
Abstract
By combining next generation whole exome sequencing and induced pluripotent stem cell (iPSC) technology we found that an Alu repeat inserted in exon 9 of the MAK gene results in a loss of normal MAK transcript and development of human autosomal recessive retinitis pigmentosa (RP). Although a relatively rare cause of disease in the general population, the MAK variant is enriched in individuals of Jewish ancestry. In this population, 1 in 55 individuals are carriers and one third of all cases of recessive RP is caused by this gene. The purpose of this study was to determine if a viral gene augmentation strategy could be used to safely restore functional MAK protein as a step toward a treatment for early stage MAK-associated RP. Patient iPSC-derived photoreceptor precursor cells were generated and transduced with viral vectors containing the MAK transcript. One week after transduction, transcript and protein could be detected via rt-PCR and western blotting respectively. Using patient-derived fibroblast cells and mak knockdown zebra fish we demonstrate that over-expression of the retinal MAK transgene restored the cells ability to regulate primary cilia length. In addition, the visual defect in mak knockdown zebrafish was mitigated via treatment with the retinal MAK transgene. There was no evidence of local or systemic toxicity at 1-month or 3-months following subretinal delivery of clinical grade vector into wild type rats. The findings reported here will help pave the way for initiation of a phase 1 clinical trial for the treatment of patients with MAK-associated RP.
Similar content being viewed by others

Introduction
Male germ cell-associated kinase (MAK) is a protein that regulates the length of the primary cilium of a variety of different cell types [1]. Interestingly, despite its original description as a gene that is important in spermatogenesis, deletion of the Mak gene in mice does not affect fertility [2]. Instead, Mak-deficient animals develop a slowly progressive form of retinal degeneration characterized by elongation of photoreceptor cell connecting cilia and an abnormal electroretinogram [3].
In 2010 we discovered that mutations in the MAK gene were responsible for approximately one third of autosomal recessive retinitis pigmentosa (RP) occurring in individuals of Jewish ancestry [1]. Clinically, MAK-associated RP is characterized by adult-onset loss of the light-sensing photoreceptor cells of the outer neural retina. The disease often begins in the inferonasal aspect of the retina resulting in a characteristic loss of visual field superotemporally [4]. The human retina expresses two different transcripts of the MAK gene. The most abundant is retina-specific and contains a 75 bp exon 12 that is absent from the canonical transcript that is expressed in a variety of cell types throughout the body. The most common disease-causing MAK genotype in RP patients is a homozygous 353 bp insertion of an Alu repeat in exon 9, which results in a translational frameshift and premature termination of translation [1]. As both the canonical and retina-specific isoforms of the MAK gene contain exon 9, MAK protein is completely absent in the cells of patients with this genotype.
Recessive diseases that are characterized by a loss of gene function are particularly amenable to viral-mediated gene replacement; delivery of full-length MAK to photoreceptor cells before they succumb to the disease could potentially slow or prevent vision loss. The fact that MAK-associated RP is an adult onset disease that is highly enriched in individuals of Jewish ancestry makes MAK-associated RP an excellent candidate for gene therapy [4].
Recent FDA approval of the adeno-associated virus (AAV)-based gene therapeutic voretigene neparvovec for the treatment of RPE65-associated Leber congenital amaurosis has invigorated the retinal gene therapy community. There are currently gene therapy clinical trials underway for the treatment of several other inherited retinal disorders. Unfortunately, there are over 100 different genes whose dysfunction cause inherited retinal disease, and more than 40 genes that cause RP [4]. So, to make significant headway against these diseases as a group, rapid simultaneous development of low-cost gene-based therapeutics will be required.
One of the most time-consuming obstacles to the development of a retinal gene therapy is the identification of a model system that recapitulates the disease phenotype faithfully enough that it can be used to demonstrate treatment efficacy. To streamline this step, we have devised an AAV testing pipeline that uses patient iPSC-derived retinal cells for this purpose. This strategy enables simultaneous testing of multiple isoforms of the gene of interest as well as several different promoter systems in human cells, without the need for a large number of animals. Once the optimal genetic isoform and promoter combination is determined the construct can be packaged into the appropriate AAV serotype under cGMP conditions and injected into wildtype animals (i.e., Sprague Dawley rats) to evaluate safety at human relevant clinical doses.
In this study, we generated constructs driving either the canonical or the retinal-specific MAK using two different promoters: a stronger Cytomegalovirus (CMV) promoter and a weaker EF1α promoter. Each of these constructs was tested for their ability to drive expression of their corresponding MAK transcript and corresponding protein in patient-iPSC-derived photoreceptor precursor cells. Further, each construct was assessed for its ability to regulate primary cilia length in both patient-derived fibroblast cells in vitro and mak knockdown zebrafish in vivo. Following identification of the optimal MAK isoform and promoter combination, clinical grade virus was packaged under current Good Manufacturing Practice (cGMP) conditions and subjected to safety analysis in wildtype Sprague Dawley rats. Animals were sacrificed 1-month and 3-months following subretinal injection and subjected to full necropsy, histopathological investigation and hematology and clinical chemistry analysis (for hematology and clinical chemistry an additional 3-days post-injection group was included). Collectively, the findings reported in this manuscript will be useful for initiation of a phase 1 clinical trial for the treatment of patients with MAK-associated RP.
Materials and methods
Ethics statement
All patients provided written, informed consent for this study, which was approved by the Institutional Review Board of the University of Iowa (project approval # 200202022) and adhered to the tenets set forth in the Declaration of Helsinki. The three patients in this study were molecularly confirmed to be homozygous for the 353 bp Alu insertion in the MAK gene, which we have previously demonstrated to cause MAK-associated retinitis pigmentosa [1, 5]. All zebrafish and rat experiments were conducted with the approval of the University of Iowa Animal Care and Use Committee (Animal welfare assurance #8071513 and #1031317, respectively) and were consistent with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the Treaty of Helsinki.
Patient-derived cells
Following informed consent, skin biopsies were collected from three patients with MAK-associated RP and used for fibroblast isolation and iPSC generation as described previously [6,7,8]. Briefly, dermal fibroblast cells were reprogrammed using the CytoTune non-integrating Sendai virus reprogramming kit according to the manufacturers protocol (Invitrogen/Thermo Fisher Scientific, Waltham, MA; CytoTune-iPS Reprogramming Kit; Cat#: A16517) [9]. Fibroblasts were plated on 6-well tissue culture plates and infected at a multiplicity of infection (MOI) of 5. At 12–16 h following transduction cells were washed and fed with fresh fibroblast cell growth media [(DMEM/F12, 5% heat inactivated FBS (Invitrogen/Thermo Fisher Scientific) and 0.2% primocin (Invivogen, San Diego, CA)]. At 7 days post-infection, cells were passaged onto 6-well LN521-coated cell culture dishes at a density of 30,000 cells/well and fed every day with E8 pluripotency media (Invitrogen/Thermo Fisher Scientific). At 3 weeks post-viral transduction, iPSC colonies were picked, passaged, and clonally expanded on fresh LN521-coated cell culture dishes. During reprogramming and maintenance of pluripotency, cells were cultured at 5% CO2, 10% O2, and 37 °C.
