
Abstract
The pathogenesis of central serous chorioretinopathy (CSCR), a pachychoroid disease, is poorly understood. While choroid hyperpermeability and retinal pigment epithelium dysfunction are cornerstones for developing CSCR, the mechanisms at the retinal, vascular, retinal pigment epithelium, and cellular level continue to be an enigma. A few preclinical studies and the development of small-sized, poorly controlled clinical trials have resulted in limited insight into the disease mechanism. Effective treatments for CSCR are still lacking as current trials have produced inconsistent results for functional and structural gains. Thus, critically evaluating the literature to explore disease mechanisms and provide an up-to-date understanding of pathophysiology can provide valuable information and avenues to new treatments. In this study, a comprehensive summary of the mechanistic insight into CSCR is presented while highlighting the shortcomings of current literature. The mechanism was divided into seven sub-categories including mechanical obstruction, inflammation, oxidative stress, paracrine factors, autonomic dysfunction, mineralocorticoid receptors activation, and medications. We implemented validated tools like the JBI and CAMARADES to objectively analyze the quality of both clinical and preclinical studies, respectively. Overall, our analysis of the literature showed that no single mechanism was populated with a large number of sufficiently sized and good-quality studies. However, compiling these studies gave hints not only to CSCR pathogenesis but also pachychoroid disease in general while providing suggestions for future exploration.
摘要
中心性浆液性脉络膜视网膜病变 (central serous chorioretinopathy, CSCR) 为肥厚性脉络膜疾病, 其发病机制尚不清楚。虽然脉络膜高通透性和视网膜色素上皮功能障碍是CSCR发生的基础, 但其在视网膜、血管、视网膜色素上皮和细胞水平上的机制仍是一个谜。一些临床前研究和小规模、控制不佳的临床试验的进展使我们对疾病机制的了解有限。由于目前的试验对CSCR的功能和结构获得的结果不一致, 因此仍然缺乏有效的治疗方法。因此, 批判性地评估文献以探索疾病机制, 并提供病理生理学方面的最新理解, 可为新的治疗方法提供有价值的信息和途径。在本研究中, 我们全面总结了CSCR的机制, 并强调了当前文献的不足之处。我们将其机制分为机械性阻塞、炎症、氧化应激、旁分泌因子、自主神经功能障碍、盐皮质激素受体激活和药物治疗等7个亚类。我们使用JBI和CAMARADES等验证工具分别客观分析临床和临床前研究的质量。总之, 我们对文献的分析表明, 缺乏大量足够大的、高质量的研究支持某个单一的机制。然而, 这些研究的汇总不仅为CSCR的发病机制提供了线索, 也提示了厚脉络疾病的一般发病机制, 同时为未来的探索提供了建议。
Similar content being viewed by others

Introduction
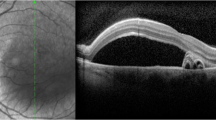
Central serous chorioretinopathy (CSCR) is a part of the pachychoroid disease spectrum characterized by the thick choroid, serous retinal detachment at the macula, and retinal pigment epithelium (RPE) detachment with subsequent atrophy (Fig. 1) [1, 2]. CSCR affects mainly middle-aged males with a mean age of 40 years and presents with blurred vision, metamorphopsia, dyschromatopsia, central scotoma, hypermetropization and micropsia [3]. Acute CSCR is self-limiting, and 84% of cases resolve in 4–6 months with almost complete resolution of symptoms without requiring treatment [4]. Episodes lasting beyond 4–6 months are referred to as persistent, non-resolving, or chronic CSCR [5]. Chronic CSCR presents with a varying degree of RPE atrophy leading to a permanent and severe decline in visual acuity, often warranting intervention (Fig. 2) [5, 6]. CSCR symptoms are often reported in one eye but bilateral involvement can be detected in up to 40% of the cases [7, 8]. Because vision loss affects the working-age group, it can result in significant financial burden, loss of independence, and a negative impact on mental/physical health, consequently increasing the burden on the healthcare system [9]. As such, a prompt and accurate diagnosis is essential for initiating appropriate management to minimize vision loss. It is also important to rule out potential vision or life-threatening conditions that mimic CSCR such as inflammatory disease (e.g. Vogt–Koyanagi–Harada [VKH] or malignancies (e.g. leukemia) [10]. Clinicians must consider various features such as disease laterality (e.g. unlike CSCR, VKH presents as bilateral and symmetrical), presence of anterior or posterior uveitis, the status of the optic disc, presence of systemic features (e.g. alopecia and vitiligo in VKH), and findings on multimodal imaging; more details about CSCR differential diagnosis is discussed by Sahoo et al. [10].

