirschner BS, et al. Premedication and infusion reactions with infliximab: results from a pediatric inflammatory bowel disease consortium. Inflamm Bowel Dis. 2005;11:442–446.
Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. J Allergy Clin Immunol. 2006;117:391–397.
Brown SG. Clinical features and severity grading of anaphylaxis. J Allergy Clin Immunol. 2004;114:371–376.
Scherer K, Brockow K, Aberer W, et al. Desensitization in delayed drug hypersensitivity reactions—an EAACI position paper of the Drug Allergy Interest Group. Allergy. 2013;68:844–852.
Fajt ML, Petrov AA. Desensitization protocol for rituximab-induced serum sickness. Curr Drug Saf. 2014;9:240–242.
Poreaux C, Bronowicki JP, Debouverie M, Schmutz JL, Waton J, Barbaud A. Clinical allergy. Managing generalized interferon-induced eruptions and the effectiveness of desensitization. Clin Exp Allergy. 2014;44:756–764.
Akbayram S, Dogan M, Akgun C, Caksen H, Oner AF. A desensitization protocol in children with L-asparaginase hypersensitivity. J Pediatr Hematol Oncol. 2010;32:e187–191.
Shah AC, Minturn JE, Li Y, et al. Carboplatin rechallenge after hypersensitivity reactions in pediatric patients with low-grade glioma. Pediatr Blood Cancer. 2016;63:21–26.
Taffet SL, Das KM. Desensitization of patients with inflammatory bowel disease to sulfasalazine. Am J Med. 1982;73:520–524.
Amoros-Reboredo P, Sanchez-Lopez J, Bastida-Fernandez C, et al. Desensitization to rituximab in a multidisciplinary setting. Int J Clin Pharm. 2015;37:744–748.
Mates M, Zevin S, Breuer GS, Navon P, Nesher G. Desensitization to hydroxychloroquine—experience of 4 patients. J Rheumatol. 2006;33:814–816.
Fam AG, Lewtas J, Stein J, Paton TW. Desensitization to allopurinol in patients with gout and cutaneous reactions. Am J Med. 1992;93:299–302.
Galvao VR, Castells MC. Hypersensitivity to biological agents—updated diagnosis, management, and treatment. J Allergy Clin Immunol Pract. 2015;3:175–185. quiz 186
Cernadas JR, Brockow K, Romano A, et al. General considerations on rapid desensitization for drug hypersensitivity—a consensus statement. Allergy. 2010;65:1357–1366.
Tucker WF, Lewis FM. Digital imaging: a diagnostic screening tool? Int J Dermatol. 2005;44:479–481.
Michele Hilton Boon JH, Karen R, Lorna T, AGREE II workshop participants for the RARE-Bestpractices Consortium. Report of an international workshop to explore the utility of the AGREE II instrument for appraisal of rare disease guidelines. Rare Dis Orphan Drugs. 2015;2:11–15.
Pai M, Iorio A, Meerpohl J, et al. Developing methodology for the creation of clinical practice guidelines for rare diseases: a report from RARE-Bestpractices. Rare Dis. 2015;3:1.
Acknowledgements
The authors thank Christine S. Brown, Executive Director of the National PKU Alliance, for her review of the guidance statements and of the manuscript. The authors also thank Farrah Rajabi of Boston Children’s Hospital; Synneva Hagen‐Lillevik of the University of Utah; Matt Fickie of Mount Auburn Hospital, Cambridge; and Jennifer Gentile of Boston Children’s Hospital, for their assistance with literature searches and analysis, and Michele Hilton-Boon, Karen Spithoff, and Melissa Brouwers for their review of the manuscript according to the AGREE II instrument. The independent secretariat comprised members of Lucid Partners, Burleighfield House, Buckinghamshire, United Kingdom. Medical writing and editorial support to the authors in the development of the manuscript was provided by Helen Jones, Hannah Tivey, and James Hadfield of Lucid Partners; financial support for these services was provided by BioMarin.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
DISCLOSURE
General statement: All authors of the manuscript were chosen as Steering Committee members based on their unique experience of treating patients with pegvaliase through the clinical trial program. All Steering Committee members were offered a discretional honorarium by BioMarin for their participation in the meetings and processes used to develop this guidance. An independent medical communications agency (Lucid) was employed to perform the role of methodology expert and secretariat throughout the process and to ensure editorial independence from the sponsor. Participants did not receive payment for completing the Delphi survey. Authors did not receive payment for development of the manuscript.