Differentiation of iPSC-derived photoreceptor precursor cells
For ease of viral transduction, patient-derived iPSCs were differentiated under 2D conditions as described previously [6, 7]. Briefly iPSCs were harvested and cultured on ultra low-binding plates (Corning Life Sciences, Tewksbury, MA) in embryoid body formation medium [DMEM F-12 (Gibco/Thermo Fisher Scientific), 10% knockout serum replacement (Gibco/Thermo Fisher Scientific), 2% B27 supplement (Gibco/Thermo Fisher Scientific), 1% N2 supplement (Cell Therapy Systems/Thermo Fisher Scientific), 1% L-glutamine (Life Technologies/Thermo Fisher Scientific), 1X NEAA (Life Technologies/Thermo Fisher Scientific), 0.2% Primocin™ (Invivogen), 1 ng/ml DKK-1 (R&D Systems, Minneapolis, MN), 1 ng/ml IGF-1 (R&D Systems), 1 ng/ml Noggin (R&D Systems) and 0.5 ng/ml bFGF (R&D Systems)] for 4–5 days. Embryoid bodies (~50/well) were plated on 6-well plates (Corning Life Sciences) coated with 25 μg/ml collagen (BD Bioscience, San Jose, CA), 50 μg/ml laminin (Life Technologies/Thermo Fisher Scientific), and 100 μg/ml fibronectin (Sigma-Aldrich) and cultured in differentiation medium one [DMEM F-12 (Life Technologies/Thermo Fisher Scientific), 2% B27 supplement (Gibco/Thermo Fisher Scientific), 1% N2 supplement (Life Technologies/Thermo Fisher Scientific), 1% L-Glutamine (Life Technologies/Thermo Fisher Scientific), 1X NEAA (Life Technologies/Thermo Fisher Scientific), 0.2% Primocin™ (Invivogen), 10 ng/ml DKK-1 (R&D Systems), 10 ng/ml IGF-1 (R&D Systems), 10 ng/ml Noggin (R&D Systems) and 5 ng/ml bFGF (R&D Systems)]. Embryoid bodies were differentiated for 10 days in differentiation medium one, six days in differentiation medium two [(differentiation media one plus 10 μM DAPT (EMD Millipore, Billerica, MA)] and 12 days in differentiation medium three [(differentiation media two plus 2 ng/ml aFGF (R&D Systems)]. To generate photoreceptor precursor cells, cultures were fed every other day for an additional 60 days in differentiation medium four [DMEM F-12 (Life Technologies/Thermo Fisher Scientific), 2% B27 supplement (Life Technologies/Thermo Fisher Scientific), 1% N2 supplement (Life Technologies/Thermo Fisher Scientific), 1% L-Glutamine (Life Technologies/Thermo Fisher Scientific), 1X NEAA (Life Technologies/Thermo Fisher Scientific), 0.2% Primocin™ (Invivogen)]. Patient-specific cells were differentiated for a total of 90 days.
Lentiviral transgene cassette cloning and packaging
For the fibroblast primary cilia rescue experiments, MAK constructs were delivered via lentiviral transduction because the efficiency of AAV-mediated transduction of human dermal fibroblast is usually quite low. Lentiviral vectors were generated as we described previously [10]. Briefly, DNA containing the coding sequence of either the human canonical MAK isoform (GenBank Accession No. NM_005906) or the human retinal-specific MAK isoform (GenBank Accession No. NM_001242957) were cloned into the Gateway® gene entry vector pENTR3C (pENTR_MAKCI or pENTR_MAKRI; Life Technologies/Thermo Fisher Scientific). HIV-1 transgene cassette plasmids expressing either the CMV- or the EF1α-promoter driving MAKCI or MAKRI were derived using three-plasmid LR recombination reactions including the Gateway® promoter entry vectors pENTR5’/CMVp or pENTR5’/EF1α (Life Technologies/Thermo Fisher Scientific), the gene entry clones pENTR_MAKCI or pENTR_MAKRI, and the Gateway® destination clone pLenti6.4/R4R2/V5-DEST (Life Technologies/Thermo Fisher Scientific) according to the manufacturer’s instructions. Third generation lentiviral vectors were produced at the University of Iowa Gene Transfer Vector Core using the Invitrogen Corporation ViraPower™ Lentivirus Expression System (Invitrogen/Thermo Fisher Scientific)). HIV-1 packaging, transgene cassette, VSV-G envelope, and Rev protein expression plasmids were co-transfected into HEK293FT cells using Lipofectamine 2000 as previously described [10]. Collected supernatants were concentrated 250:1 via overnight centrifugation and reconstituted in alpha-lactose buffer (40 mg/ml in 1X PBS). Concentrated vector was titered on HT1080s and HIV-1 genomes were quantified using TaqMan® qPCR.
Adeno-associated virus transgene cassette cloning and viral vector packaging
To generate AAV transgene cassette plasmids expressing either the canonical or retinal MAK isoform under control of either the CMV or EF1α promoter, cDNAs were cloned into the KpnI/NheI linearized pFBAAVmcsBgHpA AAV shuttle plasmid (University of Iowa Gene Transfer Vector Core). The resulting 4 plasmids carried either CMV-driven canonical MAK isoform (CMV- MAKCI), CMV-driven retina-specific MAK isoform (CMV- MAKRI), EF1α-driven canonical MAK isoform (EF1α-MAKCI)or EF1α-driven retina-specific MAK isoform (EF1α-MAKRI), which were flanked by AAV2 inverted terminal repeats. Recombinant AAV2/5 vectors were produced in the University of Iowa Gene Transfer Vector Core. Vectors were produced via triple transfection as described previously [11]. Physical titers were determined via TaqMan® qPCR. Vector purity was determined via silver staining [12].
cGMP production of clinical grade AAV5-CMV-MAKRI vectors
Production of clinical grade AAV vector was performed as we have previously described [11]. Briefly, AAV5-CMV-MAKRI was manufactured under cGMP in the Steven W. Dezii Translational Vision Research Facility (DTVRF) within the Institute for Vision Research at the University of Iowa. This facility contains two independent suites: one dedicated to autologous iPSC-derived retinal cell generation and one dedicated to AAV production. Both suites contain high-efficiency HEPA-filtered ISO class 7 (class 10,000) gowning areas and ISO class 6 (class 1000) processing rooms. The AAV processing room is equipped with 2 six-foot biosafety cabinets monitored by real time particle counters that exceed ISO class 5 (class 100) cleanliness standards, 2 copper lined heracell incubators, an iCellis bioreactor and a dedicated support room separated by a sliding glass door that contains both benchtop and ultra-centrifuges. AAV5-CMV-MAKRI was manufactured using a characterized human HEK293T master cell line and a triple transfection method. Specifically, HEK293T cells were transfected with a set of constructs encoding (1) normal human retina-specific MAKRI driven by the CMV promoter (pAAV2-CMV- MAKRI, which contains the expression cassette flanked by AAV2 ITRs) and (2) AAV (pXX2-R5C5, AAV5 packaging plasmid containing AAV5 rep and cap sequences) and helper virus-derived sequences (pHelper, containing E2A and E4 genes from adenovirus). Each of these plasmids were sequence confirmed via bidirectional sequencing before packaging in the DTVRF. HEK293T cells taken from a qualified Master Cell Bank (DTVRF HEK293T) were plated and expanded in T600 multilayer tissue culture flasks (production batch scale of 20 flasks). Upon reaching confluence, cells were simultaneously transfected with pAAV2-CMV-MAKRI, pXX2-R5C5, and pHelper. To purify AAV5-CMV-MAKRI, the following steps were performed: (1) HEK293T cultures were passaged and centrifuged to remove cell culture reagents and low molecular weight impurities; (2) HEK293T cells were lysed to release the intracellular vector, and nuclease digestion was performed to remove nucleic acid impurities; (3) cell debris was removed from the lysate via filtration, using 0.45- and 0.2-μm filters; (4) density gradient ultracentrifugation was performed to separate empty capsids (a major impurity) from the vector product; (5) affinity chromatography was performed to purify vector; (6) viral particles were concentrated via buffer exchange; and (7) working concentrations of AAV5-CMV-MAKRI were formulated in storage and injection buffer (180 mM NaCl plus 10 mM Na3PO4 [trisodium phosphate] in water for injection at pH 7.2), filtered through a 0.2-μm filter, vialed and labeled.
