
Abstract
Objectives
To establish cultures of human lacrimal gland from patient-derived, biopsy-sized, tissue specimens.
Methods
Tissue was obtained after surgical removal from patients without dry eye disease undergoing routine procedures. Samples were subjected to mechanical and enzymatic digestion and resulting cell suspensions were plated onto collagen-coated glass coverslips and grown for up to 21 days. Cultures were analysed by immunocytochemistry and light microscopy, and resultant cellular distributions were compared to those in sections of fixed human lacrimal gland tissue.
Results
Dissociation of biopsy-sized pieces of human lacrimal gland and seeding onto coated surfaces allowed development of a mixed population of cells in vitro. Within 7–14 days, cellular aggregation was observed and by 21 days many cells had organised themselves into distinct three-dimensional complexes. Immunohistochemistry revealed a heterogeneous population of cells, including epithelial, myoepithelial, mesenchymal and progenitor cells. Some of the epithelia labelled positively for lysozyme and lactoferrin.
Conclusions
Collection and dissociation of biopsy-sized pieces of human lacrimal gland leads to a cellular preparation that can proliferate in vitro and organise into three-dimensional structures. This is the first report detailing that biopsy-collected specimens of human lacrimal gland can be used to establish cell cultures.
Similar content being viewed by others

Introduction
The human lacrimal gland produces the aqueous component of the tear film with its secretory function integral in maintaining the health of the ocular surface. Dysfunction of the lacrimal gland can result in dry eye disease, a condition associated with significant ocular morbidity and reduced quality of life [1]. There are currently no curative treatment options for dry eye disease and patients suffering from this condition require lifelong supportive management [2,3,4,5].
The human lacrimal gland is composed of three major cell populations: the secretory acinar cells, ductal epithelial cells which transport aqueous secretions to the ocular surface and myoepithelial cells which assist in tear transport and secretion [6,7,8]. Lacrimal gland progenitor cells and fibroblasts are also present [9, 10]. Due to its relatively simple structure and accessibility, the human lacrimal gland represents an excellent candidate for the potential application of tissue engineering or regeneration: such approaches are currently being explored and may represent a novel approach to treating dry eye disease in the future [11,12,13,14].
Although the in vitro propagation of animal lacrimal gland cells has been reported in the literature, the culture of the equivalent human tissue remains relatively unexplored [15,16,17,18,19,20,21,22,23,24]. Previous attempts to culture cells from the human lacrimal gland have required the use of an entire gland, obtained at either the time of exenteration or post-mortem, to produce any growth in vitro [21, 24]. This approach clearly has limited translational potential, particularly since human tissue is often only available in small samples such as those obtained via biopsy. To progress in vitro studies for dissemination of lacrimal gland function or regeneration, therefore, it is necessary to first develop a method of culturing tissue from small and more practically available samples, ideally ensuring that resultant in vitro preparations at least partially recapitulate in vivo functioning.
In this study, we describe the acquisition of biopsy-sized live tissue pieces of lacrimal gland from routine surgical procedures, and show that we can consistently generate cultures in which the cells organise themselves into a three-dimensional array. This technique has significant practical implications for our study of disease processes affecting the lacrimal gland and the investigation of potential new targeted therapies.
Materials and methods
Human tissue samples
This study was conducted at the Royal Adelaide Hospital and University of Adelaide. The use of human tissue was approved by the Central Adelaide Local Health Network Human Research Ethics Committee (HREC/13/RAH/445), and all participants provided written informed consent to the use of tissue for research purposes. All participants were undergoing routine oculoplastic procedures in which small pieces of lacrimal gland tissue were excised as part of the operation and would otherwise have been discarded. No patients had a history of aqueous tear deficiency or lacrimal gland disease. In each case, the surgeon removed a small sample of healthy lacrimal gland either during a direct lacrimal gland prolapse procedure or when there was evidence of lacrimal gland prolapse during a blepharoplasty. The biopsied tissue, therefore, was healthy and without any underlying disease. Pieces of lacrimal gland were obtained unilaterally from nine separate donors (aged between 49 and 64 years; four males and five females) freshly harvested with aseptic technique and immediately placed into sterile Dulbecco’s Modified Eagle’s Medium (DMEM; Life Technologies Australia Pty Ltd, Mulgrave, VIC, Australia; #11885-084) supplemented with 100 U/ml each of Penicillin and Streptomycin (Life Technologies Australia Pty Ltd; #15140-122), 10 μg/ml Amphotericin B (Life Technologies Australia Pty Ltd; #15290-018) and 100 μg/ml Gentamicin (Sigma-Aldrich, Castle Hill, Australia). A small portion of each piece of collected tissue was fixed for 24 h in 10% (w/v) neutral-buffered formalin (NBF) before being embedded in paraffin wax and evaluated histologically to examine structure and confirm the absence of pathology.
