
Abstract
FOXP3+ regulatory T cells (Treg) are indispensable for immune homoeostasis and for the prevention of autoimmune diseases. Interleukin-2 (IL-2) signalling is critical in all aspects of Treg biology. Consequences of defective IL-2 signalling are insufficient numbers or dysfunction of Treg and hence autoimmune disorders in human and mouse. The restoration and maintenance of immune homoeostasis remain central therapeutic aims in the field of autoimmunity. Historically, broadly immunosuppressive drugs with serious side-effects have been used for the treatment of autoimmune diseases or prevention of organ-transplant rejection. More recently, ex vivo expanded or in vivo stimulated Treg have been shown to induce effective tolerance in clinical trials supporting the clinical benefit of targeting natural immunosuppressive mechanisms. Given the central role of exogenous IL-2 in Treg homoeostasis, a new and promising focus in drug development are IL-2-based approaches for in vivo targeted expansion of Treg or for enhancement of their suppressive activity. In this review, we summarise the role of IL-2 in Treg biology and consequences of dysfunctional IL-2 signalling pathways. We then examine evidence of efficacy of IL-2-based biological drugs targeting Treg with specific focus on therapeutic candidates in clinical trials and discuss their limitations.
Similar content being viewed by others

Introduction
In 1976, the supernatant of activated T cells was found to contain a potent T cell growth factor, which was cloned in 1983 as interleukin-2 (IL-2) [1,2,3]. The identification of IL-2 marked the start of substantial efforts to unravel IL-2-dependent immunological processes, to mechanistically understand IL-2 binding to its receptor and to dissect the signalling pathways downstream of receptor activation. Importantly, with the discovery of IL-2 and an increasing knowledge on IL-2 functions, immense research efforts were launched to develop IL-2-based immunotherapies to exploit its properties in cancer and autoimmune diseases. Here, we provide a brief overview on IL-2 signalling, its relevance in the biology of regulatory T cells (Treg), and detail recent advances in IL-2-based immunotherapeutics for autoimmune and inflammatory diseases predominantly in clinical stages of development.
IL-2 expression, capture and signalling
The signalling-competent IL-2 receptor (IL-2R) is expressed either as heterodimer or -trimer [4]. The dimeric IL-2R consists of the IL-2Rβ chain (CD122, shared with IL-15R) and the common γ chain (γc, CD132, shared with the receptors for IL-4/7/9/15/21) [4,5,6,7,8,9,10] and displays intermediate affinity for IL-2 (Kd~10−9 M). It can hence signal upon binding of IL-2 as well as IL-15. The trimeric IL-2R additionally includes the IL-2Rα chain (CD25) [11]. CD25 can be considered a monomeric IL-2R as it binds IL-2, however, it is not capable of signalling. Although CD25 itself displays only low affinity (Kd~10−8 M) and a high on-off rate for IL-2, it delivers IL-2 to the dimeric receptor [11] and its presence increases the affinity of the trimeric receptor for IL-2 100-fold (Kd~10−11 M), consequently providing the expressing cells with a substantial competitive advantage in IL-2 capture [12,13,14].
Various immune and non-immune cell types express the IL-2R. In humans and mice, the dimeric IL-2R is expressed at low levels by CD4 memory T cells and naïve T cells and at high levels by CD8 memory T cells [15, 16] and CD56low NK cells [17]. In mice, the trimeric IL-2R on the other hand is expressed highest on Treg [18, 19] and at lower levels on recently activated and effector CD8 T cells, ILC2, and some NKT and CD56bright NK cells [17, 20,21,22,23]. Similarly, in human peripheral blood mononuclear cells (PBMC), Treg express the highest levels of the trimeric IL-2R whereas other immune cells such as CD45ROpos CD4 T cells, most CD56high NK cells, few CD4 and CD8 naïve T cells express it at lower levels [24]. The trimeric IL-2R is also expressed by endothelial cells, with further CD25 upregulation upon IL-2 treatment [25, 26], and signalling can cause the vascular leak syndrome—a known adverse effect of high-dose IL-2 therapy in mice and patients.
IL-2 is a pleiotropic cytokine that can act in an autocrine and paracrine way, with cell type- and context-dependent positive effects on survival, population expansion or lineage stability [17, 27,28,29]. The main source of IL-2 are CD4 conventional T cells upon T cell receptor (TCR)/CD28 (co-) stimulation [30, 31]. Other immune cells such as CD8 T cells, NK(T) cells or dendritic cells can produce IL-2 as well albeit at lower quantities [24, 29]. Treg are highly dependent on exogenous IL-2 sources as FOXP3 in cooperation with other transcription factors represses Il2 transcription [32,33,34]. Yet, a sizable population of Treg in mice is capable of producing IL-2, albeit at a lower per cell level compared to Foxp3neg CD4 T cells [29]. In contrast, IL-2 expression in human peripheral blood FOXP3pos CD4 T cells is limited to a subset of cells with low expression of FOXP3 likely not representing suppressive Treg (Fig. 1) [35].

Mouse splenocytes (wildtype C57Bl6/J, n = 4) and healthy human PBMC (n = 5) were stained for CD3, TCRγδ [mouse], CD4, FOXP3 and IL-2 along with a viability dye. Representative flow plots depicting IL-2 expression in mouse (top) and human (bottom) conventional T cells (Tconv, blue) (live CD3+ TCRγδneg [mouse] CD4+ FOXP3neg) and Treg (red) (live CD3+ TCRγδneg [mouse] CD4+ FOXP3+). The frequency of IL-2+ cells of FOXP3neg and FOXP3+ cells is shown (mean ± SEM). The geometric mean fluorescence intensities of IL-2 as a measure to compare per cell protein levels between Tconv and Treg are 2958 ± 160 (mouse Tconv) vs 2130 ± 125 (mouse Treg) and 7308 ± 904 (human Tconv) vs 5555 ± 452 (human FOXP3pos cells) (mean ± SEM). Ethical approvals were obtained from the KU Leuven Animal Ethics Committee (150/2019) and the University Clinic Leuven Ethical Committee (S65883). Antibodies were purchased from BD Biosciences (564667, 566405, 624295), Biolegend (100225, 503840, 320214), Miltenyi Biotec (130–111–601), and ebioscience (65-0865-18, 56-0038-80, 48-0048-42).
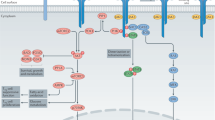
Upon binding of IL-2 to its receptor the quaternary complex is internalised, CD25 is recycled back to the cell surface while CD122 and CD132 are degraded [36, 37]. The IL-2:IL-2R complex can signal via three major pathways, each activating different downstream transcriptional regulators [27, 38, 39]. Depending on the downstream signalling pathway, PI3K (PI3K-AKT-mTOR pathway), SHC1 (MAPK pathway) or STAT (signal transducer and activator or transcription) 5 (Janus activating kinase [JAK]1/3-STAT pathway) are triggered. In T cells, and particularly Treg, phosphorylation of the IL-2Rβ chain and common γ chain by JAK1 and JAK3 and subsequent activation of STAT5 accounts for 90% of IL-2 signalling (Fig. 2)[40, 41]. In Treg, the PI3K-AKT-mTOR pathway is suppressed by PTEN (phosphatase and tensin homologue). This mechanism regulates Treg homoeostasis by negatively regulating proliferation and positively regulating lineage stability likely by increased nuclear translocation of FOXO1/FOXO3a [42,43,44]. Further, to regulate Treg homoeostasis and maintain Treg lineage fate, STAT5 activation and SOCS1 expression regulate each other in a positive inhibitory loop. IL-2 signalling induces SOCS1 expression and SOCS1 in turn attenuates IL-2R signalling by blocking JAK proteins, hence interrupting the phosphorylation of STAT5 [45,46,47,48,49].

Upon binding of IL-2 to the trimeric IL-2R, JAK1 and JAK3 phosphorylate the IL-2Rβ or IL-2R common γ (cγ) chain, respectively. STAT5 docks onto the phosphorylated residues and is then phosphorylated by JAK1/3. Phosphorylated STAT5 (pSTAT5) dimerises and translocates to the nucleus to bind its target loci (such as FoxP3/FOXP3). IL-2 signalling is critical in Treg biology. It plays a dominant role in thymic Treg development (bottom, left), during peripheral Treg functional maturation in barrier tissues (bottom, middle), and is indispensable for the survival and functional lineage stability of mature Treg in secondary lymphoid organs (SLO) (bottom, right).
Treg homoeostasis is essential to preserve the delicate balance of immune activation. Absence of Treg or decreased function will result in autoimmune diseases, while abundance of Treg will lead to overt immune suppression. These events are balanced by the exclusive IL-2 capture sensitivity of Treg, an overall high dependency of Treg on exogenous IL-2 sources and, hence, a reciprocal control between effector T cells and Treg. The preferential and high expression of the trimeric IL-2R renders Treg most sensitive to IL-2 capture thereby outcompeting other cell types. This superior efficiency in IL-2 capture is exploited by low-dose IL-2 therapy to specifically target and expand Treg [50, 51]. Further, in Treg the cooperation of the trimeric receptor and the serine/threonine phosphatase PP2A confers increased sensitivity to IL-2 [52, 53] and PP2A deficiency in Treg results in autoimmunity [54]. Consequently, 10-fold lower IL-2 levels are required for STAT5 activation in Treg compared to CD25-expressing non-Treg and optimal IL-2-dependent gene expression in Treg occurs at 100-fold lower IL-2 concentrations compared to other cell types expressing CD25 [55]. The high sensitivity to IL-2 signalling allows for sufficient signalling when available CD25 surface levels are reduced [56]. If the superior IL-2 capture is strongly compromised such as it is in CD25-deficient mice or in patients with risk alleles for CD25, systemic inflammation and/or autoimmunity are the consequence of the resulting Treg deficiency or disturbed Treg homoeostasis.
IL-2 in Treg biology and function
Initially, and with the assumption that the main function of IL-2 was the activation of effector T cells and NK cells, efforts to exploit IL-2 in immunotherapy were focused on promoting anti-tumour immunity [57]. High-dose recombinant IL-2 (aldesleukin; trade name Proleukin) was the first immunotherapy approved by the U.S. Food and Drug Administration (FDA) in 1992 [58, 59]. The activation of effector T cells as the main function of IL-2 was contested when ablation of Il2, Il2ra and Il2rb expression in mice caused lethal lymphoproliferation and autoimmunity, rather than immunodeficiency [60]. Ten years later, these observations were explained with the discovery of Treg as an immunosuppressive CD4pos T cell subset characterised by high levels of CD25 and a non-redundant function for IL-2 in many aspects of Treg biology [18, 61]. The absence or dysfunction of Treg results in fatal multiorgan autoimmunity in mice (scurfy [62]) and human (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome, IPEX [63]), and their reduced function has been reported in several systemic (auto-)inflammatory diseases [51, 64,65,66,67,68,69].