Viral transduction of cells
JK1 murine testicular stromal cells and patient iPSC-derived photoreceptor precursor cells were infected at a MOI of 104 vector genomes (vg) per cell. Patient dermal fibroblast cells were transduced at a MOI of 5. Viral transduction was performed in serum free cell culture media. Specifically, for JK1 and patient fibroblast cells, FBS was removed from the culture media and for iPSC-derived photoreceptor precursor cells transduction was performed in complete differentiation media (as described above). At 16 h post-infection, cells were washed with fresh complete medium and fed every other day for 7 days. At 7 days post-infection, cells were harvested for analysis as appropriate. For ciliogenesis assays, cells were infected as above, allowed to recover for 3 days and then serum-starved for 72 h to induce primary cilia formation prior to analysis.
Ciliogenesis assay in patient-derived dermal fibroblast cells
Dermal fibroblasts from three patients with MAK-associated RP (both with and without viral transduction) and an unaffected control individual were cultured on collagen-coated chamber slides in serum-free conditions [MEMα (Life Technologies/Thermo Fisher Scientific), 2% v/v Primocin (Life Technologies/Thermo Fisher Scientific)] for 72 h. Cells were fixed in 4% paraformaldehyde and methanol and stained with an antibody targeted against acetylated tubulin (1:200, Sigma-Aldrich). Slides were coverslipped with Polyvinyl alcohol (PVA) mounting medium containing 1,4-Diazabicyclo[2.2.2]octane (DABCO) (100 μg/ml PVA, Sigma-Aldrich; 25% v/v glycerol, Sigma-Aldrich; 0.1 M Tris-HCl, pH 8-8.5, 25 μg/ml DABCO, Sigma-Aldrich) and 4’,6-Diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma-Aldrich; 1:10,000). Following antibody labeling, slides were coded to eliminate experimenter bias. Cells and cilia were imaged and counted by two individuals who were masked to the identity of the treatment groups. Cilia were imaged using a Leica DM 2500 SPE confocal microscope (Leica Microsystems, Wetzlar, Germany). Fields of cells were found using DAPI followed by imaging acetylated tubulin. Primary cilia were counted as elongated or punctate acetylated tubulin-positive structures localized to nuclei or immediately perinuclear.
Quantification of cilia length
Cilia were measured using the Neurite tracer plugin in ImageJ as we described previously (24807808). Briefly, images of cells with primary cilia (n = 25 cells for each of three patients and a control individual) were collected via confocal microscopy using a 63× objective. Cilia displaying clear labeling of acetylated tubulin (axoneme) were traced and measurements were obtained.
Immunoblotting for MAK
Western blots were performed as described previously [1, 5, 10]. Briefly, cells were treated with 0.25% Trypsin-EDTA (Life Technologies/Thermo Fisher Scientific), homogenized in lysis buffer [50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 10 mM CaCl2, 1% triton X-100, 0.02% NaN3, (Sigma Aldrich)] and centrifuged. Supernatant protein concentrations were determined using bicinchoninic acid (BCA) according to manufacturer’s instructions (Pierce, Rockford, IL). Fifty micrograms each were subjected to SDS-PAGE (4-20% acrylamide), transferred to PVDF, and probed with rabbit anti-MAK antibody (Abcam, Cambridge, England; Cat. #: ab80536). Blots were visualized with ECL reagents (GE Healthcare Life Sciences, Pittsburgh, PA) and exposed to X-ray film (Fisher Scientific, Pittsburgh, PA).
rt-PCR
RNA was isolated from cells using a RNeasy Mini Kit (Qiagen, Venlo, Limburg, the Netherlands) and the final concentration determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific). cDNA was produced using a High Capacity cDNA Reverse Transcriptase Kit (Life Technologies/Thermo Fisher Scientific). Specific genes were then amplified using rt-PCR. The resulting DNA was characterized by electrophoresis on a 2% agarose gel for 30 min. Products were subsequently gel purified and sequence confirmed.
Subretinal injection of AAV5-CMV-MAKRI
All animal procedures were approved by the University of Iowa’s Animal Care and Use Committee. 2-month-old wildtype Sprague Dawley rats (Charles River; N = 60, 30 M and 30 F) were anesthetized via isofluorane inhalation and treated with subretinal injection, as previously described [13, 14]. Half of the animals received a single 10 μl injection of purified and concentrated clinical-grade AAV5-CMV-MAKRI (1011 vg) and half of the animals received an equal volume of AAV storage and injection buffer. Rats were sacrificed 1-month and 3-months post-injection. Hematology and clinical chemistry (N = 60, 30 M, and 30 F, performed by IDEXX, Columbia, MO) were performed at 3-day, 1-month, and 3-months post-injection. Rats sacrificed at 1-month and 3-months post-injection were evaluated with complete necropsy and histopathologic analysis following sectioning and H&E staining (N = 40, 20 M, and 20 F) were performed. All animals were examined at each time point by an ophthalmologist (ICH) to ensure no visible intraocular inflammation or other injection-related complications.
Zebrafish mak expression domains
Zebrafish mak was isolated by PCR, cloned into the pCRII-TOPO TA vector (forward primer: 5′–cgctacgacctctgtttcct-3′, reverse primer: 5′-tgctctccttttccatccat-3′), and used to generate a riboprobe for whole mount in situ hybridization. The anti-sense probe was created by linearizing the plasmid with KpnI and transcribing the gene with T7 RNA polymerase. The sense probe was created by linearizing the plasmid with NotI and transcribing the gene with SP6 RNA polymerase. The Maxiscript RNA kit (Ambion), and DIG RNA labelling mix (Roche) were used for both probes. For retina sections, embryos were post-fixed after whole mount in situ hybridization in 4% paraformaldehyde followed by incubation for 1 h each at 4 °C in 15% sucrose in sterile H2O and 30% sucrose in sterile H2O. This was followed by overnight incubation at 4 °C in optimal cutting temperature compound (OCT). Embryos were oriented and frozen in OCT for cryosectioning at 12 µm and photographed using a Zeiss Axiophot compound microscope and Axiovision software at 20×, and 63×.
Exogenous expression of Human MAK in zebrafish
Human myc-tagged MAK cDNAs were cloned into the pCS2 + expression vector. RNA generated by in vitro transcription was injected into live 1–2 cell stage zebrafish embryos. Protein lysates were isolated at selected developmental time points for Western blotting and probed with mouse anti-myc (9E10; Santa Cruz Biotechnology, Santa Cruz, CA at 1:10 000) and mouse anti-β-actin (1:2000, Sigma Clone AC-74, St Louis, MO) antibodies as previously described [15].