Immunohistochemistry
Immunohistochemistry was carried out as previously documented (see Table 1 for antibodies and dilutions) [25]. Counterstaining was achieved by using Lillie-Mayer’s haematoxylin. For evaluation of tissue histology, standard haematoxylin-eosin staining was applied. Extracellular collagen deposition was delineated by standard Masson’s trichrome staining [10].
Establishing a mixed primary culture
Freshly obtained biopsy samples of human lacrimal gland were washed in Hanks’ Balanced Salt Solution (HBSS) containing no added calcium or magnesium (HBSS; Life Technologies Australia Pty Ltd; #14175095). In a sterile petri dish, the lacrimal gland was mechanically dissected into small pieces using sterile Westcott scissors and a surgical blade (#11) while immersed in HBSS.
Tissue was subjected to enzymatic digestion at 37 °C for 70 min in HBSS containing 390 U/ml collagenase, 400 U/ml hyaluronidase and 50 U/ml deoxyribonuclease (DNAse; Sigma-Aldrich), with the mixture being gently triturated at 15 min intervals. After this time, 10 ml of cold HBSS containing 5% (v/v) fetal bovine serum (FBS; Life Technologies Australia Pty Ltd; #10100-147) was added to stop the enzyme reaction. The resultant mixture was then filtered through a 75 μm cell sieve, transferred into 2 × 15 ml tubes and centrifuged for 2 min at 450g. The supernatant was discarded, and the cell pellet disrupted into 8 ml of Dulbecco’s Modified Eagle’s Medium Nutrition Mixture F-12 (DMEM/F12; Life Technologies Australia Pty Ltd) supplemented with 10% (v/v) FBS, 100 U/ml penicillin/streptomycin and 10 μg/ml Amphotericin B.
After gently mixing the cell suspension, 1 × 106 cells/well were added to uncoated culture vessels, which were incubated at 37 °C for 24 h. During this time, fibroblasts preferentially adhered to these non-coated plates. After 24 h, unattached cells, which theoretically represented all non-fibroblast cells in the suspension, were removed and seeded into 24-well culture plates containing sterilised borosilicate glass coverslips (diameter, 13 mm) coated with 1:20 Type I Rat Tail Collagen (EMD Millipore, CA, USA; #08-115). Once cells had attached and begun to proliferate (5–7 days), growth-stimulating medium (William’s Medium E with 5 µM dexamethasone, 100 U/ml penicillin/streptomycin, 2 mM L-glutamine and 250 ng/ml epidermal growth factor) was used; this was refreshed every 3–5 days. Cells were grown at 37 °C in an incubator with saturating humidity and 5% CO2. Cultures were successful and reasonably consistent in terms of cell growth and development between tissue samples taken from individual donors.
Immunocytochemistry
After 21 days in culture, cells were fixed in 10% (w/v) NBF for 20 min and then permeabilised by immersion in phosphate-buffered saline (PBS) plus 0.1% (v/v) Triton X-100 (PBS-T) for 30 min at room temperature. Blocking was then performed at room temperature in PBS containing neonatal horse serum (3.3%, v/v; PBS-NHS) for 30 min. Each coverslip was then transferred to a humidified chamber incubated with primary antibody overnight (see Table 1 for antibody details). After this, coverslips were incubated with corresponding appropriate secondary antibodies (Vector Laboratories, Abacus ALS, Brisbane, Australia; 1:250 in PBS-NHS) for 30 min. This was followed by incubation with streptavidin-conjugated Alexafluor-594 (Molecular Probes, Invitrogen, Mulgrave, VIC, Australia; 1:1000 in PBS-NHS), for 1 h in the dark. For nuclear counterstaining, 4′,6-diamidino-2-phenylindole (DAPI; 500 ng/ml) was applied for 5 min before coverslips were mounted using anti-fade mounting medium (Dako, Sydney, New South Wales, Australia). Double-labelling immunocytochemistry was carried out as described previously [25]. Expression of different collagens in cultures was not investigated, to avoid interference by the collagen used to coat the culture vessels.
