
Abstract
Giant Cell Arteritis (GCA) is well known to be a critical ischaemic disease that requires immediate medical recognition to initiate treatment and where one in five people still suffer visual loss. The immunopathophysiology has continued to be characterised, and the influencing of ageing in the development of GCA is beginning to be understood. Recent national and international guidelines have supported the directed use of cranial ultrasound to reduce diagnostic delay and improve clinical outcomes. Immediate high dose glucocorticoids remain the standard emergency treatment for GCA, with a number of targeted agents that have been shown in clinical trials to have superior clinical efficacy and steroid sparing effects. The aim of this review was to present the latest advances in GCA that have the potential to influence routine clinical practice.
摘要
众所周知, 巨细胞动脉炎 (GCA) 是一种严重的缺血性疾病, 需要进行迅速的诊断后才能治疗, 但五分之一的患者仍会导致视力损伤。其免疫病理生理学特征正不断地被描述出来, 人们也逐渐意识到衰老对GCA进展的影响。新近的国家与国际指南中都支持直接使用颅脑超声来减少诊断过程中的延误并改善临床结局。快速使用大量糖皮质激素仍然是GCA的标准急救方法, 尽管一些靶向药物在临床实践中已经证实具有更好的临床疗效且具有类固醇的效果。本综述旨在介绍GCA的最新进展, 这可能会对日常的临床实践产生影响。
Similar content being viewed by others

Introduction
Giant cell arteritis (GCA) is the most common form of vasculitis with a pooled incidence rate of 10 per 100,000 people over the age of 50 years old [1]. The prevalence in England has been shown to be rising, with increased numbers of people being investigated for suspected GCA and increased recognition of sight loss [2]. World-wide by 2050 over 3 million people will be expected to be diagnosed with GCA and half a million are predicted to have permanent vision loss [3].
Although there has been greater awareness of GCA in recent years, its varied presentation still leads to diagnostic uncertainty amongst healthcare professionals. As such clear diagnostic criteria, accessible specialist referral pathways and informative management guidelines are vital for prompt diagnosis and appropriate initiation of treatment [4,5,6,7,8].
Epidemiology
The incidence of GCA is higher in the northern hemisphere, with the highest incidence being recorded in Scandinavia of 21.6 per 100,000 people, as compared to the European incidence being 7.3 per 100,000 [1]. Epidemiology publications on the incidence in Olmsted County, USA which have been extrapolated to reflect the incidence in the USA, may have been an overestimate as the County have a higher portion of people with Scandinavian ancestry [1, 9]. Therefore, the geographical distribution is as expected strongly linked with genetic susceptibility [10,11,12]. GCA has been reliably associated with major histocompatibility complex molecules (i.e. HLA-DR3, HLA-DR4, HLA-DR5 and HLA-DRB1) particularly with carriage of HLA-DRB1*04 alleles [13]. GCA predominantly affects people ≥50 years of age, with rising prevalence in the context of an aging population and peak in the 7th decade [14]. Women are two and half times more likely to acquire the condition than men [1, 15].
Pathophysiology
Characterised by granulomatous infiltration, GCA is a product of inappropriate T cell migration and subsequent inflammatory cytokine release into the vascular adventitia. In the simplest terms the pathogenesis of the disease can be divided into a number of different stages. Following an unknown trigger there is vascular dendritic cell activation which causes activation and polarisation of CD4 + T cells [16, 17]. The pro-inflammatory cytokines shift T-cell differentiation towards Th17 and Th1 cells [18]. The Th17 cells are reliant on Interleukin (IL)-6 and produce IL-17 (amongst other interleukins); this cluster predominants early in GCA and fluctuates with disease activity. Importantly this cluster are highly responsive to standard glucorticoid therapy [19]. Whereas IL-12 and IL-18 induce Th1cells that release interferon (IFN)-y are associated with chronic disease and more resistant to glucorticoids [18,19,20].
Ageing processes and GCA
A number of risk factors for the development of GCA have been identified such as history of vascular disease, smoking, low body mass index and early menopause [21,22,23,24], however ageing has been found to be the strongest of all risk factors [24] (Fig. 1). GCA almost exclusively affects individuals aged 50 or older [14]. Vascular ageing may play a central role in the initial immune activation in GCA. Ageing has been known to make blood vessels vulnerable to damage and inflammation, with coining of the term “inflammaging” and atherosclerosis being described as a “prototypical form” of vascular ageing [25,26,27,28]. Multiple pathways are believed to contribute to vascular ageing, including oxidative stress, mitochondrial dysfunction, chronic low-grade inflammation, cellular senescence, increased apoptosis, epigenetic alterations, genomic instability, and clonal haematopoiesis of indeterminate potential (CHIP) [27, 29].

These can be cateogorised into vascular symptoms, visual signs, rheumatology and constitutional symptoms.
Population studies have observed that ageing is associated with chronically higher circulating levels of pro-inflammatory cytokines and inflammatory markers, namely, IL-6, IL-18, IL-1ra, C-reactive protein (CRP), and fibrinogen [30]. It is unclear whether these inflammatory markers are a product of ageing alone, as many studies have associated their increased prevalence with the presence of cardiovascular risk factors, which are particularly ubiquitous in elderly populations [30, 31]. IL-6 has been associated with clonal hematopoiesis of indeterminate potential (CHIP), a pre-malignant state characterised by somatic mutations in hematologic precursor cells is another potential pathogenetic mechanism potentially implicated in development of GCA [32]. The incidence of CHIP correlates with age, and it is associated with increased levels of CRP and other classic systemic inflammatory markers [33, 34]. Preliminary works exploring a potential correlation between CHIP and the development of GCA seem to corroborate this association [35].
Another theory is the potential role of somatic variants (SV) in GCA, as the number of SVs increases with ageing. SVs are postzygotic, mutations acquired during mitosis or after exposure to endogenous (i.e. products of cellular metabolism, reactive oxygen, and nitrogen species) or exogenous factors (i.e. ultraviolet light or radiation, tobacco, and alcohol), eventually leading to mosaicisms. SVs can render immune system cells resistant to apoptosis or change their functional profile (i.e. leading to aberrant cytokine secretion), causing high-inflammatory, non-proliferative (i.e. non-neoplastic) immune disorders [36].
The link between GCA and atherosclerosis remains ambiguous. Atherosclerosis has overlapping pathophysiology with GCA as cytolytic, proteolytic and reactive oxygen species are deposited in arterial adventitia, causing chronic low-grade inflammation, angiogenesis and fibrosis, subsequently leading to arterial remodelling [37]. The remodelling process is also characterised by T-cell, macrophage and mast cell migration into the adventitia, causing collagen breakdown by Matrix metalloproteinase 9, compromising the previously immunoprivileged arterial wall [38]. Vascular remodelling may occur early in atherosclerotic disease, indeed a study on porcine coronary arteries in the context of a high cholesterol diet found adventitial vasa vasorum remodelling through neovascularization occurred prior to atheromatous plaque formation [39]. Due to the shared pathological processes, one might predict that the presence of GCA or atherosclerosis could precipitate or accelerate the development of the other, however others found that GCA incidence inversely correlates with cardiovascular risk factors (obesity, smoking, hyperglycaemia, hypercholesterolaemia), and co-existent findings of GCA and atherosclerosis are rare on temporal artery biopsies [28, 40, 41]. The underlying protective mechanism of atherosclerosis and GCA currently remains unclear, however hyperglycaemia has been speculated to impair T-cell function, suppressing the inflammatory response in GCA [28].