A Two different optical coherence tomography images showing subretinal fluid and pigment epithelial detachment. B The left panel shows a fundus photograph of a patient with chronic CSCR. The right panel is the corresponding red-free FAF image showing gravitational track region (yellow arrow) with RPE atrophy.

The schematic shows the potential mechanisms for developing CSCR. Various factors including mineralocorticoid receptor activation, vortex vein compression (venous congestion), dysregulation of complement or adrenergic pathway can result in choroidal vessel hyperpermeability or direct injury to RPE cells. Elevated hydrostatic may cause compression of choriocapillaris leading to ischemic damage to RPE cells, pigmented epithelial detachment, micro-rips or atrophy. RPE cells are overwhelmed and unable to provide adequate barrier resulting in subretinal fluid accumulation. Accumulation of subretinal fluid may be further exacerbated by changing Müller cell’s water and ion homeostasis via corticosteroids. Prolonged ischemia can stimulate the release of angiogenic factors like VEGF resulting in CNV. CNV choroidal neovascularization, HTN Hypertension, LCN2 lipocalin 2, OSA obstructive sleep apnea, PAI-1 plasminogen activator inhibitor 1, PDGF platelet-derived growth factor, PED pigmented epithelial detachment, RPE retinal pigment epithelium, SRF subretinal fluid, VEGF vascular endothelial growth factor.
An increase in CSCR research over the last decade, mainly clinical trials exploring treatment efficacies and oral medication, have shown inconsistent improvements in chorioretinal structure or visual acuity. Preclinical studies that explore CSCR pathophysiology are lacking in the literature and may relate to a paucity in the innovation of treatments while clinical trials have been small and uncontrolled. Advances in imaging modalities demonstrate that the hallmark of CSCR is choroid vascular hyperpermeability [11,12,–13]. These hyperpermeable regions localize with thickened subfoveal choroid, also known as pachychoroid, consistent with choroidal vessel dilation and congestion [13, 14]. In addition to choroidal thickening, optical coherence tomography demonstrates thinning of the inner choroid (smaller choriocapillaris) and is likely a result of direct compression by enlarged outer choroidal vessels, hyperpermeable vessels causing increased hydrostatic pressure, and atrophy of choriocapillaris [14, 15]. Overall, it is hypothesized that ischemia from choriocapillaris compression and increased choroid parenchyma hydrostatic pressure, overwhelming the RPE barrier, causes a cascade of RPE dysfunction, detachment and micro-rips resulting in fluid accumulation in the subretinal space (Fig. 2). In general, this disease mechanism is termed the “pachychoroid-driven process” and is the hallmark of pachychoroid disease which includes CSCR, pachychoroid pigment epitheliopathy (PPE), polypoidal choroidal vasculopathy, pachychoroid neovasculopathy (PNV), pachychoroid geographic atrophy (pGA) (Fig. S1); more detailed discussion can be found here [16]. CSCR is thought to represent disease progression within the continuum of pachychoroid disease. For example, PPE is considered the “forme fruste” of CSCR as it presents with pachychoroid features and RPE changes but no exudative retinal detachment [16, 17]. Chronic CSCR is often considered the progression of acute CSCR. However, only 16% of acute CSCR patients progress to chronic CSCR and a majority of chronic cases (73%) are not preceded with an acute episode, which raises the question of whether or not they share similar pathophysiology driving pachychoroid changes [18, 19]. Eventually, prolonged ischemia from pachychoroid-driven processes can cause loss of RPE and photoreceptors resulting in pGA [20]. Unlike conventional geography atrophy, which is a drusen driven inflammatory process in age-related macular degeneration (AMD), patients with pGA are younger, have thicker choroid, lack drusen, present with a smaller area of atrophy and, the AMD risk allele (ARMS2 A69S) is less common [20]. Overall, when attempting to understand the pathophysiology of CSCR it should not be treated as an individual entity but instead should be considered part of the larger pachychoroid disease spectrum.