Evaluation of histology and immunofluorescence
Histological sections were visualised with an Olympus BX-51 light microscope (Olympus, Mount Waverly, Australia) and photography was with an Olympus DP-20 digital camera (Olympus, Mount Waverly, Australia). Visualisation of fluorescent labelling was via a microscope equipped with epi-fluorescence optics (Olympus BX-61; Olympus, Mount Waverly, VIC, Australia) and photography achieved using an attached digital camera (Olympus DP-73).
For quantification, cells labelling for a particular protein antigen were counted per microscopic field in five separate cultures, as was total number of cells (via nuclear DAPI-labelling). Data were expressed as percentage cells labelling per defined antigen in relation to total number of cells per field. No statistical comparisons were carried out.
Results
Analysis of native human lacrimal gland
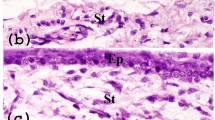
All human lacrimal gland tissue obtained from biopsies demonstrated a characteristic histological appearance when stained with haematoxylin and eosin (Fig. 1A), with epithelial cells arranged into lobes of acini (and ducts). Furthermore, staining with Masson’s trichrome revealed the presence of extracellular collagen deposits between acinar lobes (Fig. 1B). Immunohistochemistry revealed that the primary collagen present was collagen VI (Fig. 1C). Vimentin-positive mesenchymal cells were present in between acinar lobes (Fig. 1D) and were likely fibroblasts and the source of collagen. α-Smooth muscle actin (α-SMA) labelled myoepithelial cells which surrounded acinar lobes (Fig. 1E). Aquaporin 5 was detected as apical labelling in acinar epithelial cells (Fig. 1F). Pan-cytokeratin labelling was demonstrated in all epithelial cells present (acinar/ductal; Fig. 1G). Interestingly, the presence of progenitor cells was demonstrated by positive labelling of a sparsely distributed population of inter-acinar nestin-positive cells (Fig. 1H). Finally, conventional-type lysozyme was detected as intracellular granules in acinar cells with no staining of other cellular structures (Fig. 1I).

A Photomicrographs of native human lacrimal gland demonstrating tissue histology via haematoxylin and eosin staining. B Blue staining regions show deposition of extracellular collagen, via Masson’s trichrome staining. C–I Labelling of cell-specific antigens via immunohistochemistry. Immunohistochemistry was as follows: (C) collagen VI, (D) vimentin labelling of mesenchymal cells such as fibroblasts, (E) α-SMA labelling of supporting myoepithelial cells, (F) aquaporin 5 labelling of apical surfaces of acinar epithelial cells, (G) pan-cytokeratin labelling of epithelial cells, (H) nestin labelling of putative progenitor cells, (I) lysozyme labelling of secretory acinar epithelial cells. Nuclear counterstain is blue (C–I). Scale bar, 60 µm (colour figure online).
Establishment of mixed human lacrimal gland culture
Enzymatic digestion of lacrimal gland samples produced heterogeneous cellular clusters containing epithelial cells, fibroblasts and some residual red blood cells (Fig. 2A). Interspersed between this heterogeneous population of cells were some individual cells and small amounts of residual connective tissue.

Immediately after mechanical and chemical digestion, clusters of dissociated lacrimal gland epithelial cells can be seen floating in culture media (A; black arrows). After 7 days in culture (B), cells had attached and proliferated and multiple clusters of polygonal cells are seen (black arrows), interspersed with spindle-shaped cells. Epithelial cells appear polygonal (black arrows) and have become arranged in clusters by 14 days in vitro (C). By 21 days in culture (D–F), cells have apparently organised into distinct structures surrounded by large clumps of opaque, fibrous material (black arrows). Some cells (white arrows) are enlarged, clustered together and contain multiple cytoplasmic vesicles and are surrounded by elongated cells, likely myoepithelia (D–F). Images shown in (E) and (F) are magnified from image shown in (D). Scale bar shown in (D) refers to scale used in (A–D) and is 50 µm. Scale bar shown in (F) refers to scale used in (E, F) and is also 50 µm.