IL-2 signalling (via JAK-STAT5) has been demonstrated to be important for Treg thymic development, peripheral induction, lineage commitment and stability sustainability, and homoeostasis (Fig. 2). Treg development takes place in the thymus (thymic Treg, tTreg) but conventional CD4 T cells can convert into Treg upon tolerogenic stimulation in the periphery as well (peripherally-induced Treg, pTreg). IL-2 signalling is important in establishing the Treg identity alongside with TCR and TGFβ signalling [70, 71]. For tTreg development, a two-step model of TCR and cytokine signalling has been proposed in which the main driving cytokine is IL-2 and its induction of the JAK-STAT5 signalling pathway. IL-7 and IL-15 can compensate for the lack of IL-2 but in the presence of IL-2 their receptors are downregulated establishing a dominant role for IL-2 [72,73,74,75]. Defective expression of IL-2 or its receptor subunits, caused by single nucleotide polymorphisms in human or via introduced genetic modification in mice, results in a lack of functional Treg and consequently lymphoproliferation, multiorgan infiltration of activated lymphocytes and lethal autoimmunity [15, 60, 76,77,78,79,80,81]. Similarly, inappropriate regulation of IL-2 signal transduction impairs Treg homoeostasis and functional stability [42, 54, 82,83,84,85,86]. Although genetic studies using germline deletion cannot ultimately dissect the requirement for IL-2 during (thymic or peripheral) Treg development from the requirement for IL-2 during peripheral survival and expansion, several lines of evidence support both intrathymic and peripheral roles for IL-2 in Treg.
IL-2- or IL-2R-deficient mice
Autoimmunity in IL-2- or IL-2R-deficient mice can be prevented by adoptive transfer of Treg demonstrating that proficiency in IL-2-signalling in mature Treg is sufficient and necessary for peripheral tolerance even when thymic Treg development is impaired [61, 87,88,89]. Notably, Treg numbers but not their suppressive activity can be rescued in IL-2- or CD25-deficient mice by depletion of the pro-apoptotic protein Bim [90]. Also in IL-2-sufficient mice, Bim has been shown to mediate Treg apoptosis to regulate Treg numbers. A critical role for IL-2 in Treg peripheral survival is to maintain the pro-survival protein Mcl-1 [91].
Antibody-mediated neutralisation or preferential delivery of IL-2
Further, antibody-mediated neutralisation of IL-2 and studies utilising IL-2:anti-IL-2 immune complexes have illustrated the indispensable role of IL-2 in peripheral Treg maintenance and functional maturation. Neutralisation of IL-2 induces T cell-mediated autoimmunity by selectively reducing Treg numbers [92]. Conversely, the application of IL-2:anti-IL-2 immune complexes can prevent the binding of IL-2 to effector T cells and preferentially deliver it to Treg to substantially expand these [93].
Competitive advantage of IL-2RWT cells in mixed bone marrow chimeras
In Treg, IL-2-STAT5 signalling is sensed via the conserved non-coding sequence 2 (CNS2) in the FoxP3 locus sustaining FOXP3 expression and controlling stable FOXP3 expression inheritance [94]. The requirement for IL-2 in Treg development, homoeostasis and competitive fitness has further been studied in mixed bone marrow chimeric mice co-transplanted with IL-2 signalling-deficient bone marrow and wild-type bone marrow. In these mice, wild-type Treg greatly outnumbered mutant FOXP3pos CD4 T cells in the thymus and in the periphery illustrating the competitive disadvantage conferred by IL-2 signalling deficiency [19, 95]. These experimental designs, however, use bone marrow from IL-2R germline knockout mice and hence, despite analysing thymic as well as peripheral Treg, fall short on undoubtedly dissecting the role of IL-2 signalling during development versus its role in homoeostasis of Treg. Indeed, the shared signalling pathway with IL-15 and IL-7 and hence their compensatory potential [74] together with data obtained in TCR-transgenic mice [96], indicate that Treg lineage induction can be IL-2 signalling-independent.
Treg-specific rescue of IL-2 signalling
Finally, the intrinsic requirement for IL-2 signalling in Treg maintenance and fitness has been demonstrated in mice with Treg lineage-specific deficiency of CD25 or the IL-2Rβ chain presenting with decreased Treg frequencies and reduced per cell FOXP3 protein levels, and developing fatal autoimmune disease [97]. In line with the aforementioned studies, dysfunctional FOXP3low CD25neg Treg can be found in mice with germline deficiencies of IL-2 and IL-2R [19, 95, 96].
Together, these studies demonstrate the relevance for IL-2 in Treg development, maturation, and survival, and suggest that it serves a direct role in Treg suppressive function.
The role of IL-2 in Treg maturation and function can in part be attributed to a positive feedback loop between FOXP3 and CD25 expression. Upon activation of the IL-2-STAT5 signalling pathway in Treg, phosphorylated STAT5 binds the FoxP3 locus to promote its expression. FOXP3 in turn positively regulates expression of CD25 [33, 85, 98, 99]. CD25 expression constitutes part of the Treg transcriptional signature and upon loss of Treg lineage fate, CD25 gene expression is lost quickly, further illustrating the interdependency of Treg signature genes such as FoxP3 and CD25 [100]. High expression of the high-affinity trimeric IL-2R on Treg, however, is not only necessary for Treg to scavenge the low levels of IL-2 for their homoeostasis; the ability to preferentially capture IL-2 also presents an immunosuppressive mechanism by starving effector T cells and NK cells from IL-2 and hence limiting their activation and proliferation. The requirement for the IL-2-STAT5-FOXP3 axis in Treg suppressive function was elegantly demonstrated in a study using transgenic mice with Treg-specific deficiency of the IL-2R with simultaneous expression of constitutively active STAT5 [97]. Early fatal autoimmune disease otherwise observed in mice with Treg-specific IL-2R deficiency can be rescued by constitutive STAT5 signalling; however, the mice still succumbed at a later age from uncontrolled CD8 T cell activation and expansion. This demonstrated that IL-2 consumption via the high-affinity trimeric IL-2R expressed by Treg particularly controls the CD8 T cell population size and activity.
Memory Treg (mTreg)—analogous to their non-regulatory counterparts—are long-lived cells which upon secondary exposure however do not respond with proliferation and pro-inflammatory cytokine production but instead possess increased suppressive function. The induction of mTreg would hence be of therapeutic interest. They are thought to mitigate tissue damage during the rapid and heightened response of effector memory T cells upon secondary antigen exposure or to reinforce foetal tolerance during pregnancy. Accordingly, mTreg present as antigen-experienced, CCR7low cells, express high levels of anti-apoptotic BCL-2 and proliferate less (Ki67low) compared to activated Treg [101, 102].
While memory Treg may be less dependent on IL-2 for long-term maintenance, they express high levels of CD25 and expand in response to low-dose IL-2 therapy [55, 101, 103,104,105]. However, long-lived (local) tolerance induced by IL-2-based therapy relies on the pre-existence of antigen-specific mTreg or (local) induction of mTreg. Antigen therapy to induce hyposensitivity to allergens might be explained by the induction of mTreg. However, e.g. islet-specific antigen therapy alone has been disappointing in trials with type 1 diabetes patients and it has been suggested that antigen therapy must be combined with a Treg-inducing agent such as low-dose IL-2. While this presents a promising strategy for type 1 diabetes and other autoimmune diseases, clinical trials are needed to establish antigen dosing and boosting regimens, long-term efficacy and its correlation with mTreg induction and persistence.
The roles of IL-2 in Treg biology and suppressive function make IL-2 a highly attractive immunotherapeutic molecule in the context of autoimmunity and transplantation. However, its activities on different (immune) cell types also demand caution in clinical trial design and close monitoring of adverse effects. In the below chapters, we will discuss recent pre-clinical and clinical efforts to develop IL-2-based immunotherapeutic strategies that target Treg to treat autoimmune conditions characterised by low numbers or reduced suppressive activity of Treg as well as to prevent transplant rejection.
IL-2-based immunotherapies
IL-2 was the first cytokine therapy approved by the U.S. FDA [58]. The initial indications were in metastatic cancers where IL-2 had to be administered at very high doses (HD IL-2) to achieve clinical benefit. The high doses were necessary because of a very short half-life of IL-2 in vivo and for stimulation the cytotoxic effector T cells and NK cells, presumably, once the high-affinity receptor on Treg has been saturated. The approval was based on the overall objective response rates in up to 20% of patients and durable complete responses for up to 91 months [59]. However, the treatment also induced severe treatment-associated toxicities including vascular leak syndrome and clinical manifestations of a cytokine storm. Efforts to reduce toxicities by lowering the dose led to a considerable loss of therapeutic efficacy due to the expansion of immunosuppressive Treg that contain a high-affinity IL-2 receptor and thereby outcompete other cells for IL-2 [106]. HD IL-2 remains an important treatment in selected patients, either as a first-line option or in combination with new targeted and immunological therapies [107].
The preferential capture of natural IL-2 via the high-affinity IL-2R expressed by Treg is exploited in low-dose (LD) IL-2 therapy. The short half-life of IL-2 (<10 min [108]) requires daily injections of 0.5-3 million international units (MIU) in repetitive treatment courses with effects on Treg lasting days to weeks, but at the same time its quick clearance allows for fast and flexible dose adjustment to ameliorate possible adverse effects. Overall, LD IL-2 treatment is well-tolerated as documented in animal studies and clinical trials (reviewed in [50, 51]). Long-term administration in mice showed no impairment of immune responses or vaccination, nor did it increase cancer occurrence [109]. Similarly, a long-term study in children with early onset type 1 diabetes mellitus (T1D) concluded that the treatment was safe and well-tolerated [110]. However, inherent to the pleiotropic nature of IL-2, dose-dependent mild-to-moderate adverse effects are associated with LD IL-2 treatment. While high-dose IL-2 treatment can induce vascular leak syndrome, LD IL-2 may result in transient influenza-like symptoms, or in eosinophilia driven by ILC2-produced IL-5 [24, 51]. Overall, data obtained in murine disease models and clinical studies are promising with partial or complete response to treatment. Completed and ongoing clinical trials with LD IL-2 in autoimmune and rheumatic diseases are summarised elsewhere [111].
Fuelled by the therapeutic benefit of LD IL-2 and to overcome its limitations, further efforts have focused on the development of second-generation versions of IL-2 with superior pharmacokinetics and Treg selectivity. Aims of these efforts beyond target cell selectivity and reduced off-target effects are to increase the half-life of the novel molecules (at the expense of fast adjustment of dosing in case of adverse effects), less frequent administration, and increased therapeutic dose range. Several groups and pharmaceutical companies have developed PEGylated IL-2 variants [112, 113], IL-2 muteins [114,115,116], fusion proteins of IL-2 linked to CD25 [117, 118], and IL-2:anti-IL-2 antibody complexes [93, 119,120,121] that promote Treg cell expansion in vivo.
Here, we will present promising IL-2-based molecules and clinical translation thereof with focus on selected therapeutic IL-2 molecules with post-translational modifications, IL-2 muteins, fusion proteins of IL-2 with other molecules, alternative delivery methods of IL-2, and IL-2:anti-IL-2 antibody complexes (Table 1).