Zebrafish knockdown and rescue
Morpholinos (MO) were designed to target the initiator methionine codon (AUG) and two different splice sites (mak_AUG: CTTGAGTGTCGTGTAACGGTTCATT; mak_exon 3: TGTGTTCACTGAGTTTGCACCTTGA; mak_exon 4: TGATATAGAATCTCATCATACCTGT). Control MO or mak MOs (2–3 ng) were injected into 1–2 cell stage zebrafish embryos. Sequential injection was used for rescue with 1-cell stage embryos being injected with MO and then separated into groups. One MO-injected group was injected with human MAK mRNA and the other group was not injected again and served as the MO-only control. Phenotypic analysis of these groups, along with RNA only and uninjected siblings, was performed by evaluators masked to the identity of the experimental conditions.
Analysis of Kupffer’s Vesicle (KV) cilia length
Embryos at the 10-12 somite stage were fixed in 4% paraformaldehyde. Cilia were decorated with anti-acteylated tubulin (1:800, Sigma) followed with Alexa 563 Goat anti-mouse secondary (1:400, Molecular Probes). Tailbuds were mounted using Vectashield mounting medium and imaged using a Leica (SP2) confocal microscope (63X oil with 1 micron sections). Individual cilia in a MAX projection of a 10 µm optic section were traced using the Leica Confocal Software measurement tool. The lengths of all traced cilia were exported to excel for statistical analysis.
Visual startle response
Zebrafish visual function was tested at 5 days-post-fertilization (dpf) using VIZN as previously described [16, 17]. Only larva with normal morphology and swim behaviors were used for the vision assay.
Statistical analysis
One-way ANOVA with a Tukey’s post-hoc testing was used to determine statistical significance between treatment groups. P-values of less than 0.05 were considered statistically significant.
Results
Generation of AAV constructs that drive expression of canonical and retina-specific MAK
In 2010, we discovered that insertion of a 353 bp Alu repeat into exon 9 of the MAK gene is a significant cause of RP in patients of Jewish ancestry [1]. This mutation was found to cause a shift in the reading frame of the gene and creation of a premature stop codon resulting in loss of both canonical and retinal MAK expression in patient-derived photoreceptor precursor cells [1]. Loss of functional MAK protein, which normally acts as a negative regulator of cilia length, was found to cause elongation of the photoreceptor cell connecting cilia and progressive retinal degeneration in Mak knockout mice [3].
The fact that human MAK-associated RP is a late onset recessive disorder and enriched in an identifiable population [4] makes it a good candidate for clinical gene augmentation. However, MAK has several isoforms, two of which, the canonical and retina-specific versions, are expressed in the retina. To determine whether AAV-mediated delivery of either canonical (lacking exon 12) or retinal-specific MAK (including exon 12) is capable of restoring functional MAK transcript and protein, we engineered AAV constructs in which expression of MAK is driven under control of two different constitutively active promoters: the stronger Cytomegalovirus (CMV) promoter and a weaker elongation factor 1 alpha (EF1α) promoter. The resulting four vectors are depicted in Fig. 1A: (1) AAV-CMV-MAKCI (MAKCI - canonical isoform, which includes exon 9 and lacks exon 12); (2) AAV-CMV-MAKRI (MAKRI - retina-specific isoform, which includes both exons 9 and 12); (3) AAV- EF1α-MAKCI; and (4) AAV- EF1α-MAKRI.

A Schematic depicting the four AAV constructs generated in this study. Constructs were designed to deliver either the human canonical (MAKCI - lacks exon 12, constructs 1 and 3) or retina-specific MAK isoform (MAKRI - contains exon 12, constructs 2 and 4) under control of either the constitutively active CMV or EF1α promoter. B Representative image of cultured murine JK1 cells (immortalized SMA + , CD34 + testicular stromal cells, which lack endogenous expression of human MAK). Scale bar 100 μm. C, D rt-PCR analysis performed on RNA isolated from JK1 cells transduced with either AAV5-CMV-MAKCI (Construct 1), AAV5-CMV-MAKRI (Construct 2), AAV5-EF1α-MAKCI (Construct 3), or AAV5-EF1α-MAKRI (Construct 4). Each construct is capable of driving robust expression of exon-9-containing human MAK transcript (C). Unlike the MAKCI constructs, both AAV5-CMV-MAKRI and AAV5-EF1α-MAKRI constructs are capable of driving expression of the exon-12-containing retina-specific MAK transcript (D). Arrows denote rt-PCR primer positions.
To evaluate the ability of each of these constructs to drive expression of canonical or retina-specific MAK transcript, we first packaged them into AAV5 particles via triple transfection of HEK293t cells and then used them to transduce mouse JK1 testicular stromal cells (Fig. 1B), which lack endogenous expression of the human MAK gene. We then used rt-PCR with primers spanning exon 9 to evaluate the ability of each vector to drive expression of the exon-9-containing MAK transcript (Fig. 1C). As expected, MAK expression was higher in cultures transduced with vectors containing the CMV promoter than those transduced with vectors containing the weaker EF1α promoter. Interestingly, regardless of promoter used, there appeared to be increased MAK expression in cultures transduced with vectors containing the retina-specific MAK isoform that includes the 75 bp retina-specific exon 12 than those transduced with the vectors containing the canonical MAK isoform that lack exon 12. A similar trend was seen when rt-PCR primers were placed in exons 10 and 14 (Fig. 1D). As expected, rt-PCR products generated following transduction with vectors containing the retina-specific MAK isoform were 75 bp larger than the products generated following transduction with vectors containing the canonical MAK isoform. Again, MAK expression was much more robust when driven under control of the CMV promoter than the EF1α promoter. Collectively, these findings demonstrate that we have successfully engineered AAV constructs capable of driving expression of both the human canonical and retina-specific MAK isoform.
AAV5-mediated restoration of canonical and retinal-specific MAK transcript and protein in patient-specific iPSC-derived photoreceptor precursor cells
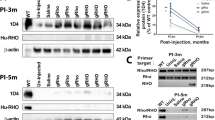
After demonstrating the ability of the AAV vectors described in Fig. 1 to drive MAK expression in a mouse cell line, we next sought to determine if we could restore MAK transcript and protein in a disease-relevant cell type, namely patient-iPSC-derived retinal photoreceptor precursor cells. As we have previously shown, iPSC-derived retinal cells generated from a patient molecularly confirmed to harbor homozygous Alu insertions in exon 9 of the MAK gene, lack expression of both exon 9- and exon 12-containing MAK transcripts and proteins [1]. For this experiment we differentiated patient-derived iPSCs for 90-days, the timepoint that we have previously shown photoreceptor precursor cells to be prominent [1, 7]. Cultures were subsequently transduced with each of the four MAK AAV constructs at a MOI of 104 vg/cell. Rt-PCR analysis using primers spanning exon 9 (i.e., exons 8–10) of the MAK gene showed that compared to untransduced cells from the same patient that lack expression of an exon 9 containing transcript, each construct was capable of restoring exon 9 expression (Fig. 2A). Rt-PCR analysis using primers spanning exon 12 (i.e., exons 10–14) revealed that only constructs carrying the retina-specific MAK isoform (MAKRI) were able to restore expression of the exon 12 containing transcript, which was also absent in the untransduced cells from the same patient (Fig. 2B). Again, MAK transcripts driven under control of the CMV promoter were expressed at higher levels than those driven under control of the EF1α promoter (Fig. 2A, B). As anticipated, restoration of MAK transcript resulted in a restoration of MAK protein. Specifically, vectors containing the canonical MAK isoform successfully restored canonical MAK protein (Fig. 2C, *P < 0.05) and vectors containing the retinal MAK isoform successfully restored the retinal MAK protein (Fig. 2C, *P < 0.001). Although a difference in protein expression between CMV and EF1α containing constructs was visible, the difference was significantly less than the difference observed at the transcriptional level and did not reach statistical significance. However, the difference in protein expression detected following transduction with vectors driving expression of the retina-specific MAK isoform versus the canonical MAK isoform was significant (Fig. 2C, ***P < 0.001).