Cells attached within 3–5 days of being plated on collagen and were seen to proliferate from 7–14 days in vitro (Fig. 2B, C). Following the initiation of cell division, confluence was reached after approximately 21 days (Fig. 2D). At all stages of the culture, multiple cell populations with distinct classes of morphologies were evident. A small population of cells with an elongated morphology were clearly present after 7–14 days (Fig. 2B, C). Despite efforts to exclude fibroblasts from the cell population grown in vitro, it is possible that a small number persisted—these likely represented the cells with the elongated morphology. In trials where no effort was made to remove fibroblasts, culture plates became confluent with fibroblasts in 5–7 days in vitro and demonstrated no growth of other lacrimal components (data not shown).
Epithelial cells were polygonal in nature and grew in clusters; as they matured the cells enlarged and the cytoplasm demonstrated accumulation of multiple vesicles (Fig. 2D–F). In a mixed culture, epithelial cells formed clusters which were surrounded by borders of fibroblast-like cells (Fig. 2E, F), mimicking the cellular arrangement in native lacrimal tissue (Fig. 1). With increasing time in vitro, the appearance of cultures became highly differentiated with clear areas of epithelial cells and surrounding mesenchymal cells. Cell cultures could be healthily maintained for approximately 35 days in culture.
Analysis of cultured human lacrimal gland
Haematoxylin-eosin staining of 21-day-old cultures revealed that cells were present in two general arrangements: monolayers and dense, multi-layered clusters (Fig. 3A–C). The presence of the thickened clustered regions was associated with eosin-dense extracellular matrix. In between these clusters, cells in monolayers arranged themselves in leaflet-like groupings based around central nodes or beams (Fig. 3B, C). These central nodes were also associated with extracellular matrix deposition shown by intense eosin staining.

Analysis of 21-day-old lacrimal gland cultures by haematoxylin and eosin (H&E) staining (A–C) and immunocytochemistry (D–I). A H&E staining of cultures revealed that cells were present in single layers and in multi-layered aggregates, the latter of which also comprised deposits of extracellular matrix (arrows). B Away from multi-layered regions, mono-layered cells formed leaflet-like clusters around spaces that superficially resembled the ducts seen in vivo (white arrows). C Magnified image showing the leaflet arrangement of cells in more detail (white arrow) and the thickening and extracellular matrix deposits in the central beams, as seen in (A; black arrow). Immunocytochemistry revealed the presence of pan-cytokeratin-positive epithelial cells (D), as well as elongated vimentin-positive mesenchymal cells (likely, fibroblasts; E), a small population of α-SMA-positive myoepithelial cells (G) and nestin-positive putative progenitor cells (H). Examination of the presence of lysozyme revealed a population of epithelial cells with punctate cytoplasmic labelling, likely acinar cells (F), were also present. Immunocytochemically labelled cells shown in (D–H) have nuclei counterstained with DAPI. Scale bars: all bars represent 50 µm; however, bar shown in (B) relates to (A and B), bar shown in (F) relates to this image only and bar shown in (I) relates to (D–I).
These heterogeneous cell cultures demonstrated clear immunoreactivity for distinct cell-types, including epithelial cells (pan-cytokeratin-positive cells, Fig. 3D), as well as elongated vimentin-positive mesenchymal cells (Fig. 3E). Clusters of polygonal epithelial cells were specifically labelled with an antibody to lysozyme (Fig. 3F); this labelling was arranged in a punctate cytoplasmic manner, likely reflecting a vesicular location. Myoepithelial cells were present and labelled with α-SMA (Fig. 3G), and a sparing population of nestin-positive progenitor cells was also identified and was randomly arranged at the periphery of the multi-layered clusters (Fig. 3H).
Vimentin-positive mesenchymal cells were arranged within the monolayers at the periphery of the leaflet-like structures, and away from the central extracellular matrix-rich nodes. This was clearly indicated by co-labelling with pan-cytokeratin-positive epithelial cells (Fig. 4A–C), or aquaporin 5-positive or lactoferrin-positive cells (Fig. 4D–F and G–I, respectively). The presence of lysozyme (Fig. 3F) and lactoferrin (Fig. 4G–I) in cells in culture suggested that cultures produced proteins that could potentially be secreted.