Clinical presentation
GCA has heterogeneous clinical features due to the overlapping spectrum of the known clinical phenotypes: cranial GCA (C-GCA), large vessel GCA (LV-GCA), and polymyalgia rheumatica (PMR) (Fig. 2) [42]. The majority of people with cranial GCA will have symptoms of new onset headache, jaw claudication and cutaneous allodynia [8, 43, 44]. Nearly half of people with GCA have symptoms of PMR (Fig. 2) while up to one-fifth of people with PMR will be diagnosed with GCA [5, 42]. There may be large vessel involvement in cranial GCA, which may be asymptomatic and revealed by diagnostic imaging alone [45, 46]. Up to 50% of people with GCA will experience constitutional symptoms such as fever, weight loss, night sweats, loss of appetite, malaise, depression [16]. These may help narrow the differential diagnosis from an ocular cause or pain syndrome (such as migraine or cluster headache) to a systemic cause, however many systemic conditions have the potential to exhibit these symptoms (Table 1).

Risk factors for GCA.
The visual symptoms of GCA are well documented and include amaurosis fugax, double vision to devastating visual loss [47, 48]. The most common ocular manifestations include anterior ischaemic optic neuropathy [49], large peripapillary cotton wool spots [50, 51], arterial occlusions (cilio-retinal artery or central artery) [50, 52], oculomotor cranial nerve palsy [47], and posterior ischaemic optic neuropathy [50], with other rarer ocular syndromes having been reported (Table 2) [53,54,55,56,57,58,59,60,61]. Initiation of treatment may result in reversal of visual loss in the minority [62, 63].
Ophthalmologists need to beware of the less common presentations of GCA, as they may be asked to examine patients who are suspected of having GCA. For example, symptoms of LV-GCA include intermittent limb claudication or absent pulses according to the vessels affected and chest or back pain if there is aortic involvement [64]. GCA can present without any symptoms of cranial or large vessel involvement, with inflammation or fever of unknown origin (IFUO), anorexia, weight loss and anaemia being the only evidence of an active disease process. Patients with constitutional GCA are at risk of significant diagnostic delay due to the large differential diagnosis of IFUO [65, 66].
Confirming a diagnosis of GCA
GCA diagnosis is made on a clinical basis, in conjunction with laboratory, temporal artery biopsy (TAB), or vascular imaging evidence, as the clinical findings can help improve pre-test probability. A key challenge in urgent clinical practice is the heterogenic presentation of GCA and the wide differential of possible diagnoses [44, 67] (Table 1). Modern publications have suggested regression, neural networks, machine learning models, or clinical scoring systems however all of these rely on complete clinical information about the individual patient. Most of these tools are yet to be validated in larger, unbiased and well-proportioned datasets [68, 69].
There are currently no diagnostic criteria for GCA, however classification criteria such as the American College of Rheumatology (ACR) are often used inappropriately for the purpose of diagnosis [70]. Such classification criteria are for research purposes and exclude symptoms that are commonly found across multiple disease entities, focussing predominantly on signs and symptoms found solely in certain disease entities and not others. Key developments in the field of GCA have now been incorporated into the 2022 ACR/European League Against Rheumatism (EULAR) classification criteria for GCA which include the advancements in ultrasound and PET imaging (Table 3) [71]. The 2022 ACR/EULAR criteria have been validated for research purposes in the Diagnostic and Classification Criteria for Vasculitis (DCVAS) data set. This was across the disease spectrum (biopsy proven-GCA versus L-GCA) and in different populations of North America and Europe [71]. The previous 1990 ACR had good sensitivity and specificity of 93.5% and 91.2%, respectively, when differentiating C-GCA from other types of vasculitis, but performed poorly when used for diagnostic purposes [70]. Indeed, a retrospective case series has shown that 25.7% of patients with a positive TAB did not meet the 1990 ACR criteria, highlighting the that these criteria are not intended for diagnostic purposes [72].
Laboratory markers
There are currently no specific routine serological markers to definitively diagnose GCA. Commonly performed blood tests to identify an inflammatory state include CRP; erythrocyte sedimentation rate (ESR) or plasma viscosity (PV); and platelet count. None are specific, however used in combination they may provide more diagnostic certainty in combination with the clinical findings, vascular imaging, or TAB [15]. The difficulty is that most inflammatory and infective aetiologies share a similar biochemical profile (Table 1). In clinical practice certain tests may not be available in the local laboratory. For example, some labs have chosen not to perform ESR and instead offer plasma viscosity as it is not affected by haematocrit variations (e.g. anaemia or polycythaemia) nor affected by a delay in analysis. The challenge here is familiarity as most publications have evaluated ESR and not PV for the diagnosis of GCA. Another diagnostic dilemma is that the ESR and CRP values have been documented as normal in people with GCA [73,74,75]. A thrombocytosis >400,000/μL has shown to be beneficial at predicting a positive biopsy result [76]. Hence, the combination of ESR, CRP, and platelet count has been recommended to provide most useful biochemical information to predict GCA probability [15].
Temporal artery examination
Clinical examination of the temporal arteries by palpation is a critical assessment. Signs of abnormality include absent or diminished pulses, tenderness or a hard “cord-like” structure [16, 67]. Temporal artery biopsy (TAB) has long been held as the “gold-standard” investigation for GCA due to its ability to provide a histopathological tissue diagnosis, with reported specificities as high as 100% [77]. To estimate the sensitivity of unilateral TAB for the diagnosis of GCA a meta-analysis based on a large sample size found the sensitivity to be 77% [78]. There are a number of reasons for the reduced sensitivity including inadequate sample length, incorrect tissue sampled and the initiation of steroids prior to biopsy [5, 8, 16]. Another widely known factor limiting the sensitivity of TAB is the presence of skip lesions in GCA [79]. Skip lesions are estimated to be present in 8–26% of cases and therefore risk false negative results if biopsies are sampled from spared segments of arteries [80, 81]. To improve the sensitivity of detecting GCA some clinicians have advocated bilateral simultaneous TABs. However there has been a wide range of discordance rates between 3% to 45% found in people undergoing bilateral simultaneous TABs [82]. One option, where frozen section is available, is to perform a unilateral biopsy and if this is positive on frozen section it avoids the contralateral biopsy [82]. The practice of performing bilateral simultaneous TAB versus unilateral TAB is known to be different worldwide [83]. Ophthalmologists can be asked to perform TAB in a person without cranial symptoms and a clinically relevant finding is that a tertiary cohort study found that only 52% of patients with LV-GCA had positive TAB results, making the investigation relatively biased towards C-GCA [84].
There has been relatively little comment in the literature regarding the criteria for which histopathologists regard as positive for GCA and their agreement, until the advent of ultrasound [77]. Indeed, there are no internationally accepted criteria for a positive temporal artery biopsy [71]. Importantly when balancing the validity of TAB histopathology assessment versus the use of US to diagnose GCA one study reporting moderate agreement between 12 trained sonographers (κ= 0.61) assessing 20 ultrasound videos and 14 pathologists (κ = 0.62) assessing 30 TAB biopsy images [77].