PEGylated IL-2 variants
PEGylation is a covalent conjugation of proteins to inert polyethylene glycol (PEG) moieties. PEGylation extends the half-life of protein therapeutics by increasing the effective molecular weight of the molecule, while the PEG moieties can also shield the proteins from digestion by proteolytic enzymes via increased steric hindrance. For example, a PEG-modified murine IL-2 increased IL-2 retention in vivo by protection from enzymatic digestion and renal clearance [122]. Although PEG is known as a safe, inert and non-immunogenic synthetic polymer, some FDA-approved drugs are associated with the development of antibodies against PEG moieties that accelerate drug clearance and loss of clinical efficacy [123, 124].
NKTR-358/LY3471851/rezpegaldesleukin (Nektar/Lilly) is recombinant human IL-2 (aldesleukin sequence) chemically conjugated with stable PEG moieties, which has an attenuated affinity for IL-2Rβ compared with recombinant human IL-2. NKTR-358 promoted selective Treg activation and increased Treg suppressive function in mice. The durability and specificity of the response was greater following a single subcutaneous administration of NKTR-358 compared to five daily administrations of IL-2, and led to disease suppression in a mouse delayed-type hypersensitivity (DTH) model [125]. Further, biweekly dosing induced preferential and sustained Treg expansion in mice and non-human primates (NHP) resulting in ameliorated disease progression in a mouse model of systemic lupus erythematosus (SLE), and in a non-human primate cutaneous hypersensitivity model [112]. The single ascending dose study in healthy volunteers (NCT04133116) and the multiple ascending dose study with three biweekly subcutaneous doses of rezpegaldesleukin versus placebo in patients with SLE (NCT03556007) yielded promising results [126]. Dose-dependent, selective, and sustained increases in percentages and absolute numbers of total CD4pos Treg and CD25bright Treg were observed, with no significant changes in conventional CD4 and CD8 T cells, and low-level increases in NK cells. At the highest dose tested, a 12–17-fold increase in CD25bright Treg over baseline was sustained for 20–30 days. Most adverse events were grade 1–2 injection-site reactions. Immunogenicity was not observed. SLE disease score was not evaluated due to study limitations, however, data for the follow-up phase 1b randomised studies in psoriasis (NCT04119557) and atopic dermatitis (NCT04081350) have been recently presented [127]. Treatment of patients with psoriasis with rezpegaldesleukin resulted in increased Treg numbers, and improved disease score (PASI, psoriasis area and severity index) versus placebo, which was maintained up to week 19 post-treatment [128]. In atopic dermatitis, biweekly subcutaneous injections of rezpegaldesleukin increased total Treg and CD25bright Treg during treatment period (12 weeks), while a dose-dependent improvement was observed in disease-relevant scores (EASI, eczema area and severity index) versus placebo up to 36 weeks following end of treatment [129]. Together with a favourable safety profile these data further support clinical development of rezpegaldesleukin in patients with atopic dermatitis [130]. Less encouraging data were reported for phase 2 ISLAND study (NCT04433585) that enroled adults with moderate-to-severe SLE. Although respegaldesleukin led to dose-dependent proliferation of Treg, the primary endpoint of the study—a four-point reduction in the SLE disease activity index (SLEDAI-2K)—was not met [131].
THOR-809/SAR444336 (Synthorx/Sanofi) is a site-specific PEGylated IL-2 variant with a PEG moiety attached to an unnatural amino acid at the IL-2Rβ interface designed to increase half-life and enhance selectivity for the trimeric IL-2R. The modified IL-2 has a reduced affinity to the IL-2Rβ chain so that the potency of trimeric IL-2R engagement relies on the IL-2Rα chain binding [132]. In mice and NHP, THOR-809 preferentially stimulated proliferation of peripheral Treg relative to effector T cells and NK cells. Expanded Treg had sustained pSTAT5 signalling and upregulated suppression markers FOXP3, CD25, ICOS and HELIOS. Furthermore, THOR-809 administration in mice led to dose-dependent expansion of highly suppressive Treg and control of skin inflammation in the DTH mouse model [133]. A phase 1 trial in healthy subjects (NCT05876767) is currently ongoing [134].
Another promising IL-2 variant with site-specific PEGylation, designated dual 31/51-20 K, similarly displayed substantially increased clearance half-life, preferentially stimulated Treg over effector T cells compared with unmodified IL-2 by selectively reducing the binding affinity for the β subunit of IL-2R, and significantly reduced disease activity and severity in mouse models of xenogeneic graft-versus-host disease (GvHD), SLE and collagen-induced arthritis. Moreover, a single subcutaneous injection of this PEGylated IL-2 did not induce anti-drug antibody formation, nor did it compromise the host defence against viral infection [113].
IL-2 muteins
The elucidation of the crystal structure of IL-2 bound to its trimeric receptor [11, 135, 136] facilitated the informed introduction of mutations into IL-2 with the aim to increase its affinity or direct its binding. Such targeted mutagenesis allows to uncouple the pleiotropic effects of IL-2 on different immune cells and to target IL-2 activity toward specific cell populations that express either the dimeric IL-2R to boost tumour immunity or the trimeric IL-2R expressed by Treg to increase tolerance in autoimmunity and to transplanted grafts. IL-2 variants with increased binding to CD25 and/or decreased binding to CD122 and/or CD132 preferentially activate and expand Treg. These cytokines are further fused to either the fragment crystalisable (Fc) domain of immunoglobulin (IgG) or the full IgG, which results in significantly extended half-life due to increased hydrodynamic radius and hence decreased renal clearance but also due to the recycling of the protein via the neonatal Fc receptor. In the following paragraphs, we discuss a selection of promising IL-2 mutein molecules, each designed for Treg selectivity and application in autoimmune or inflammatory disease, that are currently in clinical development.
AMG592 or efavaleukin alpha (Amgen) is an IL-2 mutein with V91K/C125A mutations that confers high CD25-binding affinity, and that is expressed as a fusion to the C-terminus of an Fc homodimer [137]. In a first-in-human study, efavaleukin alpha single subcutaneous administration resulted in dose-dependent Treg expansion, which was highly selective relative to conventional T cells (Tconv) and NK cells. Treg-to-Tconv ratio peaked at day 8 (4-fold vs baseline) and remained elevated up to day 29, while no increases in serum proinflammatory cytokines IL-6, TNFα or IFNγ were detected. The expanded Treg displayed increased CD25 and FOXP3 levels and were enriched for CD31pos recent thymic emigrants. Treatment was well tolerated [138] and several early-phase studies were initiated to further evaluate safety and efficacy in subjects with rheumatoid arthritis (RA) (NCT03410056), steroid-refractory chronic GvHD (NCT03422627), and SLE (NCT03451422). Amgen ended the trials in RA due to insufficient therapeutic benefit for the use of efavaleukin alpha plus standard of care therapy in the assessed study population (NCT03410056); and chronic GvHD [139]. Data from a multiple ascending dose phase 1b study in patients with SLE demonstrated that efavaleukin alpha was well tolerated and induced a robust and prolonged dose-dependent Treg expansion, with minimal changes in CD4 and CD8 Tconv, NK cells or serum levels of pro-inflammatory cytokines [140]. The biweekly administration resulted in a 50-fold increase in CD25bright Treg above baseline, and the Treg numbers remained above baseline for an average of 42 days after the last dose. Despite these promising results, a phase 2b study of efavaleukin alpha in patients with SLE (NCT04680637) has been discontinued as it met pre-defined criteria for futility, i.e., it was unlikely to achieve its objectives [141]. However, a phase 2 study in ulcerative colitis (NCT04987307) is still ongoing.
RG7835 (Roche) is a bivalent conjugate of human IL-2 mutein (T3A, N88D, C125A) and a human IgG1 with abolished binding to Fcγ receptors. Due to its reduced affinity to IL-2Rβγ, IgG-(IL-2N88D)2 has a 6–9-fold reduced ability to stimulate Treg in human whole blood pSTAT5 activation assays compared to a wild-type IL-2 dimer but had no effect on other cell types except some activity on CD56bright NK cells. Treatment of cynomolgus monkeys and humanised NSG mice (engrafted with human foetal liver CD34pos cells) with a single dose of IgG-(IL-2N88D)2 induced sustained 10-14-fold expansion of CD4pos and CD8pos CD25pos FOXP3pos Treg with no effect on other cell types. The in vivo activated and expanded cynomolgus and human Treg had demethylated epigenetic signatures for FOXP3 and CTLA4 characteristic of functionally suppressive cells. However, neither mouse disease models nor multiple-dose studies in NHP could be performed due to the immunogenicity of the molecule in both species [115]. Phase 1b study initiated to assess safety, efficacy, pharmacokinetics, and pharmacodynamics of RG7835 in patients with ulcerative colitis (NCT03943550) was terminated after 8 weeks based on the lack of robust clinical improvement in the underlying condition, according to ClinicalTrials.gov. Following the failure in ulcerative colitis, a phase 2 clinical trial designed to evaluate the effect of RG7835 on time to relapse following forced corticosteroid tapering in patients with autoimmune hepatitis (NCT04790916) was also terminated.
Using a structure-guided approach, several mutations in IL-2 were introduced that significantly decreased CD122 binding affinity in addition to other mutations that increased CD25 binding affinity (L118I, N88D, V69A, Q74P, C125S) [142]. The resulting Fc-fusion molecules, PT101/MK-6194 (Pandion/Merck), selectively activated and expanded Treg in preclinical studies in humanised NSG mice and NHP without significant effects on other immune cell types, and without eliciting proinflammatory cytokine production [143]. These expanded Treg had increased expression of FOXP3 and CD25, suggesting enhanced function and stability. In a phase 1a single ascending dose clinical trial in healthy volunteers, PT101 was safe and well-tolerated, and a dose-dependent expansion of CD25bright Treg cells was observed with a mean maximum increase of 72.5-fold for CD25bright Treg by day 8-10 (and an overall 3.6-fold increase in total Treg) [144]. Tconv and NK cells were not increased while increases in eosinophil counts were transient. A phase 1 clinical trial in ulcerative colitis (NCT04924114) was initiated by Merck & Co. in 2021 to further evaluate PT101/MK-6194.
A similar molecule, an IL-2 mutein (T3A, N88R, C125S) fused to a human IgG Fc domain, DEL106/CC-92252 (Delinia/Celgene/BMS) also preferentially binds to IL-2Rα. A single intravenous dose of the compound in cynomolgus monkeys resulted in dose-dependent and selective Treg expansion and activation, which was better compared to IL-2 [145]. An increase in total circulating Treg cells was 15-fold on day 5, while no change in the number of circulating Tconv or CD8 cells was detected. The compound also stimulated expression of suppression and proliferation markers CD25, FOXP3 and Ki67 on Treg. IL-2 induced selective STAT5 phosphorylation of Treg over a narrow dose range, also activating Tconv, CD8 T, NK and B cells; in contrast, DEL106 demonstrated over 1000-fold-greater selectivity for Treg over other immune cells. In addition, subcutaneous administration showed that DEL106 exhibited a lower serum clearance and had a longer circulating half-life than IL-2. A phase 1 first-in-human study with this molecule was conducted in three parts: as a single ascending dose or multiple ascending dose study in healthy volunteers and a multiple ascending dose study in psoriasis patients (NCT03971825). CC-92252 was found safe and well-tolerated across studies with adverse effects of mild to moderate intensity. The treatment resulted in a selective but modest (maximum 2-fold) Treg expansion in circulation of healthy participants and in skin lesions of participants with psoriasis. However, as for RG7835 (Roche), no apparent trend of clinical improvement compared to placebo was observed in patients, indicating that the achieved Treg expansion may be insufficient for robust efficacy in active disease. Mechanistic studies revealed that although highly selective, CC-92252 is a weak partial agonist with only a subset of Treg responding to this IL-2 mutein [146]. Given limited evidence for clinical efficacy, the CC-92252 programme has been discontinued, although BMS is pursuing alternative approaches to Treg selectivity with IL-2 constructs (see below, with IL-2/CD25 fusion).