A–C Pan-cytokeratin-positive epithelial cells arrange in branched leaflet-like structures which also contain some peripherally arranged vimentin-positive mesenchymal cells. D–F This is also the case with aquaporin 5-positive epithelial cells. G–I Magnified view of cultures double-labelled for the secretory protein, lactoferrin, and the mesenchymal protein, vimentin, also show the close association between central acinar-like epithelial cells and peripherally arranged vimentin-positive mesenchymal cells. J Quantification of proportion of cells present in cultures that were immunocytochemically labelled for a range of specific, identifying antibody markers. Percentage of cells labelling for each protein marker were counted in fields from 5 separate cultures. Data are expressed as mean value (n = 5) ±SD. Scale bars: all scale bars represent 50 µm; however, bar shown in (F) relates to (A–F) and bar shown in (I) relates to (G–I).
Quantification of proportion of cells present in microscopic fields that labelled for each particular protein antigen is shown in Fig. 4J. Clearly, the majority of cells label for the epithelial marker protein, pan-cytokeratin (85.4 ± 3.4% of cells). In addition, the majority of cells also labelled for aquaporin 5 (80.0 ± 3.0%), lactoferrin (82.3 ± 6.0%) and lysozyme (78.4 ± 4.2%). We were not able to determine whether these protein antigens were co-localised. A more limited population of cells labelled positively for the mesenchymal cell marker, vimentin (10.0 ± 3.2%), with smaller populations of both α-SMA (2.8 ± 2.1%) and nestin (2.4 ± 1.7%) also quantifiable. Thus, a mixture of cell types were present throughout these cultures, recapitulating the status of the in vivo lacrimal gland.
Discussion
Although human lacrimal gland cultures have previously been described, the present study is the first to demonstrate establishment of successful cultures from biopsy-sized samples of fresh human tissue. Previous studies have required entire lacrimal glands to establish culture and have focused on specific cell-types [15, 21, 24, 26]. Whilst these studies represent important practical advances in a previously unexplored area, achieving a clinically viable technique requires the use of significantly smaller samples, such as those harvested from biopsies, as well as the ability to successfully demonstrate the propagation of each cellular component of the lacrimal gland [17]. We have addressed both of these limitations. Furthermore, the expression of lysozyme and lactoferrin in a population of cells in our preparations indicates that after being established in vitro, the majority of cells still synthesise aqueous tear protein components, which could indicate that they retain secretory function.
After 14–21 days in vitro, cultured cells had organised themselves into three-dimensional structures that very superficially resembled in situ lacrimal gland tissue. Multi-layered cells in cord-like bands were detected which were rich in extracellular matrix and which surrounded regions of cellular monolayers. Cells within these monolayer regions further organised themselves into lobule or leaflet-like arrangements with a central node (Fig. 3), as suggested in a previous study [21]. The formation of these cellular arrangements in culture happened spontaneously and without any external encouragement. It is interesting to speculate at this stage as to whether the different cell-types present naturally gravitated towards this arrangement or whether progenitor cell-types were present within the cultures and these instigated the cellular modelling. Of relevance to the latter hypothesis, we showed that nestin-positive progenitor cells were indeed present in the cultures and these were based at the periphery of the cell clusters. The presence of these cells in similar cultures has also been reported previously [10, 15, 21, 27]. If these progenitor cells were the driving force behind self-organisation of cells into pseudo-gland-like structures, then they would represent, for example, a viable target for use in tissue regeneration and bioengineering [27].
Previous techniques for culturing the lacrimal gland have also been reported, concentrating on the isolation of specific cell-populations and removal of others [28,29,30]. Although isolation of single cell-types from the lacrimal gland may allow experimentation on a target cell population, the clinical utility of such techniques is more limited. The most frequently employed technique to remove unwanted cell populations is through gravitic separation, whereby cells within a mixed tissue sample are dissociated, then allowed to sit undisturbed for a variable number of hours (ranging from 2 to 24 h) [28,29,30]. In the present study, a period of 24 h provided sufficient time for preferential attachment of fibroblasts to the uncoated wells. Removal of fibroblasts from the cell preparation at this stage allows relatively uninhibited attachment and growth of lacrimal epithelial cells. That vimentin-positive mesenchymal cells were detected in the present cultures likely describes that a population of fibroblasts avoided selection-removal during culture. The presence of these cells at the periphery of the lobe-like structures in vitro, herein, recapitulates the location of fibroblasts surrounding the acini and ducts in vivo, and again, demonstrates that the heterogeneous cell population in our cultures does organise into a pseudo-appropriate arrangement.