N.L. has participated as an investigator in clinical trials sponsored by Aeglea, BioMarin Pharmaceutical Inc., Genzyme, Horizon Pharma, Protalix, Retrophin, Shire, Stealth Therapeutics, and Ultragenyx. He is on the Steering Committee for the pegvaliase clinical trial program. He has received grants from Lumos Pharma and consultancy fees from Aeglea, BioMarin Pharmaceutical Inc., Censa Pharmaceuticals, Dimension Therapeutics, Genzyme, Hemoshear, Horizon Pharma, Lumos Pharma, Moderna, Mitobridge, Pfizer, Retrophin, and Stealth Therapeutics. He has received travel fees from BioMarin Pharmaceutical Inc., Cello Health Sciences, Lumos Pharma, and SigmaTau/AlphaSigma. D.D. has (in the past 5 years) participated as an investigator in clinical trials sponsored by BioMarin Pharmaceutical Inc., Genzyme/Sanofi, and Amicus Therapeutics. He has received research support and consultancy fees from BioMarin Pharmaceutical Inc. and Demeter Therapeutics. He is a member of the Scientific and Clinical Advisory Board at Audentes Therapeutics Inc. and has served as a consultant for Ichorion Therapeutics and Complete Genomics. H.L. is involved in clinical trials sponsored by BioMarin Pharmaceutical Inc., has acted as a consultant, and has participated in advisory boards for BioMarin Pharmaceutical Inc. K.V. has been involved in clinical trials sponsored by BioMarin Pharmaceutical Inc., Horizon Pharma, and Ultragenyx. She has received consultancy fees from BioMarin Pharmaceutical Inc. and Horizon Pharma and has participated in advisory boards held by BioMarin Pharmaceutical Inc. H.B. has been involved in clinical trials sponsored by BioMarin Pharmaceutical Inc. and Ultragenyx. She has received consultancy fees from BioMarin Pharmaceutical Inc., Horizon Pharma, Cambrooke Therapeutics, and Nutricia. D.A.B. has been involved in a phase 3 study sponsored by BioMarin Pharmaceutical Inc., has participated in advisory boards held by BioMarin Pharmaceutical Inc. and Audentes Therapeutics Inc., and has received consultancy fees from BioMarin Pharmaceutical Inc. B.B. has participated as an investigator for clinical trials sponsored by by Alexion, BioMarin Pharmaceutical Inc., Genzyme Sanofi, Shire, and Ultragenyx Pharmaceuticals Inc. She has participated in advisory boards for BioMarin Pharmaceutical Inc., Horizon Pharma, Nestle, and Viking Therapeutics, and has received research grants from Shire and consultancy fees from Alexion, Shire, BioMarin Pharmaceutical Inc., and RegenxBio. C.G. is involved in clinical trials sponsored by BioMarin Pharmaceutical Inc. H.N. has been involved in clinical trials sponsored by BioMarin Pharmaceutical Inc. and The University of Texas Health Science Center, Houston, and has participated in advisory boards held by and received consultancy fees from BioMarin Pharmaceutical Inc. F.R. has been involved in clinical trials sponsored by BioMarin Pharmaceutical Inc. and has received consultancy fees from BioMarin Pharmaceutical Inc. S.S. is an investor in clinical trials sponsored by BioMarin Pharmaceutical Inc. and has participated in advisory boards held by BioMarin Pharmaceutical Inc. A.S.‐V. has participated as an investigator for clinical trials sponsored by Alexion, BioMarin Pharmaceutical Inc., Genzyme, Horizon Pharma, and Ultragenyx Pharmaceuticals Inc. She has participated in advisory boards for BioMarin Pharmaceutical Inc. and Horizon Pharma. M.S. has participated as a principal investigator in clinical trials sponsored by BioMarin Pharmaceutical Inc. J.T. has been involved in clinical trials sponsored by BioMarin Pharmaceutical Inc., and has participated in advisory boards held by BioMarin Pharmaceutical Inc. She is a member of the Steering Committee for the pegvaliase clinical program. J.V. has participated as an investigator in clinical trials sponsored by BioMarin Pharmaceutical Inc. and has received research support from BioMarin Pharmaceutical Inc. He was a speaker at the BC Childrens’ and Womens’ Hospital in Vancouver on behalf of the Canadian PKU Alliance; this event was sponsored by BioMarin Pharmaceutical Inc., Cambrooke Therapeutics, Mead Johnson, Nutricia, and Vitaflo. R.Z. has participated as an investigator in clinical trials sponsored by BioMarin Pharmaceutical Inc. and Hyperion Therapeutics Inc. and is on the Steering Committee for the pegvaliase clinical program. C.O.H. has participated as an investigator for clinical trials sponsored by BioMarin Pharmaceutical Inc. and has received consultancy fees services and research grants from BioMarin Pharmaceutical Inc., Cydan Development Inc., Dimension Therapeutics, Horizon Pharma, Pfizer, Rubius Therapeutics, StrideBio, and Synlogic.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Longo, N., Dimmock, D., Levy, H. et al. Evidence- and consensus-based recommendations for the use of pegvaliase in adults with phenylketonuria. Genet Med 21, 1851–1867 (2019). https://doi.org/10.1038/s41436-018-0403-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0403-z
Keywords
This article is cited by
-
Systematic literature review of the somatic comorbidities experienced by adults with phenylketonuria
Orphanet Journal of Rare Diseases (2024)
-
Nutrition management of PKU with pegvaliase therapy: update of the web-based PKU nutrition management guideline recommendations
Orphanet Journal of Rare Diseases (2023)
-
Genetic etiology and clinical challenges of phenylketonuria
Human Genomics (2022)
-
Expert opinion of an Italian working group on the assessment of cognitive, psychological, and neurological outcomes in pediatric, adolescent, and adult patients with phenylketonuria
Orphanet Journal of Rare Diseases (2022)
-
Neonatal screening and genotype-phenotype correlation of hyperphenylalaninemia in the Chinese population
Orphanet Journal of Rare Diseases (2021)