A, B rt-PCR analysis performed on RNA isolated from patient-specific photoreceptor precursor cells two-weeks following transduction with AAV5-CMV-MAKCI, AAV5-CMV-MAKRI, AAV5-EF1α-MAKCI, or AAV5-EF1α-MAKRI (MOI = 104vg/cell). Compared to untransduced cultures (UT), which lack expression of wildtype exon-9-containing MAK transcript, cells transduced with each of the AAV5 constructs expressed exon-9-containing MAK transcripts. Only AAV5-CMV-MAKRI and AAV5-EF1α-MAKRI were capable of driving expression of exon-12-containing retinal MAK (B). C Western blot of patient-specific photoreceptor precursor cells transduced with AAV5-CMV-MAKCI, AAV5-CMV-MAKRI, AAV5-EF1α-MAKCI, or AAV5-EF1α-MAKRI (MOI = 104vg/cell). Only the constructs driving MAK exon 12 restored expression of full-length retinal-specific MAK protein. D-G Immunocytochemical analysis of patient iPSC-derived photoreceptor precursor cells following AAV5 transduction using antibodies targeted against MAK and OTX2 (photoreceptor precursor cell marker). Although MAK was detected in cultures transduced with AAV5 vectors carrying the canonical isoform (AAV5-CMV-MAKCI (D) and AAV5-EF1α-MAKCI (F)), pronounced expression throughout the cell body and neurites was only detected in cultures transduced with vectors carrying the retinal isoform (AAV5-CMV-MAKRI (E) or AAV5-EF1α-MAKRI (G)). MAK – green, OTX2 – blue. Scale bars = 200 μm. Arrows denote rt-PCR primer positions.
To confirm these Western blotting results and evaluate the pattern of cellular expression following viral transduction we next performed immunocytochemical analysis on patient-iPSC-derived photoreceptor precursor cell cultures. In this analysis, the human specific anti-MAK antibody used in Fig. 2C and an antibody targeted against the photoreceptor precursor cell transcription factor OTX2 were used (Fig. 2D–G). Again, the highest level of MAK expression was detected in cultures transduced with the retina-specific MAK isoform under control of the CMV promoter (Fig. 2E, AAV5-CMV-MAKRI). This was followed by cultures transduced with vector containing the retinal MAK isoform under control of the EF1α promoter (Fig. 2G, AAV5-EF1α-MAKRI), the canonical MAK isoform under control of the CMV promoter (Fig. 2D, AAV5-CMV-MAKCI) and finally the canonical MAK isoform under control of the EF1α promoter (Fig. 2F, AAV5-EF1α-MAKCI). The pattern of MAK expression following transduction with each of the 4 vectors was strikingly different. Cells transduced with vectors containing the retina-specific isoforms had more robust levels of MAK expression throughout the cytoplasm extending into the neurites of OTX2-positive photoreceptor precursor cells (Fig. 2E, G). Canonical MAK protein appeared to be restricted to the nuclear/perinuclear region with minimal extension into neurites (Fig. 2D, F). Together, these results demonstrate that we were able to successfully restore full-length MAK transcript and protein in patient-iPSC-derived photoreceptor precursor cells using the MAK vectors we developed. Expression was more robust and widespread when the retina-specific MAK isoforms were delivered. Differences in protein expression levels between vectors containing the CMV and EF1α promoters were less pronounced than differences in protein expression levels observed between vectors carrying the canonical versus retinal MAK isoforms.
Viral-mediated rescue of the primary cilia defect in patient-derived dermal fibroblasts
Following confirmation that our MAK vectors were capable of driving expression of their respective MAK isoforms, we next asked if the resulting MAK proteins were functional. As indicated above, MAK functions as a negative regulator of primary cilia length. In mice, loss of MAK expression results in elongation of the connecting cilia in photoreceptor cells [3]. To determine if loss of the MAK protein in patients with MAK-associated RP had an effect on cilia length regulation, and if AAV5-mediated overexpression of MAK protein was sufficient to restore normal cilia length, an in vitro ciliogenesis assay was performed. We have previously demonstrated that serum starved patient-derived fibroblasts, whose primary cilia are readily detectible via immunocytochemical staining of acetylated tubulin, are an excellent model system for evaluating the effect of genetic defects on cilia formation and elongation [10]. In the present study, dermal fibroblasts were obtained from 3 independent patients with MAK-associated RP, each with homozygous Alu insertions in exon 9 of the MAK gene. Dermal fibroblasts isolated from a non-diseased individual were included as an unaffected control. Compared to the unffected individual, dermal fibroblasts isolated from patients with MAK-associated RP had significantly longer primary cilia (Fig. 3A vs. B, G, **P < 0.001). To determine if overexpression of canonical or retinal MAK protein could rescue the cilia length defect in patient-derived dermal fibroblasts, cultures were transduced with viral vectors driving expression of both canonical and retinal MAK under control of either the CMV or EF1α promoter as described above. As demonstrated in Fig. 3, the greatest effect on primary cilia length occurred following over-expression of the retina-specific isoform under control of the CMV promoter (Fig. 3D, G, P < 0.001), consistent with results described in Fig. 2 above. This was followed by the retina-specific isoform under control of the EF1α promoter (Fig. 3F, G, P < 0.01) and the canonical isoform under control of the CMV promoter (Fig. 3C, G, P < 0.001). Although a slight reduction in mean cilia length was detected in cells transduced with the canonical isoform driven under control of the EF1α promoter, this reduction did not reach statistical significance (Fig. 3E, G, P > 0.05). These experiments demonstrate that viral mediated delivery of retinal MAK under control of either the CMV or EF1α promoter is sufficient to restore a cell’s ability to properly regulate primary cilia length. Although canonical MAK did not significantly reduce primary cilia length when placed under control of the weaker EF1α promoter, the fact that it was capable of significantly reducing primary cilia length when placed under control of the stronger CMV promoter suggest that the function of canonical MAK in primary cilia is similar to that of the retinal-isoform. The difference in the two isoforms appear to be at the level of transcript expression and protein translation.