Aside from blood vessel-based cells, fibroblasts and progenitor cells, there are three major types of cells in a functional lacrimal gland: ductal, acinar and myoepithelial [31]. These are all types of specialised epithelial cells. The ductal cells comprise simple polarised cuboidal epithelium, whereas the acinar cells are pyramidal columnar, polarised and are rich both in rough endoplasmic reticulum and in secretory vesicles [8]. The myoepithelial cell is flattened and stellate, surrounding the base of the acinus in situ [8]. This type of cell has transdifferentiated from a traditional epithelial cell and expresses contractile elements such as α-SMA, enabling it to control secretion from glandular acinar cells [6]. In situ, the distinction between these cell types is based on histological assessment. In the cultures described in the present study, however, differentiating between them is less simple. We utilised immunocytochemistry to identify epithelium with a general marker of these cells, ie pan-cytokeratin. This protein was expressed in the majority of the cells present in the cultures, as would be expected since it is expressed by both epithelium and myoepithelial cells, which together constituted the predominant cell-types. In terms of individual types of epithelium, myoepithelial cells can be identified by positive labelling with α-SMA [6], and other epithelial cells by expression of proteins which would be secreted in tears in vivo, in this case: lysozyme and lactoferrin [32]. From Figs. 3 and 4 it is clear that, in fact, a large number of the cells present express these latter proteins. Because lysozyme and lactoferrin are both secreted in tears, in vivo, the cells expressing these proteins could be acinar in character. This is not surprising since acinar cells comprise approximately 80% of the epithelium in the lacrimal gland [33]. We were not able to identify whether both lysozyme and lactoferrin expression localised to the same cells, but the fact that epithelial cells in our cultures expressed these proteins implied that they could potentially retain a secretory function in vitro. Testing of secretory output from cultures would define whether these cells possessed this function in vitro. Were these cultures to express secretory lacrimal acinar cells, this would be in agreement with previous work [21].
We also detected expression of aquaporin 5 in cells both in vivo and in the majority of cells in vitro. Aquaporin 5 is a member of a large family of proteins responsible for transporting water across plasma membranes, a process which is vital in the production of serous tears [34]. It has previously been shown that aquaporin 5 is expressed on the apical surface of lacrimal gland acinar cells [35]. It is known that ductal cells also contribute to the aqueous content of tears [33]; in support of this, these cells have also been shown to express aquaporin 5 [36]. Labelling of cultured epithelial cells in the present study with aquaporin 5, therefore, most likely identifies both acinar cells and ductal cells.
In summary, we have applied a method of producing human lacrimal gland cultures to biopsy-sized pieces of tissue which were removed during routine surgery. These cultures are comprised of a heterogeneous population of cells, which spontaneously organise into three-dimensional arrangements in vitro. The expression of tear proteins in many of the cells suggest that these cultures may retain a rudimentary secretory function. Our procedure could prove useful in both our understanding of disease mechanisms affecting the lacrimal gland and, eventually, in the field of ophthalmic bioengineering. In terms of working towards bioengineered lacrimal gland, we have already developed a three-dimensional novel scaffold using FDA-approved chitosan which has tunable properties and has been shown to support the growth of human eyelid fibroblasts in vitro [37]. Current studies are already underway investigating the utility of this scaffold for potential lacrimal gland tissue regeneration in vitro. Subsequent studies will then focus on re-implantation of the cell-scaffold tissue complex in the subconjunctival fornices to allow direct tear delivery to the ocular surface. Ongoing studies are clearly required, but as small-sample tissue culture techniques progress and are applied to increasingly complex tissue, the potential applications become numerous.
Summary
What was known before
-
The in vitro propagation of animal lacrimal gland cells has been reported in the literature but the culture of the equivalent human tissue remains relatively unexplored. Previous attempts to culture cells from the human lacrimal gland have required the use of an entire gland, obtained at either the time of exenteration or post-mortem, to produce any growth in vitro.
What this study adds
-
In this study, we describe the acquisition of biopsy-sized live tissue pieces of lacrimal gland from routine surgical procedures, and show that we can consistently generate cultures in which the cells organise themselves into a three-dimensional array.