Histopathological features of GCA include presence of giant cells, transmural evidence of mononuclear or granulomatous medial inflammation, internal elastic lamina fragmentation, necrosis, arterial mural thickening and/or intraluminal thrombosis. The concept of perivascular adventitial inflammation alone representing a spectrum of GCA pathology has sparked much debate in recent years [83]. Perivascular inflammation is restricted to the adventitia and periadventitial structures, and encompasses small vessel vasculitis, vasa vasorum vasculitis and inflammation limited to adventitia. It has been estimated to be present in 5–9% of positive TAB biopsies and has fair specificity ranging from 81.4 to 88.1% for GCA [82,83,84,85,86,87]. However, others have reported poor positive predictive values of perivascular inflammation, associating its occurrence with anomalies of ageing, systemic inflammation, malignancy and PMR phenotypes rather than relation to GCA directly [79, 88,89,90,91,92].
Temporal and axillary artery ultrasound
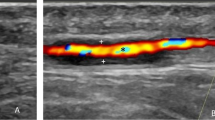
Over recent years, the availability and refinement of imaging services have improved in healthcare settings, with their rapid incorporation into diagnostic and interventional modalities for a multitude of pathologies. The same holds true for GCA; the EULAR currently recommend the use of temporal and axillary artery ultrasound (US) to confirm the diagnosis of new GCA cases, given the low invasiveness, rapid result availability, and comprehensive inflamed vessel visualisation of the imaging modality [45]. Temporal artery ultrasound has been found to be a cost-effective alternative to TAB in reducing false negatives, with US providing a £485 benefit per patient [77]. In situations where US is not available or has limited utility (e.g. thoracic aorta assessment), EULAR recommends the use of cross-sectional imaging such as MRI, CT and PET to aid GCA diagnosis in the first instance [45].
Four pathological signs are found by US in GCA: halo sign, compression sign, stenosis, and vessel occlusion [93]. When viewed using ultrasound, inflammatory tissue is hypoechoic, allowing a skilled sonographer to detect halo sign (hypoechoic artery wall thickening), and compression sign (hypoechoic vessel wall infiltrate in the presence of arterial lumen occlusion), which were initially reported to have similar sensitivity and specificity to TAB of 79% and 100%, respectively [94]. However, a recent meta-analysis comparing three GCA US signs (halo sign and temporal artery compression/stenosis) with temporal biopsy reported lower sensitivity and specificity of 68% and 81% respectively [95]. Adequate and structured training is an important consideration to improve the reliability of US in GCA diagnosis [96].
A single centre study has used halo sign thickness to develop Halo scores, which were associated with markers of systemic inflammation such as CRP, platelet count and haemoglobin, but not ESR [97]. Halo scores of ≥2 have been associated with ocular ischaemic events including anterior and posterior ischaemic optic neuropathy and the presence of a relative afferent pupillary defect (OR 12.00, p = 0.022), with scores of ≥10 conveying a specificity of 95% for GCA diagnosis, inferring their potential utility in diagnosis identifying those at risk of poor visual outcomes [97].
Ultrasonography holds great promise for diagnosing forms of GCA other than cranial. Temporal arteries can be spared in 40% of patients with LV-GCA, risking misdiagnosis when relied upon in isolation for diagnosis [98]. LV-GCA has been associated with delayed diagnosis and worse clinical outcome, with many requiring a higher cumulative glucocorticoid dose, and are at higher risk of relapse and aneurysm development [78, 99, 100]. Axillary artery involvement has been noted up to 98% in confirmed LV-GCA cases [99].
There is currently debate regarding the sensitivity of US in GCA diagnosis after starting glucocorticoid treatment. Some have found the sensitivity to decrease [92], others have found the majority of temporal and minimal numbers of axillary artery haloes take weeks to disappear [97, 101,102,103,104]. Conversely, TAB histological results remain positive for a prolonged period of time, with biopsies taken from 3 to 4 cm TAB segments showing persistent abnormal cell infiltrates in 70–75% of patients in the first 6 months, and 44% of patients within 9–12 months of starting corticosteroid therapy, making TAB the preferred investigation of choice in cases with significant delay in referral times [105]. Despite the evidence of persistent pathological features whilst receiving glucocorticoid therapy, clinicians are currently recommended to scan as early as feasible due to variability in patient response to glucocorticoid treatment [91]. There is a sizeable divergence of opinions on which test should be considered as the “gold standard” to diagnose GCA [83]. Many clinicians scrutinise the value TAB and US as separate entities, however the paradigm of one-test-to-diagnose-them-all might be considered a myopic standpoint. Appreciation for the individual test’s strengths and weaknesses, in combination with comprehensive history taking and examinations are fundamental in the work up of GCA [7, 106,107,108,109].
Rapid access GCA pathways
Early glucocorticoid treatment is associated with improved ophthalmological outcomes, with diagnostic delays risking ophthalmic ischaemic events [110]. Cranial presentations accrue a mean diagnostic delay of 7.7 weeks, with non-cranial presentations receiving longer delays of 17.6 weeks, risking permanent visual loss if glucocorticoids have not been initiated, and difficulty detecting diagnostic pathological features if they have [111].
Fast-track GCA referral pathways utilising rapid access to specialist assessment and imaging modalities within one working day were first recommended internationally for GCA diagnosis by EULAR in 2018 but have been employed by institutions since 1997 [45, 93]. The introduction of fast-track referral services have been shown to decrease rates of permanent vision loss and reduced diagnostic delay [112,113,114]. Fast track services have been shown to reduce the need for TAB by up to 93%, with the majority of TAB being performed due to inconclusive US findings [113]. Such pathways have undergone refinement since their inception. A pre-test probability score has been developed which allows risk-categorisation and algorithmic processing of referrals and has shown promise in its ability to identify non-GCA referrals, reporting a sensitivity of 100% and specificity of 48.2% in patients scored as “low risk” (≤9 points) [115]. Validation of this score is currently in early stages, however it could prove a helpful tool for referrers or diagnosticians if further studies corroborate its use. A number of UK National Health Service Trusts have fast track services, and further work is required to make these the standard of care.
Management of GCA
All treatment recommendations are well considered in the recent guidelines such as the EULAR and British Society of Rheumatology guidelines [4, 5]. High dose glucocorticoids should be started once GCA is suspected [4, 5, 8]. In a randomised control trial use of intravenous methylprednisolone versus placebo in the first 3 days of treatment in combination with oral prednisolone 40 mg/day observed faster glucocorticoid taper, reduced cumulative glucocorticoid dosing and fewer relapses in the methylprednisolone arm, as compared to placebo [116]. It is worthy of note that those with visual loss were excluded from this trial [116]. Guideline groups have debated the use of intravenous glucocorticoids for initiation of therapy but there is a lack of good evidence to conclusively recommend their mandated used [4, 5]. Relative contraindications to intravenous glucocorticoid therapy may include uncontrolled hyperglycaemia, diabetes mellitus, osteoporosis and facture, and other medical conditions that the clinician would need to weigh up the relevance such as a recent history of pancreatitis, uncontrolled mental health disorders, or congestive heart failure [5, 117, 118].
The burden of side effects, and their management, from long-term glucocorticoids in GCA are well known [119,120,121]. Gradual and controlled glucocorticoid reduction is imperative, in order to balance the risk of flare or relapse versus the risk of metabolic side effects related to their use [119,120,121]. However, there is a mismatch between recommended steroid tapering regimens and real world data, where cumulative doses of glucocorticoids have been three times higher than recommended [122, 123].