A–F Immunocytochemical analysis using an antibody targeted against anti-acetylated tubulin (primary cilia marker) to stain dermal fibroblasts isolated from an unaffected control individual (A – control) and a patient with molecularly confirmed MAK-associated RP (B–F). Cells from the patient with MAK-associated RP were transduced with viral vectors driving canonical (C, E) or retinal MAK (D and G. Untransduced cultures of patient derived cells were used as a disease phenotype control (B). G Histogram comparing mean primary cilia length between a normal individual (n = 25 cells) and 3 independent patients with MAK-associated RP before and after transduction with viral vectors driving either canonical or retinal MAK (n = 75 cells, 25 cells per patient for each of 3 patients). The primary cilium in fibroblasts isolated from patients with MAK-associated RP was significantly longer than that of an unaffected control individual. Transduction with retinal MAK under control of either the CMV or EF1α promoter reduced the primary cilium length to that of the control. A significant reduction in primary cilium length was detected following transduction of with canonical MAK driven by the CMV promoter only. (**p < 0.01, ****P < 0.001). Scale bars = 100 μm.
Injection of MAK mRNA restores Kupffer’s vesicle cilia length and response to a visual stimulus in a zebrafish model of MAK-associated RP
To test whether delivery of canonical or retina-specific MAK mRNA could restore cilia length in vivo, a mak-knockdown zebrafish model was created. First, we evaluated the endogenous activity of zebrafish mak. Antisense morpholino oligonucleotides directed against the translation start site and splice junctions were used to knockdown zebrafish mak gene function (morphants). To measure cilia length, we assessed cilia within Kupffer’s vesicle (a small ciliated organ that is transiently present during early zebrafish embryogenesis). As in human patient-derived dermal fibroblasts, loss of mak resulted in significant elongation of the primary cilia (Fig. 4A vs B & E, p < 0.01). Three independent morpholinos directed against zebrafish mak all showed an increased cilia length defect.

A–D Immunocytochemical labeling of primary cilia with an anti-acetylated tubulin antibody within Kupffer’s vesicle in uninjected (A), mak morpholino-injected (B), and morpholino-injected zebrafish that simultaneously received canonical MAKCI (D) or retinal MAKRI mRNA (D). E Quantification of mean cilia length measured in each treatment group shown in A–D. MAK morphants displayed significantly longer cilia compared to uninjected siblings (uninjected vs MO only; p = 0.0004). Sequential injection of retinal MAKRI mRNA or canonical MAKCI mRNA significantly shortened primary cilia length (MO only vs MO + RNA; p < 0.0001 for both). F–H Light micrographs of wildtype (F), MAK morpholino-injected (G), and MAK morpholino and retinal MAK mRNA injected (H) demonstrating normal overall morphology among groups. I Histogram depicting the number of responses in the vision startle assay of wild-type, MAK morpholino-injected (MAK Mutant) and MAK morpholino/MAKRI mRNA injected (Mutant + MAKRI mRNA). MAK morphants had a significant decrease in average number of responses compared to wild-type fish (Wildtype vs MAK Mutant; p < 0.05). Injection of retinal MAK mRNA into MAK mutants partially rescued visual responses (i.e. no significant difference between wild-type and treatment groups). Kruskal-Wallis test with Dunn’s multiple comparisons. Scale bars in A-D = 10 μm.
To determine if overexpression of human MAK was capable of rescuing the Kupffer’s vesicle cilia length defect in mutant zebrafish, myc-tagged human MAK mRNA was generated. Sequential injection (at the single cell stage) of morphant zebrafish with human MAK mRNA demonstrated that the canonical and retina-specific MAK isoforms were capable of significantly reducing cilia length (Fig. 4B–D & E, p < 0.01). Consistent with the enriched mak expression domains in zebrafish, morphants displayed Kupffer’s vesicle cilia defects with no other gross morphological differences observed between wild-type (Fig. 4F), mak morpholino-injected (Fig. 4G), and MAK mRNA-injected fish (Fig. 4H). These results demonstrate that exogenous human MAK is sufficient to suppress the cilia length defect detected in mak mutant zebrafish without causing global morphological changes.
To determine if mak knockdown in zebrafish alters visual function, behavioral analysis of the vision-dependent startle response was performed. This assay is based on the observation that zebrafish display a specific swimming behavior when exposed to a rapid change in light that is lost in visually impaired fish [15, 18, 19]. As shown in Fig. 4I, when compared to wildtype fish, mak mutants display a significantly reduced startle response (p < 0.01). As described in the Kupffer’s vesicle experiment above, to evaluate rescue mRNA was injected into morphant zebrafish at the single cell stage. As injected mRNA is halved with each cell division, we expected that MAK protein would be diluted beyond the level of detection at the point we test for visual function at day 5. To our surprise, compared to mak morphants, zebrafish that were sequentially injected with retina-specific human MAK mRNA showed a significant increase in the average number of responses to visual stimuli, suggesting partial recovery of visual function (Fig. 4I, p < 0.05).
Safety profile of clinical grade AAV5-CMV-MAK RI
Following analysis in patient-derived cells in vitro and mak morphant zebrafish in vivo, it became clear that of the four constructs we developed, AAV5-CMV-MAKRI produced the most consistent and robust expression of functional MAK protein. This construct was therefore advanced through our clinical AAV production and preclinical local and systemic toxicity analysis pipeline. Following production under cGMP conditions, 10 μl of AAV5-CMV-MAKRI viral particles (1013 vg/ml, 1011 vg per 10 μl injection) were injected into the subretinal space of 20 (10 M and 10 F) normal Sprague Dawley rats. As a control, an additional 20 (10 M and 10 F) animals received subretinal injections of an equal volume of AAV storage and injection buffer (vehicle). Half of the animals (i.e., 10 AAV5-CMV-MAKRI treated (5 M and 5 F) and 10 vehicle control (5 M and 5 F)) were sacrificed 1-month following injection and the other half were sacrificed 3-months following injection. Following sacrifice, necropsy was performed, and gross findings recorded. The tissues listed in Supplemental Table 1 were collected, fixed, paraffin embedded, sectioned, stained with hematoxylin and eosin (H&E) and subjected to histopathological analysis by a certified veterinarian pathologist (please see Supplemental Fig. 1 for example H&E stained sections obtained from an animal at 3-months following subretinal injection of AAV5-CMV-MAKRI). As shown in Table 1, there were no significant AAV5-CMV-MAKRI related findings noted at the time of necropsy. No significant AAV5-CMV-MAKRI induced difference in mean organ weight or total body weight between treated and vehicle control animals at either of the timepoints tested were detected. Finally, no significant AAV5-CMV-MAKRI related adverse findings were reported following complete histopathological analysis.
To further demonstrate systemic tolerability, hematology, and clinical chemistry analysis were also performed for the 40 animals sacrificed at 1-month and 3-month post-injection, in addition to another set of 20 animals (10 M and 10 F, injected in the same manner as above) at a 3-day post-injection time point. At the time of sacrifice, 2 ml of blood was collected from every animal. One milliliter was used for hematology and the remainder was centrifuged for serum collection and clinical chemistry. The hematology and clinical chemistry parameters listed in Supplemental Table 2 were evaluated. As shown in Table 2, no significant AAV5-CMV-MAKRI induced events were detected between treated and vehicle control animals at any of the timepoints tested for any of the hematology or clinical chemistry parameters evaluated. Collectively, these findings indicate that subretinal injection of AAV5-CMV-MAKRI (1011 vg) is well-tolerated in a mammalian model.