Data availability
Data comprising the study are available from the corresponding author on reasonable request.
Change history
20 January 2022
The figures were changed to color figures in pdf.
References
The epidemiology of dry eye disease: report of the Epidemiology Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf. 2007;5:93–107.
Messmer EM. The pathophysiology, diagnosis, and treatment of dry eye disease. Dtsch Arztebl Int. 2015;112:71–81. quiz 82.
Fowler SB. A review of best evidence for dry eye. Insight. 2014;39:5–9.
Al-Saedi Z, Zimmerman A, Bachu RD, Dey S, Shah Z, Baugh R, et al. Dry eye disease: present challenges in the management and future trends. Curr Pharm Des. 2016;22:4470–90.
Calonge M. The treatment of dry eye. Surv Ophthalmol. 2001;45 Suppl 2:S227–239.
Makarenkova HP, Dartt DA. Myoepithelial cells: their origin and function in lacrimal gland morphogenesis, homeostasis, and repair. Curr Mol Biol Rep. 2015;1:115–23.
Lorber M. Gross characteristics of normal human lacrimal glands. Ocul Surf. 2007;5:13–22.
Obata H. Anatomy and histopathology of the human lacrimal gland. Cornea. 2006;25:S82–89.
Xi X, McMillan DH, Lehmann GM, Sime PJ, Libby RT, Huxlin KR, et al. Ocular fibroblast diversity: implications for inflammation and ocular wound healing. Invest Ophthalmol Vis Sci. 2011;52:4859–65.
You S, Kublin CL, Avidan O, Miyasaki D, Zoukhri D. Isolation and propagation of mesenchymal stem cells from the lacrimal gland. Invest Ophthalmol Vis Sci. 2011;52:2087–94.
Hirayama M, Ogawa M, Oshima M, Sekine Y, Ishida K, Yamashita, K K, et al. Functional lacrimal gland regeneration by transplantation of a bioengineered organ germ. Nat Commun. 2013;4:2497.
Jeong SY, Choi WH, Jeon SG, Lee S, Park JM, Park M, et al. Establishment of functional epithelial organoids from human lacrimal glands. Stem Cell Res Ther. 2021;12:247.
Lu Q, Yin H, Grant MP, Elisseeff JH. An in vitro model for the ocular surface and tear film system. Sci Rep. 2017;7:6163.
Xiao S, Zhang Y. Establishment of long-term serum-free culture for lacrimal gland stem cells aiming at lacrimal gland repair. Stem Cell Res Ther. 2020;11:20.
Lin H, Liu Y, Yiu S. Three dimensional culture of potential epithelial progenitor cells in human lacrimal gland. Transl Vis Sci Technol. 2019;8:32.
Lin H, Sun G, He H, Botsford B, Li M, Elisseeff JH, et al. Three-dimensional culture of functional adult rabbit lacrimal gland epithelial cells on decellularized scaffold. Tissue Eng Part A. 2016;22:65–74.
Massie I, Spaniol K, Barbian A, Geerling G, Metzger M, Schrader S. Development of lacrimal gland spheroids for lacrimal gland tissue regeneration. J Tissue Eng Regen Med. 2018;12:e2001–9.
Rismondo V, Gierow JP, Lambert RW, Golchini K, Feldon SE, Mircheff AK. Rabbit lacrimal acinar cells in primary culture: Morphology and acute responses to cholinergic stimulation. Investigative Ophthalmol Vis Sci. 1994;35:1176–83.
Schechter JE, Warren DW, Mircheff AK. A lacrimal gland is a lacrimal gland, but rodent’s and rabbit’s are not human. Ocul Surf. 2010;8:111–34.
Stevenson D, Schechter JE, Nakamuro T, Chang D, Chang NY, Pidgeon M, et al. A new model system for studying lacrimal physiology using cultured lacrimal gland acinar cells on Matrigel® rafts. Adv Exp Med Biol. 2002;506:159–63.
Tiwari S, Ali MJ, Balla MMS, Naik MN, Honavar SG, Reddy VAP, et al. Establishing human lacrimal gland cultures with secretory function. PLoS ONE 2012;7:e29458.
Ueda Y, Karasawa Y, Satoh Y, Nishikawa S, Imaki J, Ito M. Purification and characterization of mouse lacrimal gland epithelial cells and reconstruction of an acinarlike structure in three-dimensional culture. Invest Ophthalmol Vis Sci. 2009;50:1978–87.