A number of different conventional synthetic and biologic disease-modifying anti-rheumatic drugs (DMARD) have been trialled in GCA [67, 124]. Low-dose methotrexate demonstrated a modest reduction in relapse and cumulative glucocorticoid dose at meta-analysis [125], and is used routinely in clinical practice in the UK and Europe [4, 5]. Leflunomide, another conventional synthetic DMARD suppresses the production of pro-inflammatory cytokines through the activation of dendritic cells and modifies the action of the T-cell response in GCA [126]. A number of studies support its use, but as of yet there is no randomised controlled evidence for its directed use [127,128,129,130].
There remains no conclusive evidence from controlled trials to determine the safety and efficacy of low-dose aspirin as an adjunctive treatment in GCA. A portion of people with GCA will be on aspirin at time of the diagnosis, and aspirin does not need to be discontinued [5]. There is not good enough evidence to consider the use of low-dose aspirin as an adjunctive treatment for GCA and clinicians must recognise the established haemorrhagic risks associated with aspirin, especially in the context of concurrent treatment with glucocorticoids [131].
Targeted treatment with subcutaneous Tocilizumab (TCZ) has shown significant glucocorticoid-sparing effects in new-onset and relapsing patients with GCA [132,133,134]. TCZ is a monoclonal antibody directed against the IL-6 receptor that inhibits signalling by the pro-inflammatory cytokine IL-6 [135]. In the landmark study Giant-Cell Arteritis Actemra (GiACTA), 249 patients with new onset GCA or refractory disease were enrolled and randomised to one of four arms: weekly (TCZ QW) or fortnightly (TCZ 2QW) dosing of TCZ with a 26-week prednisone taper or placebo plus a 26-week or 52-week prednisone taper. At 52 weeks, patients in the TCZ groups were significantly more likely to have achieved sustained remission as compared with both the 26-week and 52-week glucocorticoid taper groups, and at just over half the cumulative glucocorticoid dose [133]. The data from the GiACTA at 52 weeks showed that the outcomes of patients with new-onset disease at baseline who were randomly assigned to TCZ Q2W did not clearly differ from the outcomes of patients who received TCZ QW, and TCZ QW was more effective than TCZ Q2W for these outcomes in patients with relapsing disease at baseline [133]. At 3 years following the GiACTA trial and open label follow-up, treatment once weekly (TCZ QW) overall delayed time to flare and reduced glucocorticoid exposure in patients with both new-onset and relapsing GCA as compared to those treated TCZ 2QW [136, 137].
Replicating the GiACTA trial results in the real world may be challenging as both placebo arms had a significantly faster glucocorticoid taper than used in routine clinical practice, and one third of patients had a diagnosis of GCA based on large vessel imaging, not ultrasound [133]. It is important also to note that use of intravenous glucocorticoid therapy was a specific exclusion criterion in GiACTA, which would have negatively biased against enrolling those with visual loss. Delivery of TCZ in the UK is devolved to the four nations [138, 139], and new data from Scotland possibly suggests under-utilisation of TCZ in terms of those with relapsing disease or those with high-risk comorbidities [140]. Conway et al. [141] thoughtfully discuss the Scottish findings in the context of prescribing confidence of a newly licensed biological agent and on the backdrop of the COVID-19 pandemic which changed many GCA pathways and practices [142, 143].
Visual outcomes in GCA
Visual outcomes in those with visual loss secondary to GCA is poor, with little chance of recovery [62, 144]. While intravenous methylprednisolone has been used since the 1990s, there is not good enough evidence to determine if it actually prevents visual loss. One early case series concluded that use of intravenous methylprednisolone was no better than using high dose oral prednisone [145]. In another study where patients were treated with a standard protocol of 1 g of intravenous methylprednisolone daily for 3 days followed by oral prednisone 60 or 80 mg (depending on patient weight), visual deterioration was noted in 27% of eyes, with the greatest risk of deterioration observed within the first 6 days [62].
An enduring clinical concern in immediate or long-term follow-up is what is the ongoing risk of visual loss in a person with GCA and concurrent treatment, and when treatment has ceased. A recent longitudinal study found the incidence of permanent visual loss to be around 2.2%, which was corroborated by the pooled incidence in the literature of 2.8% [146]. Those at risk are people with an established ischaemic event (such as contralateral visual loss from anterior ischaemic optic neuropathy), and it appears that the risk may be higher at initiation of treatment but can also occur when glucocorticoids are tapered. More data is required to understand the beneficial impact of targeted therapies, such as TCZ on rates of visual complications [63, 133, 147].
Conclusion
In this global health care environment different attitudes, variable access to medicines and what is recommended by country specific guidelines informs the clinician and indeed the literature moving forward [4,5,6]. The challenge for Ophthalmologists who routinely investigate and manage GCA, is whether they have optimised their treatment for each individual patient whether it be early in the disease or further down the line. The importance of collaborative working with Rheumatology specialists, who have in depth experience of second line therapies, cannot be over-estimated [5, 67]. Targeted treatment remains an individualised approach that is required to balance the burden of treatment against its effectiveness at reducing relapses and inducing sustained remission [148]. There remain many unanswered questions, particularly pertaining to visual loss in GCA and whether this can be minimised or ideally even prevented.
References
Li KJ, Semenov D, Turk M, Pope J. A meta-analysis of the epidemiology of giant cell arteritis across time and space. Arthritis Res Ther. 2021;23:82.
Mollan SP, Begaj I, Mackie S, O’Sullivan EP, Denniston AK. Increase in admissions related to giant cell arteritis and polymyalgia rheumatica in the UK, 2002-13, without a decrease in associated sight loss: potential implications for service provision. Rheumatol (Oxf). 2015;54:375–7.
De Smit E, Palmer AJ, Hewitt AW. Projected Worldwide Disease Burden from Giant Cell Arteritis by 2050. J Rheumatol. 2015;42:119–25.
Hellmich B, Agueda A, Monti S, Buttgereit F, de Boysson H, Brouwer E, et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis. 2020;79:19–30.
Mackie SL, Dejaco C, Appenzeller S, Camellino D, Duftner C, Gonzalez-Chiappe S, et al. British Society for Rheumatology guideline on diagnosis and treatment of giant cell arteritis. Rheumatology 2020;59:e1–e23.
Maz M, Chung SA, Abril A, Langford CA, Gorelik M, Guyatt G, et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Giant Cell Arteritis and Takayasu Arteritis. Arthritis Rheumatol. 2021;73:1349–65.
Mollan SP, Grech O, O’Sullivan E, Mackie SL. Practice points for ophthalmologists from the 2020 British Society for Rheumatology Giant Cell Arteritis guidelines. Eye 2021;35:699–701.
Mollan SP, Paemeleire K, Versijpt J, Luqmani R, Sinclair AJ. European Headache Federation recommendations for neurologists managing giant cell arteritis. J Headache Pain. 2020;21:28.
Chandran A, Udayakumar P, Crowson C, Warrington K, Matteson E. The incidence of giant cell arteritis in Olmsted County, Minnesota, over a 60-year period 1950–2009. Scand J Rheumatol. 2015;44:215–8.
Borchers AT, Gershwin ME. Giant cell arteritis: a review of classification, pathophysiology, geoepidemiology and treatment. Autoimmun Rev. 2012;11:A544–54.
Carmona FD, Vaglio A, Mackie SL, Hernández-Rodríguez J, Monach PA, Castañeda S, et al. A Genome-wide Association Study Identifies Risk Alleles in Plasminogen and P4HA2 Associated with Giant Cell Arteritis. Am J Hum Genet. 2017;100:64–74.
Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. The HLA-DRB1 locus as a genetic component in giant cell arteritis. Mapping of a disease-linked sequence motif to the antigen binding site of the HLA-DR molecule. J Clin Investig. 1992;90:2355–61.
Carmona FD, González-Gay MA, Martín J. Genetic component of giant cell arteritis. Rheumatol (Oxf). 2014;53:6–18.
Gonzalez-Gay MA, Vazquez-Rodriguez TR, Lopez-Diaz MJ, Miranda-Filloy JA, Gonzalez-Juanatey C, Martin J, et al. Epidemiology of giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatism. 2009;61:1454–61.
Van Der Geest KSM, Sandovici M, Brouwer E, Mackie SL. Diagnostic Accuracy of Symptoms, Physical Signs, and Laboratory Tests for Giant Cell Arteritis. JAMA Intern Med. 2020;180:1295.
Al-Mousawi AZ, Gurney SP, Lorenzi AR, Pohl U, Dayan M, Mollan SP. Reviewing the Pathophysiology Behind the Advances in the Management of Giant Cell Arteritis. Ophthalmol Ther. 2019;8:177–93.
Ma-Krupa W, Jeon M-S, Spoerl S, Tedder TF, Goronzy JRJ, Weyand CM. Activation of Arterial Wall Dendritic Cells and Breakdown of Self-tolerance in Giant Cell Arteritis. J Exp Med. 2004;199:173–83.
Deng J, Younge BR, Olshen RA, Goronzy JRJ, Weyand CM. Th17 and Th1 T-Cell Responses in Giant Cell Arteritis. Circulation 2010;121:906–15.
Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. 2013;9:731–40.
Dejaco C, Brouwer E, Mason JC, Buttgereit F, Matteson EL, Dasgupta B. Giant cell arteritis and polymyalgia rheumatica: current challenges and opportunities. Nat Rev Rheumatol. 2017;13:578–92.
Larsson K, Mellström D, Nordborg C, Odén A, Nordberg E. Early menopause, low body mass index, and smoking are independent risk factors for developing giant cell arteritis. Ann Rheum Dis. 2006;65:529–32.
Li L, Neogi T, Jick S. Giant cell arteritis and vascular disease-risk factors and outcomes: a cohort study using UK Clinical Practice Research Datalink. Rheumatol (Oxf). 2017;56:753–62.
Weyand CM, Goronzy JJ. Giant-Cell Arteritis and Polymyalgia Rheumatica. N Engl J Med. 2014;371:50–7.
Weyand CM, Liao YJ, Goronzy JJ. The Immunopathology of Giant Cell Arteritis. J Neuro-Ophthalmol. 2012;32:259–65.
Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505–22.
Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14:576–90.
Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of Vascular Aging. Circ Res. 2018;123:849–67.
Watanabe R, Hashimoto M. Aging-Related Vascular Inflammation: Giant Cell Arteritis and Neurological Disorders. Front Aging Neurosci. 2022;14:843305.
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377:111–21.
Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub DD, et al. The origins of age-related proinflammatory state. Blood 2005;105:2294–9.
Bermudez EA, Rifai N, Buring J, Manson JE, Ridker PM. Interrelationships Among Circulating Interleukin-6, C-Reactive Protein, and Traditional Cardiovascular Risk Factors in Women. Arterioscler Thromb Vasc Biol. 2002;22:1668–73.
Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol. 2021;18:58–68.
Jaiswal S. Clonal hematopoiesis and nonhematologic disorders. Blood 2020;136:1606–14.
Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science 2019;366:eaan4673.
Papo M, Friedrich C, Delaval L, Boysson H, Viallard JF, Bachmeyer C, et al. Myeloproliferative neoplasms and clonal haematopoiesis in patients with giant cell arteritis: a case-control and exploratory study. Rheumatol (Oxf). 2022;61:775–80.
Van Horebeek L, Dubois B, Goris A. Somatic Variants: New Kids on the Block in Human Immunogenetics. Trends Genet. 2019;35:935–47.
Michel J-B, Thaunat O, Houard X, Meilhac O, Caligiuri G, Nicoletti A. Topological Determinants and Consequences of Adventitial Responses to Arterial Wall Injury. Arterioscler Thromb Vasc Biol. 2007;27:1259–68.
Hollestelle SCG, De Vries MR, Van Keulen JK, Schoneveld AH, Vink A, Strijder CF, et al. Toll-Like Receptor 4 Is Involved in Outward Arterial Remodeling. Circulation 2004;109:393–8.
Kwon HM, Sangiorgi G, Ritman EL, McKenna C, Holmes DR, Schwartz RS, et al. Enhanced coronary vasa vasorum neovascularization in experimental hypercholesterolemia. J Clin Investig. 1998;101:1551–6.
Tomasson G, Bjornsson J, Zhang Y, Gudnason V, Merkel P. Cardiovascular risk factors and incident giant cell arteritis: a population-based cohort study. Scand J Rheumatol. 2019;48:213–7.
Wadström K, Jacobsson L, Mohammad AJ, Warrington KJ, Matteson EL, Turesson C. Negative associations for fasting blood glucose, cholesterol and triglyceride levels with the development of giant cell arteritis. Rheumatol (Oxf). 2020;59:3229–36.
Kaushik M, Ponte C, Mollan SP. Current advances in giant cell arteritis. Curr Opin Neurol. 2021;34:133–41.
Gajree S, Borooah S, Dhillon N, Goudie C, Smith C, Aspinall P, et al. Temporal artery biopsies in south-east Scotland: a five year review. J R Coll Physicians Edinb. 2017;47:124–8.
Mollan SP, Virdee JS, Bilton EJ, Thaller M, Krishan A, Sinclair AJ. Headache for ophthalmologists: current advances in headache understanding and management. Eye 2021;35:1574–86.
Dejaco C, Ramiro S, Duftner C, Besson FL, Bley TA, Blockmans D, et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann Rheum Dis. 2018;77:636–43.
Henry IM, Fernández Fernández E, Peiteado D, Balsa A, de Miguel E. Diagnostic validity of ultrasound including extra-cranial arteries in giant cell arteritis. Clin Rheumatol. 2022. https://doi.org/10.1007/s10067-022-06420-8.
Castillejo Becerra CM, Crowson CS, Koster MJ, Warrington KJ, Bhatti MT, Chen JJ. Population-based Rate and Patterns of Diplopia in Giant Cell Arteritis. Neuroophthalmology. 2022;46:75–9.
Denniston AK, Murray P. Oxford Handbook of Ophthalmology. 3 ed. Oxford: Oxford University Press; 2014.
Salvarani C, Cimino L, Macchioni P, Consonni D, Cantini F, Bajocchi G, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum. 2005;53:293–7.
Hayreh SS, Podhajsky PA, Zimmerman B. Ocular manifestations of giant cell arteritis. Am J Ophthalmol. 1998;125:509–20.
Virdee J, Horsburgh J, Roque M, Mollan S. A giant escape. Postgrad Med J. 2019;95:346.
Singh AG, Kermani TA, Crowson CS, Weyand CM, Matteson EL, Warrington KJ. Visual Manifestations in Giant Cell Arteritis: Trend over 5 Decades in a Population-based Cohort. J Rheumatol. 2015;42:309–15.
Barricks ME, Traviesa DB, Glaser JS, Levy IS. Ophthalmoplegia in cranial arteritis. Brain 1977;100:209–21.