Discussion
Inherited retinal degeneration can be caused by mutations in any of more than 100 different genes [4]. More than 75% of the retinal-disease-causing genes that behave in a recessive fashion [67/84, [4]] are small enough to be packaged into an AAV making them ideal candidates for clinical gene augmentation therapy. Unfortunately, the paucity of model systems that faithfully recapitulate disease phenotypes and the costs associated with manufacturing and testing clinical gene therapy vectors make it challenging to carry a gene therapy candidate from bench to bedside in a rapid cost-effective manner. To make significant progress toward treating all patients with inherited retinal degeneration, the time and cost associated with the development of clinical grade gene-based therapeutics both need to be reduced.
To address the lack of model systems suitable for evaluating treatment efficacy we have focused our attention on the use of patient-iPSC-derived retinal cells. Unlike animal models, which take significant time and money to create, the human iPSC field has evolved to the point where disease-specific cell types can be readily generated, and disease phenotypes evaluated quickly and inexpensively [recently reviewed by Mullin et al. [20]]. In this study, we used iPSCs generated from three independent patients with molecularly-confirmed MAK-associated RP as a model system to evaluate AAV-mediated MAK gene replacement at the transcriptional, translational, immunocytochemical, and functional levels in human cells.
We previously found that the 75 bp exon 12 of the MAK gene is expressed exclusively in the retina and has been highly conserved since first appearing in reptiles nearly 400 million years ago [1]. Interestingly, both the canonical and retinal-specific versions of the MAK protein are expressed in the human retina [1] and when present in isolation, either isoform has the ability to regulate cilia length, suggesting that the gene’s primary function is independent of exon 12. It was somewhat surprising to find that inclusion of the retina-specific exon in our gene transfer constructs dramatically increased expression of the MAK transcript and protein regardless of the strength of the promoter used to drive the gene. This led us to believe initially that exon 12 was acting as a transcriptional enhancer element. However, when equimolar amounts of canonical and retina-specific MAK mRNAs were injected directly into developing zebrafish, bypassing the transcriptional machinery, the retina-specific isoform was still expressed more robustly than the canonical one (Supplemental Fig. 2). These findings suggest that exon 12 may play a role in stabilizing the MAK transcript or promoting its translation. Regardless of mechanism, it seems likely that the primary evolutionary advantage of MAK exon 12 is to ensure robust expression of the protein in photoreceptor cells where integrity of the connecting cilia is critically important for cell viability. Unlike most cell types in which primary cilia are small sensory organelles that account for a minute portion of the cell’s total surface area, the photoreceptor outer segment is a modified primary cilium that makes up the majority of the cell’s mass [21,22,23,24]. On average, a rod photoreceptor’s outer segment is completely recycled every ten days requiring massive amount of protein to be shuttled between the photoreceptor inner and outer segment via the connecting cilium [25]. In the absence of efficient protein trafficking, outer segment turnover and phototransduction are disrupted resulting in visual dysfunction. As such, it is not surprising that many of the genes that cause RP [e.g., BBS genes, CEP290, IFT88, etc. [4, 26, 27]] are required for proper protein trafficking through the connecting cilium.
After determining that CMV-driven retinal-MAK promoted the greatest degree of phenotypic correction, the next step was to demonstrate the safety of a dose equal to or greater than what would be proposed as the maximum clinical dose in a phase 1 human trial. To calculate a human equivalent dose, one must consider the number of cells being targeted in both the animal model being used and the eventual human patients. Although large animal models with eye sizes similar to humans (e.g., pigs) allow one to deliver exactly the same volume of drug to the subretinal space as would be used in humans, the cost associated with housing a large number of pigs for at least 3 months, which is the current post-injection survival time being requested by the FDA, is prohibitive. In addition, few facilities have the space to maintain more than a handful of large animals at any given time making simultaneous testing of multiple therapeutic vectors challenging. Because they are relatively inexpensive to house, mice have been used extensively for preclinical analysis of novel gene-based therapeutics. Unfortunately, the volume of drug that can be delivered to the subretinal space of the mouse is very small, typically less than 1ul. In addition, the amount of blood that can be obtained following injection for hematology and clinical chemistry analysis is so low that it often requires duplication of study animals, which significantly increases the cost and time needed to perform the study. We have found the normal adult Sprague Dawley rat to be an ideal model system for safety analysis. Sprague Dawley rats are readily available, relatively inexpensive, and have been very well characterized with many strain specific lesions and hematologic findings thoroughly described. At just two months of age, Sprague Dawley rats are large enough that one can reliably inject a dose of AAV into the subretinal space that would be equivalent to the maximum clinical dose delivered in human without needing virus of exceptionally high titer. In addition, rats have significantly more total blood volume than mice, which allows one to perform a complete hematology and blood chemistry panel on each animal. Finally, the Sprague Dawley rat is small enough that it can be housed in sufficient numbers to perform all of the post-injection survival timepoints required for testing multiple products at the same time.
As indicated above, in this manuscript we report development of a pipeline specifically designed to allow for rapid evaluation of novel gene therapy candidates (Fig. 5). As a gene therapy product progresses from synthesis through to filing of an IND application, the cost associated with each stage within the pipeline increases significantly. We have incorporated several go/no go quality control check points (Table S3), at which the decision to progress to the next stage of production and testing or to return to research and development must be made. To keep cost low, early expression, cytotoxicity, and efficacy testing are done using research grade virus produced in the lab using cGMP compatible protocols and reagents. Once confident that we are able to safely overexpress the gene of interest and in turn restore functional protein in a relevant model system, the next step is to proceed with clinical vector production and detailed toxicity analysis, which is where a majority of the cost associated with this process is incurred. It is at this point where we recommend engaging with the FDA in the form of a pre-IND application and meeting (i.e., to get their opinion on suitability of data in hand and proposed or ongoing preclinical safety testing for initiation of a phase 1/2 safety trial). Following completion of clinical vector release testing and preclinical safety analysis, with the support of FDA consultants data are packaged and submitted to the FDA in an investigator initiated IND application.

Using this strategy we were able to develop, test, and validate enough product to treat 500 patients (at a maximum clinical dose of 1 × 1011v.g.) with MAK associated RP for less than $500,000 USD (i.e., ~$1000 USD/patient). v.g. = viral genomes. QC = quality control. Example QC analysis performed at each step is provided. For a complete list of QC analysis performed see Supplemental Table 3.
One of the greatest misconceptions among patients diagnosed with retinitis pigmentosa is that restoration of functional gene expression will result in restoration of vision. While this is true in the rare circumstance in which the disease is caused by dysfunction of a living cell, for the majority of patients with an inherited retinal disease successful gene augmentation will at best arrest the progression of the disease. The most important factor for successful therapy is likely to be early intervention, before so many rod photoreceptors have been lost that there is an insufficient amount of critical rod-derived retinal viability factors and too high a level of intra-retinal oxygen for the remaining cone photoreceptors to survive long term. MAK-associated RP has two favorable avenues for early intervention. First, this disease is a relatively mild form of RP and many patients still have a large number of photoreceptors when they are first diagnosed in early adulthood. Second, the Jewish population is more likely to undergo preconception genetic testing to avoid devastating disorders like Tay Sachs disease that are common in this population [28]. If the Alu insertion in MAK were added to Jewish disease panels the incidence of new cases of the disease could be reduced. However, such testing would also identify some completely presymptomatic affected adults near the beginning of their third decade of life who would have the greatest possible chance for a good outcome from gene augmentation therapy.