Vanaken H, Vercaeren I, Claessens F, De Vos R, Dewolf-Peeters C, Vaerman JP, et al. Primary rat lacrimal cells undergo acinar-like morphogenesis on reconstituted basement membrane and express secretory component under androgen stimulation. Exp Cell Res. 1998;238:377–88.
Yoshino K. Establishment of a human lacrimal gland epithelial culture system with in vivo mimicry and its substrate modulation. Cornea. 2000;19:S26–S36.
Mammone T, Chidlow G, Casson RJ, Wood JPM. Expression and activation of mitogen-activated protein kinases in the optic nerve head in a rat model of ocular hypertension. Mol Cell Neurosci. 2018;88:270–91.
Tiwari S, Nair RM, Vamadevan P, Ali MJ, Naik MN, Honavar SG, et al. Establishing and characterizing lacrispheres from human lacrimal gland for potential clinical application. Graefes Arch Clin Exp Ophthalmol. 2018;256:717–27.
Basova L, Parfitt GJ, Richardson A, Delcroix V, Umazume T, Pelaez D, et al. Origin and lineage plasticity of endogenous lacrimal gland epithelial stem/progenitor cells. iScience. 2020;23:101230.
Guo Z, Azzarolo AM, Schechter JE, Warren DW, Wood RL, Mircheff AK, Kaslow HR. Lacrimal gland epithelial cells stimulate proliferation in autologous lymphocyte preparations. Exp Eye Res. 2000;71:11–22.
Schönthal AH, Warren DW, Stevenson D, Schecter JE, Azzarolo AM, Mircheff AK, et al. Proliferation of lacrimal gland acinar cells in primary culture. Stimulation by extracellular matrix, EGF, and DHT. Exp Eye Res. 2000;70:639–49.
Schechter J, Stevenson D, Chang D, Chang N, Pidgeon M, Nakamura T, et al. Growth of purified lacrimal acinar cells in matrigel raft cultures. Exp Eye Res. 2002;74:349–60.
Garg A, Zhang X. Lacrimal gland development: from signaling interactions to regenerative medicine. Dev Dyn. 2017;246:970–80.
Bron AJ. Eyelid secretions and the prevention and production of disease. Eye (Lond). 1988;2:164–71.
Toth-Molnar E, Ding C. New insight into lacrimal gland function: role of the duct epithelium in tear secretion. Ocul Surf. 2020;18:595–603.
Schey KL, Wang Z, Qi LWJ,Y. Aquaporins in the eye: expression, function, and roles in ocular disease. Biochim Biophys Acta. 1840;2014:1513–23.
Hamann S, Zeuthen T, La Cour M, Nagelhus EA, Ottersen OP, Agre P, et al. Aquaporins in complex tissues: distribution of aquaporins 1–5 in human and rat eye. Am J Physiol. 1998;274:C1332–1345.
Sasaki Y, Tsubota K, Kawedia JD, Menon AG, Yasui M. The difference of aquaporin 5 distribution in acinar and ductal cells in lacrimal and parotid glands. Curr Eye Res. 2007;32:923–9.
Sun MT, O’Connor AJ, Milne I, Biswas D, Casson R, Wood J, et al. Development of macroporous chitosan scaffolds for eyelid tarsus tissue engineering. Tissue Eng Regen Med. 2019;16:595–604.
Funding
This study was funded by a Project Grant from The Hospital Research Foundation, Adelaide, Australia and an Ideas Grant from the National Health and Medical Research Council, Australia (APP1183278).
Author information
Authors and Affiliations
Contributions
L.A.H.: Conceived and carried out experimental work; drafted manuscript. J.P.M.W.: Conceived and carried out experimental work; edited and revised manuscript. G.C.: Carried out experimental work; edited and revised manuscript. R.J.C.: Edited and revised manuscript. D.S. and M.T.S.: Conceived study; edited and revised manuscript. All authors approved final version and agreed to be accountable for all aspects of the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Halliday, L.A., Wood, J.P.M., Chidlow, G. et al. Establishing human lacrimal gland cultures from biopsy-sized tissue specimens. Eye 37, 62–68 (2023). https://doi.org/10.1038/s41433-021-01872-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-021-01872-9