The therapeutic molecule CUG252 (Cugene/Abbvie) is an IL-2 mutein (L19H, C125I, Q126E) Fc-fusion protein designed for biased binding activity to IL-2Rα but attenuated binding to the IL-2Rβγ complex [147]. In mice and cynomolgus monkeys, administration of CUG252 resulted in dose-dependent increases in Treg expansion by 10- to 30-fold, with largely abolished activities in effector T cells and NK cells [148]. Treg had enhanced expression of functional and inhibitory markers (CD25, FOXP3, PD-1, CTLA-4, TIM3 and ICOS) and increased suppressive capacity in DTH. In T cell-dependent antibody response models, CUG252 strongly inhibited antigen-driven inflammation, B cell maturation, and antibody production. The molecule is currently in phase 1 study, which aims to evaluate the safety and tolerability of single escalating subcutaneous doses of CUG252 in healthy adult subjects, and multiple escalating subcutaneous doses of CUG252 in patients with mild to moderate SLE (NCT05328557).
MDNA209 (Medicenna) is an IL-2 mutein (L18R, Q22E, Q126T, S130R) with increased affinity to the IL-2Rβ and greatly decreased affinity for IL-2Rγ, resulting in attenuated IL-2Rβγ heterodimerization and reduced signalling. The design of MDNA209 is based on the scaffold of IL-2 ‘superkine’ variants that had an increased affinity for the β chain of the IL-2 receptor [114]. Rather than triggering IL-2 signalling, however, MDNA209 acts as an antagonist, blocking the receptor and preventing it from transmitting the signal. When targeted to T cell subsets, this IL-2 variant could be clinically translated in the context of controlling T-cell mediated (auto)immune disorders where it is essential to prevent effector T cell activation and expansion resulting in effector cell-mediated tissue damage, such as during acute GvHD. The mutein and its Fc-fusion version have been characterised ex vivo and in vivo. MDNA209 prevented IL-2- and IL-15-induced signalling via STAT5 and blocked proliferation of CD8pos T cells and NK cells, while inhibiting helper T cell type (TH) 1, TH9 and Treg cells but promoting TH17 cell differentiation. Mice treated with an Fc-fusion version of MDNA209 for 10 days showed prolonged survival in a full MHC-mismatched acute GvHD model compared to control IgG [149].
Other IL-2 fusion proteins
An alternative approach to increase the selectivity of IL-2 for Treg is through fusion with CD25. The mouse IL-2/CD25 fusion protein forms a tight inactive dimer that slowly releases the active monomer to stimulate the high-affinity IL-2R [117]. The long-acting biologic expands Treg in vivo more potently than IL-2, but also increases their activation and migration into lymphoid tissues as well as non-lymphoid tissues as shown for the pancreas and its inhibition of anti-insulin autoantibodies. Moreover, the IL-2/CD25 fusion protein was effective in treating diabetes and inhibiting lupus nephritis in mouse models [118, 150]. The human version of the compound is a full agonist, which maintains high selectivity on Treg over other cell types in whole blood pSTAT5 assays [151]. The human IL-2/CD25 had a prolonged half-life and induced a dose-dependent selective increase in Treg in cynomolgus monkeys compared to IL-2 or IL-2 mutein Fc-fusion molecules [146]. The first-in-human study is still ongoing, but preliminary single-dose pharmacodynamics data confirm robust and prolonged Treg induction in humans with no expansion of inflammatory CD8 or Tconv cells [152, 153].
CUE-401 (Cue Biopharma) is a tolerogenic IL-2/TGFβ Fc-fusion protein designed to activate and induce FOXP3 expression in CD4 T cells (iTreg). In mouse CD4 T cells, it induces FOXP3 expression in vitro (iTreg). Also, in human CD4 T cells from healthy donors, inflammatory bowel disease and RA patients, it results in increased number of FOXP3-expressing cells, however, induction of FOXP3 expression versus preferential expansion of containing Treg has not been dissected. The in vitro induced/expanded iTreg suppress polyclonal T cell proliferation and express comparable phenotypic markers as iTreg induced with combination of TGFβ and IL-2 (CD25, CTLA-4, PD-1, GITR, CD38, CD73, GARP). A single dose of CUE-401 administered to TxA23 mice with ongoing autoimmune gastritis increased FOXP3pos CD4 T cells in blood and lymph nodes and inhibited autoreactive T cell proliferation in gastric lymph nodes [154, 155].
TNF signalling via TNFR2 enhances expansion, function and stability of Treg [156]. A dual-acting fusion protein, with IL-2 fused to a TNFR2-selective TNF mutein (IL2-EHD2-sc-mTNFR2) promoted strong activation and expansion of CD4 and CD8 Treg cells in vitro compared to either IL-2 or TNFR2 stimulation alone, with both components necessary for superior biological activity [157]. The combination of IL-2 and a TNFR2 agonist is therefore a promising approach for selective Treg expansion in vivo [158].
Alternative IL-2 delivery methods
The therapeutic IL-2 molecules described above are expressed in living cells and are administered as formulations of recombinant protein. Novel technologies are instead based on the in situ expression of encoded proteins and include lipid nanoparticle (LNP)-mediated mRNA delivery, DNA vaccines and gene transfer using viral vectors. Nucleic acid therapeutics are considered safe, well-tolerated and efficacious with major advantages over protein-based therapeutics including simple and cost-effective production processes and opportunities to improve the drug characteristics [159, 160]. However, several challenges remain. The greatest challenge for mRNA nanomedicine is immunogenicity both against the LNP itself as well as against the mRNA-encoded proteins. With gene therapies, which are designed for permanent integration of the viral vector into genome, the uncertainty about delayed adverse events remains the greatest risk factor [160].
mRNA-6231 (Moderna) is a lipid nanoparticle (LNP)-encapsulated mRNA encoding a Treg -specific IL-2 mutein fused to human serum albumin (HSA). Two triple-mutant molecules (V69A/Q74P/N88D or V69A/Q74P/V91K) showed the highest difference in pSTAT5 signal between Treg and other cell subsets in human PBMC in vitro and selectively activated and expanded Treg in mice. LNP-formulated mRNA encoding HSA fused to wild-type IL-2 elevated the percentage of Treg in cynomolgus monkeys and was also effective in preclinical models of murine acute GvHD and collagen-induced rat arthritis [161, 162]. The first-in-human trial of mRNA-6231 in healthy adult participants (NCT04916431) was stopped after early clinical data became available [163].
A tolerogenic immunotherapy NNC0361-0041 (Novo Nordisk) involves a DNA plasmid which encodes for pre-proinsulin (PPI), TGFβ1, IL-10, and IL-2 [164]. The combination of antigen (PPI) with the three immune response modifiers (TGFβ1, IL-10, and IL-2) is intended to induce antigen-specific Treg accumulating in the pancreas, and to preserve beta cell function in type 1 diabetes (T1D). The safety and efficacy of treatment was demonstrated in NOD mice as assessed by delayed disease progression, necessity of both antigen and IL-2 for increased efficacy and robustness, and tolerability of chronic dosing [165]. However, no pharmacodynamic-related measurements such as Treg activity or cytokine expression were performed. The phase 1 trial in adults with recent-onset T1D is currently recruiting and will evaluate safety, tolerability, and pharmacokinetics of the therapy (NCT04279613).
Adeno-associated viral (AAV) vector-mediated gene transfer for systemic and continuous IL-2 production has been investigated using a single administration of an AAV-IL-2 vector in mice. The treatment enabled sustained stimulation and expansion of Treg without inducing effector T cell activation while preventing diabetes in NOD mice [109] or alleviating Alzheimer’s disease in APP/PS1ΔE9 mice with established pathology [166]. Moreover, the long-term IL-2 expression did not impair immune responses to infections, vaccination or cancer [109]. However, this approach does not allow to interrupt or stop the treatment in case of adverse events. A tissue-specific gene-delivery approach of IL-2 for the treatment of neuroinflammatory pathologies has been developed by Yshii et al. Treg constitute a small resident cell population in the brain, where low levels of IL-2 are thought to limit the natural anti-inflammatory processes. Tissue-specific IL-2 expression targeted to astrocytes via an AAV vector induced a local and transient expansion of the Treg cell population in the mouse brain, which led to beneficial effects in mouse models of traumatic brain injury, multiple sclerosis and stroke [167]. Both the tissue-specific IL-2 delivery system as well as the ability to control the encoded protein expression are promising approaches to improve the clinical translation of gene therapy.
IL-2/anti-IL-2 antibody complexes
Coupling of IL-2 to specific monoclonal antibodies can modify the interaction of IL-2 with its receptor leading to a targeted and longer-lasting in vivo biological activity compared with soluble IL-2 [93, 119]. Depending on the antibody-binding site on IL-2, the IL-2:antibody complex (IL-2c) can preferentially activate either the cells expressing high levels of CD122, such as memory CD8 T cells and NK cells, or CD25-expressing cells such as Treg. A prominent and well-studied example is the complex of mouse IL-2 bound to the anti-mouse IL-2 antibody JES6-1. JES6-1 binding sterically obstructs mouse IL-2 interaction with the IL-2Rβγ heterodimer to block the signalling on IL-2Rαlow effector cells. Thereupon, IL-2 is preferentially delivered to the trimeric receptor via a unique allosteric exchange mechanism, where the IL-2Rα subunit displaces the JES6-1 antibody allowing IL-2 to initiate signalling via the IL-2βγ subunits. This complex prolonged the in vivo half-life of IL-2 and led to selective expansion of murine Treg in a murine dextran sodium sulphate colitis model [119, 168]. The efficacy of this approach has been further demonstrated in various experimental models of autoimmune diseases or other inflammatory settings as exemplified below:
-
enhanced allograft survival in a murine model of islet transplantation and experimental autoimmune encephalomyelitis (EAE) prevention in combination with rapamycin [169],
-
markedly attenuated acute GvHD while preserving graft-versus-leukaemia activity after allo-hematopoietic cell transplantation at higher efficacy than tacrolimus treatment [170],
-
survival of fully MHC-mismatched skin allograft: IL-2c failed to augment the survival of skin allografts as monotherapy but initial treatment with anti-IL-6 monoclonal antibody followed by supplementation with rapamycin led to graft survival and elevated intra-graft Treg levels [171],
-
attenuation of CNS inflammation and neurological deficits in EAE [172],
-
suppression of experimental myasthenia gravis [173],
-
inhibition of collagen-induced arthritis [174],
-
attenuation of atherosclerosis in apolipoprotein E-deficient mice [175],
-
decreased myofiber injury in murine muscular dystrophy model [176].