In summary, in this manuscript, we report the development of a novel, clinical grade gene augmentation vector for the treatment of MAK-associated RP and used patient-iPSC-derived retinal cells, a zebrafish morphant model, and normal Sprague Dawley rats to demonstrate safety and efficacy in vitro and vivo. Importantly, the pipeline used here is readily adaptable to many other heritable retinal degenerations and should allow us and others to accelerate the development of treatments for these conditions.
References
Tucker BA, Scheetz TE, Mullins RF, DeLuca AP, Hoffmann JM, Johnston RM, et al. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc Natl Acad Sci USA. 2011;108:E569–76.
Shinkai Y, Satoh H, Takeda N, Fukuda M, Chiba E, Kato T, et al. A testicular germ cell-associated serine-threonine kinase, MAK, is dispensable for sperm formation. Mol Cell Biol. 2002;22:3276–80.
Omori Y, Chaya T, Katoh K, Kajimura N, Sato S, Muraoka K, et al. Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc Natl Acad Sci USA. 2010;107:22671–6.
Stone EM, Andorf JL, Whitmore SS, DeLuca AP, Giacalone JC, Streb LM, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017;124:1314–31.
Burnight ER, Gupta M, Wiley LA, Anfinson KR, Tran A, Triboulet R, et al. Using CRISPR-Cas9 to generate gene-corrected autologous iPSCs for the treatment of inherited retinal degeneration. Mol Ther. 2017;25:1999–2013.
Tucker BA, Anfinson KR, Mullins RF, Stone EM, Young MJ. Use of a synthetic xeno-free culture substrate for induced pluripotent stem cell induction and retinal differentiation. Stem Cells Transl Med. 2013;2:16–24.
Tucker BA, Mullins RF, Streb LM, Anfinson K, Eyestone ME, Kaalberg E, et al. Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. Elife. 2013;2:e00824.
Wiley LA, Burnight ER, DeLuca AP, Anfinson KR, Cranston CM, Kaalberg EE, et al. cGMP production of patient-specific iPSCs and photoreceptor precursor cells to treat retinal degenerative blindness. Sci Rep. 2016;6:30742.
Lieu PT, Fontes A, Vemuri MC, Macarthur CC. Generation of induced pluripotent stem cells with CytoTune, a non-integrating Sendai virus. Methods Mol Biol. 2013;997:45–56.
Burnight ER, Wiley LA, Drack AV, Braun TA, Anfinson KR, Kaalberg EE, et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther. 2014;21:662–72.
Wiley LA, Burnight ER, Drack AV, Banach BB, Ochoa D, Cranston CM, et al. Using patient-specific induced pluripotent stem cells and wild-type mice to develop a gene augmentation-based strategy to treat CLN3-associated retinal degeneration. Hum Gene Ther. 2016;27:835–46.
Gao G, Qu G, Burnham MS, Huang J, Chirmule N, Joshi B, et al. Purification of recombinant adeno-associated virus vectors by column chromatography and its performance in vivo. Hum Gene Ther. 2000;11:2079–91.
Han IC, Burnight ER, Ulferts MJ, Worthington KS, Russell SR, Sohn EH, et al. Helper-dependent adenovirus transduces the human and rat retina but elicits an inflammatory reaction when delivered subretinally in rats. Hum Gene Ther. 2019;30:1371–84.
Han IC, Cheng JL, Burnight ER, Ralston CL, Fick JL, Thomsen GJ et al. Retinal tropism and transduction of adeno-associated virus varies by serotype and route of delivery (Intravitreal, Subretinal, or Suprachoroidal) in rats. Hum Gene Ther. 2020.
DeLuca AP, Whitmore SS, Barnes J, Sharma TP, Westfall TA, Scott CA, et al. Hypomorphic mutations in TRNT1 cause retinitis pigmentosa with erythrocytic microcytosis. Hum Mol Genet. 2016;25:44–56.
Brastrom LK, Scott CA, Dawson DV, Slusarski DC. A high-throughput assay for congenital and age-related eye diseases in Zebrafish. Biomedicines. 2019; 7.
Scott CA, Marsden AN, Slusarski DC. Automated, high-throughput, in vivo analysis of visual function using the zebrafish. Dev Dyn. 2016;245:605–13.
Baye LM, Patrinostro X, Swaminathan S, Beck JS, Zhang Y, Stone EM, et al. The N-terminal region of centrosomal protein 290 (CEP290) restores vision in a zebrafish model of human blindness. Hum Mol Genet. 2011;20:1467–77.
Pretorius PR, Aldahmesh MA, Alkuraya FS, Sheffield VC, Slusarski DC. Functional analysis of BBS3 A89V that results in non-syndromic retinal degeneration. Hum Mol Genet. 2011;20:1625–32.
Mullin NK, Voigt AP, Cooke JA, Bohrer LR, Burnight ER, Stone EM et al. Patient derived stem cells for discovery and validation of novel pathogenic variants in inherited retinal disease. Prog Retin Eye Res. 2020:100918.
Besharse JC, Forestner DM, Defoe DM. Membrane assembly in retinal photoreceptors. III. Distinct membrane domains of the connecting cilium of developing rods. J Neurosci. 1985;5:1035–48.
De Robertis E. Electron microscope observations on the submicroscopic organization of the retinal rods. J Biophys Biochem Cytol. 1956;2:319–30.
De Robertis E. Some observations on the ultrastructure and morphogenesis of photoreceptors. J Gen Physiol. 1960;43:1–13.
Tokuyasu K, Yamada E. The fine structure of the retina studied with the electron microscope. IV. Morphogenesis of outer segments of retinal rods. J Biophys Biochem Cytol. 1959;6:225–30.
Young RW. The renewal of photoreceptor cell outer segments. J Cell Biol. 1967;33:61–72.
Sheffield VC, Nishimura D, Stone EM. The molecular genetics of Bardet-Biedl syndrome. Curr Opin Genet Dev. 2001;11:317–21.
Walia S, Fishman GA, Jacobson SG, Aleman TS, Koenekoop RK, Traboulsi EI, et al. Visual acuity in patients with Leber’s congenital amaurosis and early childhood-onset retinitis pigmentosa. Ophthalmology. 2010;117:1190–8.
Levin M. Screening Jews and genes: a consideration of the ethics of genetic screening within the Jewish community: challenges and responses. Genet Test. 1999;3:207–13.
Author information
Authors and Affiliations
Contributions
BAT, EMS, RFM: Conceived and/or designed the work that led to the submission, acquired data, and/or played an important role in interpreting the results, drafted and revised the manuscript, approved the final version of the manuscript, and are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. ERB, LAW, ICH, KCG, DCS: Acquired data and played an important role in interpreting the results, drafted and revised the manuscript. CMC, MJU, MAL, TW, CAS, AM, LAW: Acquired data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tucker, B.A., Burnight, E.R., Cranston, C.M. et al. Development and biological characterization of a clinical gene transfer vector for the treatment of MAK-associated retinitis pigmentosa. Gene Ther 29, 259–288 (2022). https://doi.org/10.1038/s41434-021-00291-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41434-021-00291-5

{kind=link}
{kind=link}