Bayar SA, Gokmen O, Pinarci EY, Altinors DD, Gedik S. Corneal endothelial decompansation and ocular hypotony in a case with temporal arteritis. J Neuroophthalmol. 2012;32:385.
Cavallini GM, Volante V, Bigliardi MC, Mascia MT, Forlini M. Bilateral posterior scleritis as a presenting manifestation of giant cell arteritis: A case report. Can J Ophthalmol. 2014;49:e141–3.
Foroozan R, Buono LM, Savino PJ, Sergott RC. Tonic pupils from giant cell arteritis. Br J Ophthalmol. 2003;87:510–2.
Huna-Baron R, Mizrachi IB, Glovinsky Y. Intraocular pressure is low in eyes with giant cell arteritis. J Neuroophthalmol. 2006;26:273–5.
Kopsachilis N, Pefkianaki M, Marinescu A, Sivaprasad S. Giant Cell Arteritis Presenting as Choroidal Infarction. Case Rep Ophthalmol Med. 2013;2013:1–3.
Papathanassiou M, Elezoglu A, Nikita E, Theodossiadis PG, Vergados I. A rare case of peripheral ulcerative keratitis in temporal arteritis. Eur J Ophthalmol. 2009;19:866–9.
Prasad S, Baccon J, Galetta SL. Mydriatic pupil in giant cell arteritis. J Neurol Sci. 2009;284:196–7.
Radda TM, Bardach H, Riss B. Acute ocular hypotony. A rare complication of temporal arteritis. Ophthalmologica 1981;182:148–52.
Danesh-Meyer H, Savino PJ, Gamble GG. Poor prognosis of visual outcome after visual loss from giant cell arteritis. Ophthalmology 2005;112:1098–103.
Svasti-Salee CR, Mollan SP, Morgan AW, Quick V. Rapid visual recovery following intravenous tocilizumab in glucocorticoid resistant refractory giant cell arteritis. BMJ Case Rep. 2019;12:e229236.
Dejaco C, Duftner C, Buttgereit F, Matteson EL, Dasgupta B. The spectrum of giant cell arteritis and polymyalgia rheumatica: revisiting the concept of the disease. Rheumatol (Oxf). 2017;56:506–15.
de Boysson H, Liozon E, Ly KH, Dumont A, Delmas C, Aouba A. The different clinical patterns of giant cell arteritis. Clin Exp Rheumatol 2019;37:57–60.
Desmet GD, Knockaert DC, Bobbaers HJ. Temporal arteritis: the silent presentation and delay in diagnosis. J Intern Med. 1990;227:237–40.
Lyons HS, Quick V, Sinclair AJ, Nagaraju S, Mollan SP. A new era for giant cell arteritis. Eye 2020;34:1013–26.
Ing EB, Miller NR, Nguyen A, Su W, Bursztyn LL, Poole M, et al. Neural network and logistic regression diagnostic prediction models for giant cell arteritis: development and validation. Clin Ophthalmol. 2019;13:421–30.
Laskou F, Coath F, Mackie SL, Banerjee S, Aung T, Dasgupta B. A probability score to aid the diagnosis of suspected giant cell arteritis. Clin Exp Rheumatol. 2019;37:104–8.
Hunder GG, Bloch DA, Michel BA, Stevens MB, Arend WP, Calabrese LH, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33:1122–8.
Grayson PC, Ponte C, Suppiah R, Robson JC, Gribbons KB, Judge A, et al. 2022 American College of Rheumatology/EULAR classification criteria for Takayasu arteritis. Ann Rheum Dis. 2022;81:1654–60.
Murchison AP, Gilbert ME, Bilyk JR, Eagle RC Jr, Pueyo V, Sergott RC, et al. Validity of the American College of Rheumatology criteria for the diagnosis of giant cell arteritis. Am J Ophthalmol. 2012;154:722–9.
Kermani TA, Schmidt J, Crowson CS, Ytterberg SR, Hunder GG, Matteson EL, et al. Utility of erythrocyte sedimentation rate and C-reactive protein for the diagnosis of giant cell arteritis. Semin Arthritis Rheum. 2012;41:866–71.
Laria A, Zoli A, Bocci M, Castri F, Federico F, Ferraccioli GF. Systematic review of the literature and a case report informing biopsy-proven giant cell arteritis (GCA) with normal C-reactive protein. Clin Rheumatol. 2012;31:1389–93.
Zweegman S, Makkink B, Stehouwer CD. Giant-cell arteritis with normal erythrocyte sedimentation rate: case report and review of the literature. Neth J Med. 1993;42:128–31.
Walvick MD, Walvick MP. Giant cell arteritis: laboratory predictors of a positive temporal artery biopsy. Ophthalmology 2011;118:1201–4.
Luqmani R, Lee E, Singh S, Gillett M, Schmidt WA, Bradburn M, et al. The Role of Ultrasound Compared to Biopsy of Temporal Arteries in the Diagnosis and Treatment of Giant Cell Arteritis (TABUL): a diagnostic accuracy and cost-effectiveness study. Health Technol Assess. 2016;20:1–238.
Rubenstein E, Maldini C, Gonzalez-Chiappe S, Chevret S, Mahr A. Sensitivity of temporal artery biopsy in the diagnosis of giant cell arteritis: a systematic literature review and meta-analysis. Rheumatology 2020;59:1011–20.
Chakrabarty AF. AJ Temporal artery biopsy: is there any value in examining biopsies at multiple levels? J Clin Pathol. 2000;53:131–6.
Klein RG, Campbell RJ, Hunder GG, Carney JA. Skip lesions in temporal arteritis. Mayo Clin Proc. 1976;51:504–10.
Poller DN, van Wyk Q, Jeffrey MJ. The importance of skip lesions in temporal arteritis. J Clin Pathol. 2000;53:137–9.
Cohen DA, Chen JJ, Neth BJ, Sabbagh N, Hodge D, Warrington KJ, et al. Discordance Rate Among Bilateral Simultaneous and Sequential Temporal Artery Biopsies in Giant Cell Arteritis: Role of Frozen Sectioning Based on the Mayo Clinic Experience. JAMA Ophthalmol. 2021;139:406–13.
Lee AG, Sinclair AJ, Sadaka A, Berry S, Mollan SP. Neuro-Ophthalmology: global trends in diagnosis, treatment and management. Springer; Switzerland; 2019.
Muratore F, Kermani TA, Crowson CS, Green AB, Salvarani C, Matteson EL, et al. Large-vessel giant cell arteritis: a cohort study. Rheumatology. 2015;54:463–70.
Galli E, Muratore F, Boiardi L, Restuccia G, Cavazza A, Catanoso M, et al. Significance of inflammation restricted to adventitial/periadventitial tissue on temporal artery biopsy. Semin Arthritis Rheum. 2020;50:1064–72.
Cavazza A, Muratore F, Boiardi L, Restuccia G, Pipitone N, Pazzola G, et al. Inflamed Temporal Artery: Histologic Findings in 354 Biopsies, With Clinical Correlations. Am J Surg Pathol. 2014;38:1360–70.
Hernández-Rodríguez J, Murgia G, Villar I, Campo E, Mackie SL, Chakrabarty A, et al. Description and Validation of Histological Patterns and Proposal of a Dynamic Model of Inflammatory Infiltration in Giant-cell Arteritis. Med (Baltim). 2016;95:e2368.