These results motivate the investigation of IL-2-based therapies in inflammatory diseases or conditions that are not caused by autoimmune or alloimmune reactions.
A fully human anti-IL-2 antibody F5111.2 that resembles the exchange mechanism observed for the anti-mouse IL-2 antibody JES6-1, was developed by Trotta et al. [120]. Comparison of the crystal structure of IL-2c with the IL-2/IL-2R quaternary structure revealed that F5111.2 sterically obstructs the binding of human IL-2 to IL-2Rβ and allosterically reduces the affinity of the cytokine to IL-2Rα. Administration of F5111.2-hIL-2 complex results in the preferential STAT5 phosphorylation of Treg in vitro and selective expansion of Treg in vivo. When complexed with human IL-2, F5111.2 induced remission of T1D in the NOD mouse model, reduced disease severity in a model of EAE and protected mice against xenogeneic GvHD [120].
Another anti-human IL-2Rα-biased IL-2 antibody, UFKA-20, uses a similar mechanism to selectively target Treg [121]. The IL-2 bound to UFKA-20 fails to induce cell activation via the dimeric IL-2R unless the cells also express CD25. Once the IL-2/UFKA-20 complex is bound to CD25, the antibody dissociates from IL-2 and allows the formation of high affinity quaternary IL-2/IL-2R structure that leads to intracellular signalling. Consequently, the IL-2/UFKA-20 complexes efficiently and preferentially stimulated CD4pos Treg in freshly isolated human T cells ex vivo and in mice and rhesus macaques in vivo [121].
The clinical translation of the IL-2/antibody complex approach is complicated by the instability of the cytokine/antibody complex and the need to optimise dosing ratios, as dissociation would lead to off-target effects and rapid clearance. Genetically fusing IL-2 and the antibody should circumvent these drawbacks [168, 177]. A single-chain hIL-2/F5111 antibody-fusion protein has been engineered that demonstrated selective Treg bias and showed efficacy in mouse models of colitis and checkpoint inhibitor-induced diabetes mellitus [178].
Conclusions
IL-2 is central in the biology of Treg during development, functional maturation, lineage stability, peripheral homoeostasis, and function. The consequence of the dependency of Treg on IL-2 is the development of autoimmunity in the absence of IL-2 signalling. Treg compensate for the dependency with an exceptional IL-2 capture sensitivity that outcompetes that of other cell types. The necessity for IL-2 signalling and the high expression of the high-affinity trimeric IL-2R make the IL-2 signalling pathway a prime-candidate for Treg-targeting therapeutic approaches in autoimmune and inflammatory diseases as well as in the prevention of transplant rejection.
Despite its high efficacy, given the limitations of low-dose IL-2, numerous approaches have been developed to increase the targeting specificity of IL-2 and hence to avoid binding of the new IL-2-based biologicals to non-Treg cells. Informed by structural and empirical studies, modified IL-2-based molecules are being tested in pre-clinical studies as well as in clinical trials. Yet, informed design may not entirely predict therapeutic success as illustrated by insufficient efficacy and incomplete translation of pre-clinical data in clinical trials for some candidates. However, despite the requirement for thorough clinical assessment of therapeutic benefit in each disease, recent successes in clinical trials for several modified IL-2-based molecules in various autoimmune contexts are representative of the promising therapeutic perspective of IL-2-based immunotherapeutics.
The further possibility to target (modified) IL-2 to Treg subsets of particular prevalence in a disease context by the use of additional moieties may expand the drug development toolbox in the future. Similarly, combinatorial therapy, such as with rapamycin, may prove beneficial but will require assessment in clinical trials. Finally, and undoubtedly, an increasing understanding of structural modifications and their functional consequences will further the design of IL-2-based molecules to increase targeting efficiency as well as to minimise risk for off-target activity and hence maximise safety and efficacy.
References
Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science.1976;193:1007–8. https://doi.org/10.1126/SCIENCE.181845.
Robb RJ, Kutny RM, Chowdhry V. Purification and partial sequence analysis of human T-cell growth factor. Proc Natl Acad Sci USA. 1983;80:5990–4. https://doi.org/10.1073/PNAS.80.19.5990.
Taniguchi T, Matsui H, Fujita T, Takaoka C, Kashima N, Yoshimoto R, et al. Structure and expression of a cloned cDNA for human interleukin-2. Nature. 1983;302:305–10. https://doi.org/10.1038/302305A0.
Nakamura Y, Russell SM, Mess SA, Friedmann M, Erdos M, Francois C, et al. Heterodimerization of the IL-2 receptor β- and γ-chain cytoplasmic domains is required for signalling. Nature. 1994;369:330–3. https://doi.org/10.1038/369330A0.
Russell SM, Keegan AD, Harada N, Nakamura Y, Noguchi M, Leland P, et al. Interleukin-2 receptor γ chain: a functional component of the interleukin-4 receptor. Science.1993;262:1880–3. https://doi.org/10.1126/SCIENCE.8266078.
Noguchi M, Nakamura Y, Russell SM, Ziegler SF, Tsang M, Cao X, et al. Interleukin-2 receptor γ chain: a functional component of the interleukin-7 receptor. Science. 1993;262:1877–80. https://doi.org/10.1126/SCIENCE.8266077.
Kondo M, Takeshita T, Ishii N, Nakamura M, Watanabe S, Arai K, et al. Sharing of the interleukin-2 (IL-2) receptor γ chain between receptors for IL-2 and IL-4. Science. 1993;262:1874–7. https://doi.org/10.1126/SCIENCE.8266076.
Kimura Y, Takeshita T, Kondo M, Ishii N, Nakamura M, Van Snick J, et al. Sharing of the IL-2 receptor γ chain with the functional IL-9 receptor complex. Int Immunol. 1995;7:115–20. https://doi.org/10.1093/INTIMM/7.1.115.
Giri JG, Ahdieh M, Eisenman J, Shanebeck K, Grabstein K, Kumaki S, et al. Utilization of the beta and gamma chains of the IL-2 receptor by the novel cytokine IL-15. EMBO J. 1994;13:2822–30. https://doi.org/10.1002/J.1460-2075.1994.TB06576.X.
Asao H, Okuyama C, Kumaki S, Ishii N, Tsuchiya S, Foster D, et al. Cutting edge: the common γ-chain is an indispensable subunit of the IL-21 receptor complex1. J Immunol. 2001;167:1–5. https://doi.org/10.4049/JIMMUNOL.167.1.1.
Stauber DJ, Debler EW, Horton PA, Smith KA, Wilson IA. Crystal structure of the IL-2 signaling complex: paradigm for a heterotrimeric cytokine receptor. Proc Natl Acad Sci USA. 2006;103:2788–93. https://doi.org/10.1073/PNAS.0511161103.
Wang HM, Smith KA. The interleukin 2 receptor. Functional consequences of its bimolecular structure. J Exp Med. 1987;166:1055–69. https://doi.org/10.1084/JEM.166.4.1055.
Lowenthal JW, Greene WC. Contrasting interleukin 2 binding properties of the alpha (p55) and beta (p70) protein subunits of the human high-affinity interleukin 2 receptor. J Exp Med. 1987;166:1156–61. https://doi.org/10.1084/JEM.166.4.1156.
Taniguchi T, Minami Y. The IL-2IL-2 receptor system: a current overview. Cell. 1993;73:5–8. https://doi.org/10.1016/0092-8674(93)90152-G.
Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. 2018;18:648–59. https://doi.org/10.1038/S41577-018-0046-Y.
Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Rα expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. 2010;32:91–103. https://doi.org/10.1016/J.IMMUNI.2009.11.010.
Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–79. https://doi.org/10.1146/ANNUREV.IMMUNOL.26.021607.090357.
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. https://doi.org/10.4049/jimmunol.155.3.1151.
Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–51. https://doi.org/10.1038/NI1263.
Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: biology, design and application. Trends Immunol. 2015;36:763–77. https://doi.org/10.1016/J.IT.2015.10.003.
Bessoles S, Fouret F, Dudal S, Besra GS, Sanchez F, Lafont V. IL-2 triggers specific signaling pathways in human NKT cells leading to the production of pro- and anti-inflammatory cytokines. J Leukoc Biol. 2008;84:224–33. https://doi.org/10.1189/JLB.1007669.
Caldirola MS, Rodríguez Broggi MG, Gaillard MI, Bezrodnik L, Zwirner NW. Primary immunodeficiencies unravel the role of IL-2/CD25/STAT5b in human natural killer cell maturation. Front Immunol. 2018;9:1429. https://doi.org/10.3389/FIMMU.2018.01429.
Roediger B, Kyle R, Tay SS, Mitchell AJ, Bolton HA, Guy TV, et al. IL-2 is a critical regulator of group 2 innate lymphoid cell function during pulmonary inflammation. J Allergy Clin Immunol. 2015;136:1653–1663.e7. https://doi.org/10.1016/J.JACI.2015.03.043.
Hernandez R, Põder J, LaPorte KM, Malek TR. Engineering IL-2 for immunotherapy of autoimmunity and cancer. Nat Rev Immunol. 2022;22:614–28. https://doi.org/10.1038/S41577-022-00680-W.
Krieg C, Létourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci USA. 2010;107:11906–11. https://doi.org/10.1073/PNAS.1002569107/.
Kim DW, Zloza A, Broucek J, Schenkel JM, Ruby C, Samaha G, et al. Interleukin-2 alters distribution of CD144 (VE-cadherin) in endothelial cells. J Transl Med. 2014;12:113. https://doi.org/10.1186/1479-5876-12-113.
Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38:13–25. https://doi.org/10.1016/J.IMMUNI.2013.01.004.
Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol. 2018;36:411–33. https://doi.org/10.1146/ANNUREV-IMMUNOL-042617-053352.
Whyte CE, Singh K, Burton OT, Aloulou M, Kouser L, Veiga RV, et al. Context-dependent effects of IL-2 rewire immunity into distinct cellular circuits. J Exp Med. 2022;219:e20212391. https://doi.org/10.1084/JEM.20212391.
Powell JD, Ragheb JA, Kitagawa-Sakakida S, Schwartz RH. Molecular regulation of interleukin-2 expression by CD28 co-stimulation and anergy. Immunol Rev. 1998;165:287–300. https://doi.org/10.1111/J.1600-065X.1998.TB01246.X.
June CH, Ledbetter JA, Gillespie MM, Lindsten T, Thompson CB. T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol Cell Biol. 1987;7:4472–81. https://doi.org/10.1128/MCB.7.12.4472-4481.1987.
Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–9. https://doi.org/10.1038/NATURE05673.
Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 2006;126:375–87. https://doi.org/10.1016/J.CELL.2006.05.042.
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. https://doi.org/10.7717/peerj.15261.
Bendfeldt H, Benary M, Scheel T, Steinbrink K, Radbruch A, Baumgrass R. IL-2 Expression in activated human memory FOXP3+ cells critically depends on the cellular levels of FOXP3 as well as of four transcription factors of T cell activation. Front Immunol. 2012;3. https://doi.org/10.3389/FIMMU.2012.00264.
Hémar A, Subtil A, Lieb M, Morelon E, Hellio R, Dautry-Varsat A. Endocytosis of interleukin 2 receptors in human T lymphocytes: distinct intracellular localization and fate of the receptor alpha, beta, and gamma chains. J Cell Biol. 1995;129:55–64. https://doi.org/10.1083/JCB.129.1.55.
Yu A, Malek TR. The proteasome regulates receptor-mediated endocytosis of interleukin-2. J Biol Chem. 2001;276:381–5. https://doi.org/10.1074/JBC.M007991200.
Gaffen SL. Signaling domains of the interleukin 2 receptor. Cytokine. 2001;14:63–77. https://doi.org/10.1006/CYTO.2001.0862.
Hoxhaj G, Manning BD. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20:74–88. https://doi.org/10.1038/S41568-019-0216-7.
Bensinger SJ, Walsh PT, Zhang J, Carroll M, Parsons R, Rathmell JC, et al. Distinct IL-2 receptor signaling pattern in CD4+CD25+ regulatory T cells. J Immunol. 2004;172:5287–96. https://doi.org/10.4049/JIMMUNOL.172.9.5287.
Ross SH, Rollings C, Anderson KE, Hawkins PT, Stephens LR, Cantrell DA. Phosphoproteomic analyses of interleukin 2 signaling reveal integrated JAK kinase-dependent and -independent networks in CD8+ T cells. Immunity. 2016;45:685–700. https://doi.org/10.1016/J.IMMUNI.2016.07.022.
Walsh PT, Buckler JL, Zhang J, Gelman AE, Dalton NM, Taylor DK, et al. PTEN inhibits IL-2 receptor–mediated expansion of CD4+ CD25+ Tregs. J Clin Investig. 2006;116:2521–31. https://doi.org/10.1172/JCI28057.
Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, et al. Stability and function of regulatory T cells is maintained by a neuropilin-1–semaphorin-4a axis. Nature. 2013;501:252–6. https://doi.org/10.1038/NATURE12428.
Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. 2015;16:188–96. https://doi.org/10.1038/NI.3077.
Sporri B, Kovanen PE, Sasaki A, Yoshimura A, Leonard WJ. JAB/SOCS1/SSI-1 is an interleukin-2–induced inhibitor of IL-2 signaling. Blood. 2001;97:221–6. https://doi.org/10.1182/BLOOD.V97.1.221.
McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–23. https://doi.org/10.1016/S1074-7613(02)00280-7.
Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, et al. Foxp3-dependent MicroRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. https://doi.org/10.1016/J.IMMUNI.2008.11.010.
Takahashi R, Nakatsukasa H, Shiozawa S, Yoshimura A. SOCS1 is a key molecule that prevents regulatory T cell plasticity under inflammatory conditions. J Immunol. 2017;199:149–58. https://doi.org/10.4049/JIMMUNOL.1600441.
Fujimoto M, Naka T. Regulation of cytokine signaling by SOCS family molecules. Trends Immunol. 2003;24:659–66. https://doi.org/10.1016/J.IT.2003.10.008.
Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol. 2015;15:283–94. https://doi.org/10.1038/NRI3823.
Kolios AGA, Tsokos GC, Klatzmann D. Interleukin-2 and regulatory T cells in rheumatic diseases. Nat Rev Rheumatol. 2021;17:749–66. https://doi.org/10.1038/S41584-021-00707-X.
Ding Y, Yu A, Tsokos GC, Malek TR. CD25 and protein phosphatase 2A cooperate to enhance IL-2R signaling in human regulatory T cells. J Immunol. 2019;203:93–104. https://doi.org/10.4049/JIMMUNOL.1801570.
Sharabi A, Li H, Kasper IR, Pan W, Meidan E, Tsokos MG, et al. PP2A enables IL-2 signaling by preserving IL-2Rβ chain expression during Treg development. JCI Insight. 2019;5. https://doi.org/10.1172/JCI.INSIGHT.126294.
Sharabi A, Kasper IR, Tsokos GC. The serine/threonine protein phosphatase 2A controls autoimmunity. Clin Immunol. 2018;186:38–42. https://doi.org/10.1016/J.CLIM.2017.07.012.
Yu A, Snowhite I, Vendrame F, Rosenzwajg M, Klatzmann D, Pugliese A, et al. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes. 2015;64:2172–83. https://doi.org/10.2337/DB14-1322.
Hayes ET, Hagan CE, Khoryati L, Gavin MA, Campbell DJ. Regulatory T cells maintain selective access to IL-2 and immune homeostasis despite substantially reduced CD25 function. J Immunol. 2020;205:2667–78. https://doi.org/10.4049/JIMMUNOL.1901520.
Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313:1485–92. https://doi.org/10.1056/NEJM198512053132327.
Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192:5451–8. https://doi.org/10.4049/JIMMUNOL.1490019.
Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907–13. https://doi.org/10.1001/JAMA.1994.03510360033032.
Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–61. https://doi.org/10.1016/0092-8674(93)80067-O.
Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rβ-deficient mice: implications for the nonredundant function of IL-2. Immunity. 2002;17:167–78. https://doi.org/10.1016/S1074-7613(02)00367-9.
Godfrey VL, Wilkinson JE, Russell LB. X-linked lymphoreticular disease in the scurfy (sf) mutant mouse. Am J Pathol. 1991;138:1379–87.
Moraes-Vasconcelos D, Costa-Carvalho BT, Torgerson TR, Ochs HD. Primary immune deficiency disorders presenting as autoimmune diseases: IPEX and APECED. J Clin Immunol. 2008;28:11–9. https://doi.org/10.1007/S10875-008-9176-5.
Haufe S, Haug M, Schepp C, Kuemmerle-Deschner J, Hansmann S, Rieber N, et al. Impaired suppression of synovial fluid CD4+CD25− T cells from patients with juvenile idiopathic arthritis by CD4+CD25+ Treg cells. Arthritis Rheum. 2011;63:3153–62. https://doi.org/10.1002/ART.30503.
Guo H, Zheng M, Zhang K, Yang F, Zhang X, Han Q, et al. Functional defects in CD4+ CD25high FoxP3+ regulatory cells in ankylosing spondylitis. Sci Rep. 2016;6:37559. https://doi.org/10.1038/SREP37559.
Slobodin G, Rimar D. Regulatory T cells in systemic sclerosis: a comprehensive review. Clin Rev Allergy Immunol. 2017;52:194–201. https://doi.org/10.1007/S12016-016-8563-6.
Frantz C, Auffray C, Avouac J, Allanore Y. Regulatory T cells in systemic sclerosis. Front Immunol. 2018;9. https://doi.org/10.3389/FIMMU.2018.02356.
Grunewald J, Grutters JC, Arkema EV, Saketkoo LA, Moller DR, Müller-Quernheim J. Sarcoidosis. Nat Rev Dis Primers. 2019;5. https://doi.org/10.1038/S41572-019-0096-X.
Wang J, Zhang SX, Hao YF, Qiu MT, Luo J, Li YY, et al. The numbers of peripheral regulatory T cells are reduced in patients with psoriatic arthritis and are restored by low-dose interleukin-2. Ther Adv Chronic Dis. 2020;11. https://doi.org/10.1177/2040622320916014.
Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27:786–800. https://doi.org/10.1016/J.IMMUNI.2007.09.010.
Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol. 2018;19:291–301. https://doi.org/10.1038/S41590-018-0051-0.
Lio CWJ, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity.2008;28:100–11. https://doi.org/10.1016/J.IMMUNI.2007.11.021.
Burchill MA, Yang J, Vang KB, Moon JJ, Chu HH, Lio CWJ, et al. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity. 2008;28:112–21. https://doi.org/10.1016/J.IMMUNI.2007.11.022.
Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA. IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol. 2008;181:3285–90. https://doi.org/10.4049/JIMMUNOL.181.5.3285.
Herppich S, Toker A, Pietzsch B, Kitagawa Y, Ohkura N, Miyao T, et al. Dynamic imprinting of the treg cell-specific epigenetic signature in developing thymic regulatory T cells. Front Immunol. 2019;10:2382. https://doi.org/10.3389/FIMMU.2019.02382.
Sadlack B, Löhler J, Schorle H, Klebb G, Haber H, Sickel E, et al. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol. 1995;25:3053–9. https://doi.org/10.1002/EJI.1830251111.
Schorle H, Holtschke T, Hünig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 1991;352:621–4. https://doi.org/10.1038/352621A0.
Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–30. https://doi.org/10.1016/1074-7613(95)90180-9.
Suzuki H, Kündig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. 1995;268:1472–6. https://doi.org/10.1126/SCIENCE.7770771.
Abbas AK, Trotta E, R Simeonov D, Marson A, Bluestone JA. Revisiting IL-2: Biology and therapeutic prospects. Sci Immunol. 2018;3. https://doi.org/10.1126/SCIIMMUNOL.AAT1482.
Sharfe N, Dadi HK, Shahar M, Roifman CM. Human immune disorder arising from mutation of the α chain of the interleukin-2 receptor. Proc Natl Acad Sci USA. 1997;94:3168–71. https://doi.org/10.1073/PNAS.94.7.3168.
Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. 2015;16:178–87. https://doi.org/10.1038/NI.3076.
Essig K, Hu D, Guimaraes JC, Alterauge D, Edelmann S, Raj T, et al. Roquin suppresses the PI3K-mTOR signaling pathway to inhibit T helper cell differentiation and conversion of Treg to Tfr cells. Immunity. 2017;47:1067–1082.e12. https://doi.org/10.1016/J.IMMUNI.2017.11.008.
Yi Z, Lin WW, Stunz LL, Bishop GA. The adaptor TRAF3 restrains the lineage determination of thymic regulatory T cells by modulating signaling via the receptor for IL-2. Nat Immunol. 2014;15:866–74. https://doi.org/10.1038/NI.2944.
Yao Z, Kanno Y, Kerenyi M, Stephens G, Durant L, Watford WT, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–75. https://doi.org/10.1182/BLOOD-2006-11-055756.
Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349:1139–47. https://doi.org/10.1056/NEJMOA022926.
Wolf M, Schimpl A, Hünig T. Control of T cell hyperactivation in IL-2-deficient mice by CD4+CD25– and CD4+CD25+ T cells: evidence for two distinct regulatory mechanisms. Eur J Immunol. 2001;31:1637–45. https://doi.org/10.1002/15214141.
Almeida ARM, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T Cells: IL-2Rα and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. 2002;169:4850–60. https://doi.org/10.4049/JIMMUNOL.169.9.4850.
Furtado GC, de Lafaille MAC, Kutchukhidze N, Lafaille JJ. Interleukin 2 signaling is required for CD4+ regulatory T cell function. J Exp Med. 2002;196:851–7. https://doi.org/10.1084/JEM.20020190.
Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O’Gorman WE, et al. Cutting edge: mechanisms of IL-2–dependent maintenance of functional regulatory T cells. J Immunol. 2010;185:6426–30. https://doi.org/10.4049/JIMMUNOL.0903940.
Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3+ regulatory T cells. Nat Immunol. 2013;14:959–65. https://doi.org/10.1038/ni.2649.
Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3 + CD25+ CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201:723–35. https://doi.org/10.1084/JEM.20041982.
Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006;311:1924–7. https://doi.org/10.1126/SCIENCE.1122927.
Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell. 2014;158:749–63. https://doi.org/10.1016/J.CELL.2014.07.031.
Bayer AL, Yu A, Malek TR. Function of the IL-2R for thymic and peripheral CD4+CD25+ Foxp3+ T regulatory cells. J Immunol. 2007;178:4062–71. https://doi.org/10.4049/JIMMUNOL.178.7.4062.
D’Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol. 2005;6:1152–9. https://doi.org/10.1038/NI1264.
Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. 2016;17:1322–33. https://doi.org/10.1038/NI.3540.
Passerini L, Allan SE, Battaglia M, Di Nunzio S, Alstad AN, Levings MK, et al. STAT5-signaling cytokines regulate the expression of FOXP3 in CD4+CD25+ regulatory T cells and CD4+CD25− effector T cells. Int Immunol. 2008;20:421–31. https://doi.org/10.1093/INTIMM/DXN002.
Camperio C, Caristi S, Fanelli G, Soligo M, Porto PD, Piccolella E. Forkhead transcription factor FOXP3 upregulates CD25 expression through cooperation with RelA/NF-κB. PLoS One. 2012;7. https://doi.org/10.1371/JOURNAL.PONE.0048303.
Junius S, Mavrogiannis AV, Lemaitre P, Gerbaux M, Staels F, Malviya V, et al. Unstable regulatory T cells, enriched for naïve and Nrp1neg cells, are purged after fate challenge. Sci Immunol. 2021;6. https://doi.org/10.1126/SCIIMMUNOL.ABE4723.
Rosenblum MD, Way SS, Abbas AK. Regulatory T cell memory. Nat Rev Immunol. 2016;16:90–101. https://doi.org/10.1038/nri.2015.1.
Khantakova JN, Bulygin AS, Sennikov SV. The regulatory-T-cell memory phenotype: what we know. Cells 2022;11:1687. https://doi.org/10.3390/cells11101687. Jan
Dong S, Hiam-Galvez KJ, Mowery CT, Herold KC, Gitelman SE, Esensten JH, et al. The effect of low-dose IL-2 and Treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight. 2021;6:e147474. https://doi.org/10.1172/jci.insight.147474.
Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transnl Med. 2013;5:179ra43–179ra43. https://doi.org/10.1126/scitranslmed.3005265.
Rosenzwajg M, Churlaud G, Mallone R, Six A, Dérian N, Chaara W, et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun. 2015;58:48–58. https://doi.org/10.1016/j.jaut.2015.01.001.
Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+CD25hi Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107:2409–14. https://doi.org/10.1182/blood-2005-06-2399.
Dutcher JP, Schwartzentruber DJ, Kaufman HL, Agarwala SS, Tarhini AA, Lowder JN, et al. High dose interleukin-2 (Aldesleukin)—expert consensus on best management practices-2014. J Immunother Cancer. 2014;2:26. https://doi.org/10.1186/s40425-014-0026-0.
Lotze MT, Frana LW, Sharrow SO, Robb RJ, Rosenberg SA. In vivo administration of purified human interleukin 2. I. Half-life and immunologic effects of the Jurkat cell line-derived interleukin 2. J Immunol. 1985;134:157–66.
Churlaud G, Jimenez V, Ruberte J, Amadoudji Zin M, Fourcade G, Gottrand G, et al. Sustained stimulation and expansion of Tregs by IL2 control autoimmunity without impairing immune responses to infection, vaccination and cancer. Clin Immunol. 2014;151:114–26. https://doi.org/10.1016/J.CLIM.2014.02.003.
Rosenzwajg M, Salet R, Lorenzon R, Tchitchek N, Roux A, Bernard C, et al. Low-dose IL-2 in children with recently diagnosed type 1 diabetes: a phase I/II randomised, double-blind, placebo-controlled, dose-finding study. Diabetologia. 2020;63:1808–21. https://doi.org/10.1007/S00125-020-05200-W.
Graßhoff H, Comdühr S, Monne LR, Müller A, Lamprecht P, Riemekasten G, et al. Low-Dose IL-2 therapy in autoimmune and rheumatic diseases. Front Immunol. 2021;12. https://doi.org/10.3389/FIMMU.2021.648408.
Dixit N, Fanton C, Langowski JL, Kirksey Y, Kirk P, Chang T, et al. NKTR-358: A novel regulatory T-cell stimulator that selectively stimulates expansion and suppressive function of regulatory T cells for the treatment of autoimmune and inflammatory diseases. J Transl Autoimmun. 2021;4. https://doi.org/10.1016/J.JTAUTO.2021.100103.
Zhang B, Sun J, Wang Y, Ji D, Yuan Y, Li S, et al. Site-specific PEGylation of interleukin-2 enhances immunosuppression via the sustained activation of regulatory T cells. Nat Biomed Eng. 2021;11:1288–305. https://doi.org/10.1038/s41551-021-00797-8.
Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484:529–33. https://doi.org/10.1038/NATURE10975.
Peterson LB, Bell CJM, Howlett SK, Pekalski ML, Brady K, Hinton H, et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J Autoimmun. 2018;95:1–14. https://doi.org/10.1016/J.JAUT.2018.10.017.
Khoryati L, Pham MN, Sherve M, Kumari S, Cook K, Pearson J, et al. An IL-2 mutein engineered to promote expansion of regulatory T cells arrests ongoing autoimmunity in mice. Sci Immunol. 2020;5. https://doi.org/10.1126/SCIIMMUNOL.ABA5264.
Ward NC, Yu A, Moro A, Ban Y, Chen X, Hsiung S, et al. IL-2/CD25: a long-acting fusion protein that promotes immune tolerance by selectively targeting the IL-2 receptor on regulatory T cells. J Immunol. 2018;201:2579–92. https://doi.org/10.4049/JIMMUNOL.1800907.
Ward NC, Lui JB, Hernandez R, Yu L, Struthers M, Xie J, et al. Persistent IL-2 receptor signaling by IL-2/CD25 fusion protein controls diabetes in NOD mice by multiple mechanisms. Diabetes. 2020;69:2400–13. https://doi.org/10.2337/DB20-0186.
Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to interleukin-2 elicit selective T cell subset potentiation through distinct conformational mechanisms. Immunity. 2015;42:815–25. https://doi.org/10.1016/J.IMMUNI.2015.04.015.
Trotta E, Bessette PH, Silveria SL, Ely LK, Jude KM, Le DT, et al. A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat Med. 2018;24:1005–14. https://doi.org/10.1038/s41591-018-0070-2.
Karakus U, Sahin D, Mittl PRE, Mooij P, Koopman G, Boyman O. Receptor-gated IL-2 delivery by an anti-human IL-2 antibody activates regulatory T cells in three different species. Sci Transl Med. 2020;12. https://doi.org/10.1126/SCITRANSLMED.ABB9283.
Wu K, Ma J, Bai W, Cui X, Han T, Wang S, et al. Short-term intratracheal use of PEG-modified IL-2 and glucocorticoid persistently alleviates asthma in a mouse model. Sci Rep. 2016;6:1–12. https://doi.org/10.1038/srep31562.
Lipsky PE, Calabrese LH, Kavanaugh A, Sundy JS, Wright D, Wolfson M, et al. Pegloticase immunogenicity: the relationship between efficacy and antibody development in patients treated for refractory chronic gout. Arthritis Res Ther. 2014;16. https://doi.org/10.1186/AR4497.
Berkhout LC, Vogelzang EH, Hart MM, Loeff FC, Dijk L, Derksen NIL, et al. The effect of certolizumab drug concentration and anti-drug antibodies on TNF neutralisation. Clin Exp Rheumatol. 2020;38:306–13. https://doi.org/10.55563/CLINEXPRHEUMATOL/NLR4R8.
Langowski JL, Kirk P, Addepalli M, Chang T, Dixit V, Kim G, et al. NKTR-358: a selective, first-in-class IL-2 pathway agonist, which increases number and suppressive function of regulatory T cells for the treatment of immune in ammatory disorders [abstract]. Arthritis Rheumatol. 2017;69(suppl 10).
Fanton C, Furie R, Chindalore V, Levin R, Diab I, Dixit N, et al. Selective expansion of regulatory T cells by NKTR-358 in healthy volunteers and patients with systemic lupus erythematosus. J Transl Autoimmun. 2022;5. https://doi.org/10.1016/J.JTAUTO.2022.100152.
Nektar therapeutics presents data for Rezpegaldesleukin (LY3471851) in patients with atopic dermatitis and psoriasis from two separate clinical studies at 2022 European Academy of Dermatology (EADV) Congress. 2022. https://ir.nektar.com/news-releases/news-release-details/nektar-therapeutics-presents-data-rezpegaldesleukin-ly3471851.
Forman S, Schmitz C, Budelsky A, Benschop R, Jackson K, Zou H, et al. Efficacy and safety of a selective regulatory T-cell inducing IL-2 conjugate (LY3471851) in the treatment of psoriasis: a phase 1 randomised study. EADV Congress 2022.
Schleicher S, Schmitz C, Budelsky A, Benschop R, Jackson K, Zou H, et al. in the Treatment of atopic dermatitis: a phase 1 randomised study. EADV Congress 2022.
Nektar Announces Promising New and Corrected Rezpegaldesleukin Efficacy Data Which Were Previously Reported in 2022 and Incorrectly Calculated by Former Collaborator Eli Lilly & Company. 2023. https://ir.nektar.com/news-releases/news-release-details/nektar-announces-promising-new-and-corrected-rezpegaldesleukin.
Nektar Therapeutics Announces Phase 2 Topline Data for Rezpegaldesleukin in Patients with Systemic Lupus Erythematosus. 2023. https://ir.nektar.com/news-releases/news-release-details/nektar-therapeutics-announces-phase-2-topline-data.
Ptacin J, Caffaro C, Ma L, Joseph I, Chen D, Ismaili T, et al. THOR-809: An IL-2 engineered from an expanded genetic alphabet for the potential treatment of autoimmune disorders [abstract]. Arthritis Rheumatol. 2019;71 (suppl 10).
Immunology Investor Evenb. 2022. https://www.sanofi.com/en/investors/financial-results-and-events/investor-presentations/Immunology-Investor-Event-2022
Sanofi pipelinb. 2022. https://www.sanofi.com/en/science-and-innovation/research-and-development/rd-pipeline
Wang X, Rickert M, Garcia KC. Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gammac receptors. Science. 2005;310:1159–63. https://doi.org/10.1126/SCIENCE.1117893.
Rickert M, Wang X, Boulanger MJ, Goriatcheva N, Garcia KC. The structure of interleukin-2 complexed with its alpha receptor. Science. 2005;308:1477–80. https://doi.org/10.1126/SCIENCE.1109745.