Chatelain D, Duhaut P, Loire R, Bosshard S, Pellet H, Piette JC, et al. Small-vessel vasculitis surrounding an uninflamed temporal artery: a new diagnostic criterion for polymyalgia rheumatica? Arthritis Rheum. 2008;58:2565–73.
Corcoran GM, Prayson RA, Herzog KM. The significance of perivascular inflammation in the absence of arteritis in temporal artery biopsy specimens. Am J Clin Pathol. 2001;115:342–7.
Jia L, Couce M, Barnholtz-Sloan JS, Cohen ML. Is all inflammation within temporal artery biopsies temporal arteritis? Hum Pathol. 2016;57:17–21.
Le Pendu C, Meignin V, Gonzalez-Chiappe S, Hij A, Galateau-Sallé F, Mahr A. Poor Predictive Value of Isolated Adventitial and Periadventitial Infiltrates in Temporal Artery Biopsies for Diagnosis of Giant Cell Arteritis. J Rheumatol. 2017;44:1039–43.
Restuccia G, Cavazza A, Boiardi L, Pipitone N, Macchioni P, Bajocchi G, et al. Small-vessel vasculitis surrounding an uninflamed temporal artery and isolated vasa vasorum vasculitis of the temporal artery: Two subsets of giant cell arteritis. Arthritis Rheumatism. 2012;64:549–56.
Schmidt WA. Ultrasound in the diagnosis and management of giant cell arteritis. Rheumatol (Oxf). 2018;57:ii22–ii31.
Aschwanden M, Daikeler T, Kesten F, Baldi T, Benz D, Tyndall A, et al. Temporal Artery Compression Sign - A Novel Ultrasound Finding for the Diagnosis of Giant Cell Arteritis. Ultraschall der Med - Eur J Ultrasound 2012;34:47–50.
Rinagel M, Chatelus E, Jousse-Joulin S, Sibilia J, Gottenberg JE, Chasset F, et al. Diagnostic performance of temporal artery ultrasound for the diagnosis of giant cell arteritis: a systematic review and meta-analysis of the literature. Autoimmun Rev. 2019;18:56–61.
Chrysidis S, Terslev L, Christensen R, Fredberg U, Larsen K, Lorenzen T, et al. Vascular ultrasound for the diagnosis of giant cell arteritis: a reliability and agreement study based on a standardised training programme. RMD Open. 2020;6:e001337.
Van Der Geest KSM, Borg F, Kayani A, Paap D, Gondo P, Schmidt W, et al. Novel ultrasonographic Halo Score for giant cell arteritis: assessment of diagnostic accuracy and association with ocular ischaemia. Ann Rheum Dis. 2020;79:393–9.
Brack A, Martinez-Taboada V, Stanson A, Goronzy JJ, Weyand CM. Disease pattern in cranial and large-vessel giant cell arteritis. Arthritis Rheum. 1999;42:311–7.
Koster MJ, Matteson EL, Warrington KJ. Large-vessel giant cell arteritis: diagnosis, monitoring and management. Rheumatology 2018;57:ii32–ii42.
Nuenninghoff DM, Hunder GG, Christianson TJ, McClelland RL, Matteson EL. Incidence and predictors of large-artery complication (aortic aneurysm, aortic dissection, and/or large-artery stenosis) in patients with giant cell arteritis: a population-based study over 50 years. Arthritis Rheum. 2003;48:3522–31.
Aschwanden M, Schegk E, Imfeld S, Staub D, Rottenburger C, Berger CT, et al. Vessel wall plasticity in large vessel giant cell arteritis: an ultrasound follow-up study. Rheumatol (Oxf). 2019;58:792–7.
De Miguel E, Roxo A, Castillo C, Peiteado D, Villalba A, Martín-Mola E. The utility and sensitivity of colour Doppler ultrasound in monitoring changes in giant cell arteritis. Clin Exp Rheumatol. 2012;30:S34–8.
Karahaliou M, Vaiopoulos G, Papaspyrou S, Kanakis MA, Revenas K, Sfikakis PP. Arthritis Research & Therapy. 2006;8:R116.
Schmidt WA, Kraft HE, Vorpahl K, Völker L, Gromnica-Ihle EJ. Color Duplex Ultrasonography in the Diagnosis of Temporal Arteritis. N Engl J Med. 1997;337:1336–42.
Maleszewski JJ, Younge BR, Fritzlen JT, Hunder GG, Goronzy JJ, Warrington KJ, et al. Clinical and pathological evolution of giant cell arteritis: a prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod Pathol. 2017;30:788–96.
Croft AP, Thompson N, Duddy MJ, Barton C, Khattak F, Mollan SP, et al. Cranial ultrasound for the diagnosis of giant cell arteritis. A retrospective cohort study. J R Coll Physicians Edinb. 2015;45:268–72.
Mackie S, Neill L, Byrne D, Mollan S, Dasgupta B, Dejaco C. Comment on: British Society for Rheumatology guideline on diagnosis and treatment of giant cell arteritis: reply. Rheumatol (Oxf). 2020;59:e163–e4.
Mackie SL, Dejaco C, Appenzeller S, Camellino D, Duftner C, Gonzalez-Chiappe S, et al. British Society for Rheumatology guideline on diagnosis and treatment of giant cell arteritis: executive summary. Rheumatol (Oxf). 2020;59:487–94.
Mollan SP, Mackie SL. British Society for Rheumatology guideline for diagnosis and treatment of giant cell arteritis. Pr Neurol. 2020;20:474–5.
Héron E, Sedira N, Dahia O, Jamart C. Ocular Complications of Giant Cell Arteritis: An Acute Therapeutic Emergency. J Clin Med. 2022;11:1997.
Prior JA, Ranjbar H, Belcher J, Mackie SL, Helliwell T, Liddle J, et al. Diagnostic delay for giant cell arteritis – a systematic review and meta-analysis. BMC Med. 2017;15:120.
Diamantopoulos AP, Haugeberg G, Lindland A, Myklebust G. The fast-track ultrasound clinic for early diagnosis of giant cell arteritis significantly reduces permanent visual impairment: towards a more effective strategy to improve clinical outcome in giant cell arteritis? Rheumatol (Oxf). 2016;55:66–70.
Monti S, Bartoletti A, Bellis E, Delvino P, Montecucco C. Fast-Track Ultrasound Clinic for the Diagnosis of Giant Cell Arteritis Changes the Prognosis of the Disease but Not the Risk of Future Relapse. Front Med (Lausanne). 2020;7:589794.
Patil P, Williams M, Maw WW, Achilleos K, Elsideeg S, Dejaco C, et al. Fast track pathway reduces sight loss in giant cell arteritis: results of a longitudinal observational cohort study. Clin Exp Rheumatol. 2015;33:S-103-6.
Sebastian A, Tomelleri A, Kayani A, Prieto-Pena D, Ranasinghe C, Dasgupta B. Probability-based algorithm using ultrasound and additional tests for suspected GCA in a fast-track clinic. RMD Open. 2020;6:e001297.
Mazlumzadeh M, Hunder GG, Easley KA, Calamia KT, Matteson EL, Griffing WL, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum. 2006;54:3310–8.
Seneviratne DR, Mollan SP, Elsherbiny S, Worstmann T. Giant cell arteritis complicated by acute pancreatitis: a case report. J Med Case Rep. 2008;2:346.
Mukhtyar C, Cate H, Graham C, Merry P, Mills K, Misra A, et al. Development of an evidence-based regimen of prednisolone to treat giant cell arteritis - the Norwich regimen. Rheumatol Adv Pract. 2019;3:rkz001.