Gavin MA, Kannan G, Li L, Pearson JT, Karow M. Interleukin-2 muteins for the expansion of T-regulatory cells. EP 2970423 B1, 2019.
Tchao N, Gorski KS, Yuraszeck T, Sohn SJ, Ishida K, Wong H, et al. PS7:135 Amg 592 is an investigational il-2 mutein that induces highly selective expansion of regulatory t cells [poster]. Lupus Sci Med. 2018;5. https://doi.org/10.1136/LUPUS-2018-ABSTRACT.178.
Amgen reports fourth quarter and full year 2021 financial results. 2022. https://www.amgen.com/newsroom/press-releases/2022/02/amgen-reports-fourth-quarter-and-full-year-2021-financial-results.
Tchao N, Amouzadeh H, Sarkar N, Chow V, Hu X, Kroenke M, et al. Efavaleukin alfa, a novel IL-2 mutein, selectively expands regulatory T cells in patients with SLE: Interim results of a phase 1b multiple ascending dose study [abstract]. Arthritis Rheumatol. 2021;73 (suppl 10).
Efficacy and safety of Efavaleukin alfa in subjects with active systemic lupus. 2023. https://clinicaltrials.gov/study/NCT04680637.
Higginson-Scott N, Viney JL, Visweswaraiah J, Sampson ER, Otipoby KL. IL-2 muteins and uses thereof. US 10946068 B2, 2021.
Visweswaraiah J, Sampson ER, Kiprono T, Petaipimol P, Monsef A, Kis-Toth K, et al. Generation of PT101, a highly selective IL-2 mutein for treatment of autoimmune diseases [abstract]. J Immunol. 2021;206.
Sundy JS, Otipoby KL, Higginson-Scott N, Visweswaraiah J, Sampson E, Kis-Toth K, et al. AB0282 Safety, tolerability and selective expansion of regulatory T cells by a single dose of the novel IL-2 mutein PT101 in a phase 1 study in healthy volunteers [abstract]. Ann Rheum Dis. 2021;80:1167–1167. https://doi.org/10.1136/ANNRHEUMDIS-2021-EULAR.1200.
Greve J. IL-2 variants for the treatment of psoriasis. US 11077172 B2, 2017.
Ramirez-Valle F. Found in translation: lessons from an IL-2 mutein [presentation]. In Proceedings of the 4th Treg Directed Therapies Summit, Boston, May 17–19 2022.
Li Y-S, Rui L, Xu J. Interleukin-2 variants and methods of uses thereof. WO 2019/246404 A1, 2019.
Hsieh YT, Hubeau C, Massa V, LI W, Frei S, Capraro B, et al. OP0316 Emerging best-in-class IL-2 variant highlights Treg-directed therapy for autoimmune disease [abstract]. Ann Rheum Dis. 2020;79:195–195. https://doi.org/10.1136/ANNRHEUMDIS-2020-EULAR.1999.
Mitra S, Ring AM, Amarnath S, Spangler JB, Li P, Ju W, et al. Interleukin-2 activity can be fine tuned with engineered receptor signaling clamps. Immunity. 2015;42:826–38. https://doi.org/10.1016/J.IMMUNI.2015.04.018.
Xie JH, Zhang Y, Loubeau M, Mangan P, Heimrich E, Tovar C, et al. Mouse IL-2/CD25 fusion protein induces regulatory T cell expansion and immune suppression in preclinical models of systemic lupus erythematosus. J Immunol. 2021;207:34–43. https://doi.org/10.4049/JIMMUNOL.2100078.
Struthers M, Davis JH, Doyle ML, Madia PA. Interleukin-2/interleukin-2 receptor alpha fusion proteins and methods of use. WO 2019/191295 A1, 2019.
In the pipeline. Bristol Myers Squibb. October 26, n.d. https://www.bms.com/researchers-and-partners/in-the-pipeline.html.
Struthers M. Circulating slow-release of IL-2R agonist for Treg expansion. The promise of interleukin-2 therapy; September 14–17; Paris.
Seidel I, Chaparro RJ, Ross JF, Low CM. TGF-β polypeptides. WO 2021/081258 A1, 2021.
Tweedle J, Jackson N, Saggu G, Yeung K, Spaulding E, Histed A, et al. CUE-401: A novel IL-2/TGF-beta fusion protein for the induction of CD4+FOXP3+ regulatory T cells [poster]. FOCIS 2021, Virtual Meeting, June 8-11, 2021.
Zaragoza B, Chen X, Oppenheim JJ, Baeyens A, Gregoire S, Chader D, et al. Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat Med. 2016;22:16–7. https://doi.org/10.1038/NM.4019.
Padutsch T, Sendetski M, Huber C, Peters N, Pfizenmaier K, Bethea JR, et al. Superior Treg-expanding properties of a novel dual-acting cytokine fusion protein. Front Pharmacol. 2019;10. https://doi.org/10.3389/FPHAR.2019.01490.
Chaudrhy A, Ouyang W. Dual interleukin-2/TNF receptor agonist for use in therapy. WO 2021/127262 A1, 2021.
Gene therapy needs a long-term approach. Nat Med. 2021;27:563. https://doi.org/10.1038/s41591-021-01333-6.
Garber K. mRNA pioneers refocus on therapeutics. Nat Rev Drug Discov. 2022;21:699–701. https://doi.org/10.1038/d41573-022-00156-5.
de Picciotto S, DeVita N, Hsiao CJ, Honan C, Tse SW, Nguyen M, et al. Selective activation and expansion of regulatory T cells using lipid encapsulated mRNA encoding a long-acting IL-2 mutein. Nat Commun. 2022;13. https://doi.org/10.1038/s41467-022-31130-9.
Huang EY-C, Iacovelli J, de Picciotto S, Tse S-W, Kenney L mRNAs encoding immune modulating polypeptides and uses thereof. WO 2021/076805 A1, 2021.
Quarterly results. 2022. https://investors.modernatx.com/financials/quarterly-results/default.aspx.
Pagni PP, Chaplin J, Wijaranakula M, Wesley JD, Granger J, Cracraft J, et al. Multicomponent plasmid protects mice from spontaneous autoimmune diabetes. Diabetes. 2021;71:157–69. https://doi.org/10.2337/db21-0327.
Chaplin J, Wijaranakula M. Tolerogenic DNA vaccine. WO 2018/083111 A1, 2018.
Alves S, Churlaud G, Audrain M, Michaelsen-Preusse K, Fol R, Souchet B, et al. Interleukin-2 improves amyloid pathology, synaptic failure and memory in Alzheimer’s disease mice. Brain. 2017;140:826–42. https://doi.org/10.1093/brain/aww330.
Yshii L, Pasciuto E, Bielefeld P, Mascali L, Lemaitre P, Marino M, et al. Astrocyte-targeted gene delivery of interleukin 2 specifically increases brain-resident regulatory T cell numbers and protects against pathological neuroinflammation. Nat Immunol. 2022;23:878–91. https://doi.org/10.1038/S41590-022-01208-Z.
Spangler JB, Trotta E, Tomala J, Peck A, Young TA, Savvides CS, et al. Engineering a single-agent cytokine/antibody fusion that selectively expands regulatory T cells for autoimmune disease therapy. J Immunol. 2018;201:2094–106. https://doi.org/10.4049/JIMMUNOL.1800578.
Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, et al. In vivo expansion of Treg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med. 2009;206:751–60. https://doi.org/10.1084/JEM.20082824.
Song Q, Wang X, Wu X, Qin H, Li Y, Riggs AD, et al. Tolerogenic anti-IL-2 mAb prevents graft-versus-host disease while preserving strong graft-versus-leukemia activity. Blood. 2021;137:2243–55. https://doi.org/10.1182/BLOOD.2020006345.
Pilat N, Wiletel M, Weijler AM, Steiner R, Mahr B, Warren J, et al. Treg-mediated prolonged survival of skin allografts without immunosuppression. Proc Natl Acad Sci USA. 2019;116:13508–16. https://doi.org/10.3390/ijms24021752.
Hao J, Campagnolo D, Liu R, Piao W, Shi S, Hu B, et al. Interleukin-2/interleukin-2 antibody therapy induces target organ natural killer cells that inhibit central nervous system inflammation. Ann Neurol. 2011;69:721–34. https://doi.org/10.1002/ANA.22339.
Liu R, Zhou Q, la Cava A, Campagnolo DI, van Kaer L, Shi FD. Expansion of regulatory T cells via IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia. Eur J Immunol. 2010;40:1577–89. https://doi.org/10.1002/EJI.200939792.
Yokoyama Y, Iwasaki T, Kitano S, Satake A, Nomura S, Furukawa T, et al. IL-2-anti-IL-2 monoclonal antibody immune complexes inhibit collagen-induced arthritis by augmenting regulatory T cell functions. J Immunol. 2018;201:1899–906. https://doi.org/10.4049/JIMMUNOL.1701502.
Dinh TN, Kyaw TS, Kanellakis P, To K, Tipping P, Toh BH, et al. Cytokine therapy with interleukin-2/anti-interleukin-2 monoclonal antibody complexes expands CD4+CD25+Foxp3+ regulatory T cells and attenuates development and progression of atherosclerosis. Circulation. 2012;126:1256–66. https://doi.org/10.1161/CIRCULATIONAHA.112.099044.
Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, et al. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci Transl Med. 2014;6:258ra142. https://doi.org/10.1126/SCITRANSLMED.3009925.
Sahin D, Arenas-Ramirez N, Rath M, Karakus U, Hümbelin M, van Gogh M, et al. An IL-2-grafted antibody immunotherapy with potent efficacy against metastatic cancer. Nat Commun. 2020;11. https://doi.org/10.1038/S41467-020-20220-1.
VanDyke D, Iglesias M, Tomala J, Young A, Smith J, Perry JA, et al. Engineered human cytokine/antibody fusion proteins expand regulatory T cells and confer autoimmune disease protection. Cell Rep. 2022;41:111478. https://doi.org/10.1016/j.celrep.2022.111478.
Funding
VL is funded by a Baekeland mandate from the Flanders Innovation & Entrepreneurship Agency (VLAIO) (HBC.2019.2214). VM is supported by the Research Foundation Flanders (FWO) (Fundamental Research PhD fellowship, 11l7523N). Research in the SMS lab is supported by Research Foundation Flanders (FWO) (Fundamental Research Grant, G054722N), KU Leuven (C1 grant, C14/20/106), and Flanders Innovation & Entrepreneurship Agency (VLAIO) (HBC.2021.0529). Research in the SHB lab is supported by the KU Leuven (STG/19/032).
Author information
Authors and Affiliations
Contributions
SMS and VL conceived and wrote the manuscript. VL and VM prepared figures and tables. SHB contributed to the writing and revision of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lykhopiy, V., Malviya, V., Humblet-Baron, S. et al. IL-2 immunotherapy for targeting regulatory T cells in autoimmunity. Genes Immun 24, 248–262 (2023). https://doi.org/10.1038/s41435-023-00221-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41435-023-00221-y