Duftner C, Dejaco C, Sepriano A, Falzon L, Schmidt WA, Ramiro S. Imaging in diagnosis, outcome prediction and monitoring of large vessel vasculitis: a systematic literature review and meta-analysis informing the EULAR recommendations. RMD Open. 2018;4:e000612.
Kermani TA, Sreih AG, Cuthbertson D, Carette S, Hoffman GS, Khalidi NA, et al. Evaluation of damage in giant cell arteritis. Rheumatol (Oxf). 2018;57:322–8.
Wilson JC, Sarsour K, Collinson N, Tuckwell K, Musselman D, Klearman M, et al. Incidence of outcomes potentially associated with corticosteroid therapy in patients with giant cell arteritis. Semin Arthritis Rheum. 2017;46:650–6.
Dasgupta B. Concise guidance: diagnosis and management of giant cell arteritis. Clin Med (Lond). 2010;10:381–6.
Petri H, Nevitt A, Sarsour K, Napalkov P, Collinson N. Incidence of giant cell arteritis and characteristics of patients: data-driven analysis of comorbidities. Arthritis Care Res (Hoboken). 2015;67:390–5.
Piccus R, Hansen MS, Hamann S, Mollan SP. An update on the clinical approach to giant cell arteritis. Clin Med (Lond). 2022;22:107–11.
Mahr AD, Jover JA, Spiera RF, Hernández-García C, Fernández-Gutiérrez B, Lavalley MP, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. 2007;56:2789–97.
Hočevar A, Ješe R, Rotar Ž, Tomšič M. Does leflunomide have a role in giant cell arteritis? An open-label study. Clin Rheumatol. 2019;38:291–6.
Adizie T, Christidis D, Dharmapaliah C, Borg F, Dasgupta B. Efficacy and tolerability of leflunomide in difficult-to-treat polymyalgia rheumatica and giant cell arteritis: a case series. Int J Clin Pr. 2012;66:906–9.
Diamantopoulos AP, Hetland H, Myklebust G. Leflunomide as a corticosteroid-sparing agent in giant cell arteritis and polymyalgia rheumatica: a case series. Biomed Res Int. 2013;2013:120638.
Kramarič J, Rotar Ž, Tomšič M, Hočevar A. Performance of leflunomide as a steroid-sparing agent in giant cell arteritis: A single-center, open-label study. Front Med (Lausanne). 2022;9:1069013.
Tengesdal S, Diamantopoulos AP, Myklebust G. Leflunomide versus methotrexate in treatment of giant cell arteritis: comparison of efficacy, safety, and drug survival. Scand J Rheumatol. 2019;48:333–5.
Mollan SP, Sharrack N, Burdon MA, Denniston AK. Aspirin as adjunctive treatment for giant cell arteritis. Cochrane Database Syst Rev. 2014;3:Cd010453.
Antonio AA, Santos RN, Abariga SA. Tocilizumab for giant cell arteritis. Cochrane Database Syst Rev. 2021;8:Cd013484.
Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of Tocilizumab in Giant-Cell Arteritis. N Engl J Med. 2017;377:317–28.
Villiger PM, Adler S, Kuchen S, Wermelinger F, Dan D, Fiege V, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2016;387:1921–7.
Mollan SP, Horsburgh J, Dasgupta B. Profile of tocilizumab and its potential in the treatment of giant cell arteritis. Eye Brain. 2018;10:1–11.
Stone JH, Han J, Aringer M, Blockmans D, Brouwer E, Cid MC, et al. Long-term effect of tocilizumab in patients with giant cell arteritis: open-label extension phase of the Giant Cell Arteritis Actemra (GiACTA) trial. Lancet Rheumatol. 2021;3:e328–e36.
Stone JH, Spotswood H, Unizony SH, Aringer M, Blockmans D, Brouwer E, et al. New-onset versus relapsing giant cell arteritis treated with tocilizumab: 3-year results from a randomized controlled trial and extension. Rheumatol (Oxf). 2022;61:2915–22.
NICE. Tocilizumab for treating giant cell arteritis: technology appraisal guidance [TA518]: National Institute for Health and Care Excellence; 2018; [cited 2022 30th December]. https://www.nice.org.uk/guidance/ta518/chapter/1-Recommendations.
SMC. Tocilizumab, 162 mg solution for injection in pre-filled syringe and pre-filled pen (RoActemra®). Scottish Medicines Consortium; 2018; [cited 2022 30th December]. https://www.scottishmedicines.org.uk/media/3693/tocilizumab-roactemra-final-august-2018-for-website.pdf.
Cronin O, Preston H, Fahmy H, Kuske B, Singh M, Scott N, et al. Tocilizumab for the treatment of giant cell arteritis in Scotland: a report on behalf of the Scottish Society for Rheumatology standards subgroup. Rheumatol Adv Pract. 2022;6:rkac017.
Conway R, Putman MS, Mackie SL. Benchmarking tocilizumab use for giant cell arteritis. Rheumatol Adv Pract. 2022;6:rkac037.
Mackie SL, Brouwer E, Conway R, van der Geest KSM, Mehta P, Mollan SP, et al. Clinical pathways for patients with giant cell arteritis during the COVID-19 pandemic: an international perspective. Lancet Rheumatol. 2021;3:e71–e82.
Nguyen AAK, Sammel AM, Mollan SP, Subramanian PS, Fraser CL. Giant Cell Arteritis Incidence During the COVID Pandemic. J Neuroophthalmol. 2022.
Foroozan R, Deramo VA, Buono LM, Jayamanne DG, Sergott RC, Danesh-Meyer H, et al. Recovery of visual function in patients with biopsy-proven giant cell arteritis. Ophthalmology 2003;110:539–42.
Cornblath WT, Eggenberger ER. Progressive visual loss from giant cell arteritis despite high-dose intravenous methylprednisolone. Ophthalmology 1997;104:854–8.
Curumthaullee MF, Liozon E, Dumonteil S, Gondran G, Fauchais AL, Ly KH, et al. Features and risk factors for new (secondary) permanent visual involvement in giant cell arteritis. Clin Exp Rheumatol. 2022;40:734–40.
Unizony S, McCulley TJ, Spiera R, Pei J, Sidiropoulos PN, Best JH, et al. Clinical outcomes of patients with giant cell arteritis treated with tocilizumab in real-world clinical practice: decreased incidence of new visual manifestations. Arthritis Res Ther. 2021;23:8.
Dejaco C, Kerschbaumer A, Aletaha D, Bond M, Hysa E, Camellino D, et al. Treat-to-target Recommendations in Giant Cell Arteritis and Polymyalgia Rheumatica. Ann Rheumatic Dis. 2023, in press.
Author information
Authors and Affiliations
Contributions
EJB: Literature review, data analysis, and first draft of the paper. SPM: Study concept and design, literature review, supervision and critical review of the paper.
Corresponding author
Ethics declarations
Competing interests
Dr EJB has no competing interests to declare. Professor SPM reports consultancy fees (Invex Therapeutics); advisory board fees (Invex therapeutics; Gensight) and speaker fees (Heidelberg engineering; Chugai-Roche Ltd; Allergan; Santen; Chiesi; and Santhera).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bilton, E.J., Mollan, S.P. Giant cell arteritis: reviewing the advancing diagnostics and management. Eye 37, 2365–2373 (2023). https://doi.org/10.1038/s41433-023-02433-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-023-02433-y