While the pachychoroid-driven process in CSCR is well appreciated with multimodal imaging, how this process occurs remains elusive. Emerging evidence shows that CSCR is a complex multifactorial disease likely involving venous congestion, inflammation, ischemia, and changes in hormone and the biochemical milieu involving factors such as catecholamine, corticosteroids, or inflammatory cytokines [12, 21,22,23,24,25,26]. This review has the goal of updating the current understanding of the pathophysiology of CSCR to assist founding ideas for new treatments. In addition, the plethora of publications dissecting the mechanism of CSCR will also help shed light on the etiopathology of pachychoroid disease. The authors have synthesized the literature pertaining to CSCR disease pathogenesis, and objectively assessed the quality of studies using the validated Joanna Briggs Institute (JBI), and Collaborative Approach to Meta-Analysis and Review of Animal Data from Experimental Studies (CAMARADES) tool for clinical (observational/experimental) and preclinical studies [27].
Methods
Literature search
A thorough literature search was completed using PubMed and Medline using the terms “central serous retinopathy” and “central serous chorioretinopathy’ with other key phrases including “pathophysiology,” “pathogenesis,” “etiology” and “animal studies” and “animal model” from 1981 to 2021 (Table S1). After the initial review of literature, the etiology of CSCR was categorized into seven subheadings: mechanical obstruction; inflammation; oxidative stress; paracrine factors; autonomic system dysfunction; mineralocorticoid receptors activation, and medication. The search criteria were further streamlined to include “mineralocorticoid receptor antagonist,” “corticosteroid or steroid,” “paracrine factors or biomarkers,” “oxidative stress,” “inflammation,” “sympathetic or autonomic regulation/dysregulation,” “vortex vein,” “venous congestion” and “gene association” (Fig. S2). Key article references were also identified using meta-analysis or original articles that were not found using the search strategy.
Quality assessment
The JBI checklist (Tables S2–S7) was used to assess the quality of all experimental, observational, and descriptive clinical studies by at least two independent authors (PK and AG) [27, 28]. To remain objective, we included studies that supported or undermined a mechanism for CSCR. The JBI could not be applied to assess the quality of genetic association studies. Studies not addressing mechanisms were not subjected to JBI quality assessment. For each individual criteria on the JBI checklist, to maintain objectivity, the following guidelines were used. If a study appropriately addressed the criteria (i.e., consistency in implementation, explained its rationale), then, it was given “yes”. If a study mentioned the criteria but did not explain its rationale or did not subsequently apply it, then, it was given unclear. Otherwise, if a study omitted the criteria from its methods altogether, then, it was given “no”. Each study was then given a rating score based on the number of “Yes” responses to each question in the checklist and was expressed as a percentage; very poor quality if 0–25% Yes to questions; poor quality if 26–50% and; fair quality if 51–75%; good quality if 76–100%. The scores from the two independent assessments were averaged for the final report. The search came across many studies showing potential links between corticosteroids and CSCR in either endogenous or exogenous use, however, not all studies could be discussed using quality assessment. When necessary, quality assessment of specific clinically relevant evidence was discussed, such as treatments targeting the pathway for mineralocorticoid receptors activation. Summaries of meta-analyses that discussed the association between steroids and CSCR were also included. To note, interpretation of results from interventional case series/reports (or poorly controlled trials) needed to be exercised with caution as improvement can be due to spontaneous disease resolution rather than intervention.
Similarly, the CAMARADES checklist was used to assess the quality of preclinical studies (Table S8) [29]. In general, the higher the score, the higher the quality of the publication and less likely to introduce biases.
Results
The quality of clinical and preclinical studies reporting on the etiology of CSCR were assessed using the JBI and CAMARADES checklists, respectively (Tables S2–S8). Quality assessment was performed on a total of 51 clinical studies from 2003 to 2021 with 8% case controls, 12% cohort, 10% case reports, 31% case series, 16% cross-sectional analysis, and 23% RCTs (Table 1; Fig. 2 summarizes the mechanisms in a diagram). As shown in Fig. 3, most studies were observational/descriptive (predominated by case-series) and 50% of RCTs targeted the mineralocorticoid receptors (MR) pathway. Overall, 38%, 38%, and 24% of observational/descriptive studies had a JBI score range of 0–50% (very poor-to-poor quality), 51–75% (fair quality), and 76–100% (good quality), respectively (Fig. 3, Tables 1 and S2–S6). For RCTs, 34% of studies were very poor-to-poor quality, 58% fair quality, and 8% good quality (Fig. 3, Tables 1 and S7).

The graph shows the quality assessment score in percentage for case reports (CR), case-control (CC), case series (CS), cohort (Ch) studies, cross-sectional (CrS) studies, and randomized controlled trial (RCT). The mechanism of each study is colour coded as shown in the figure legend. MR mineralocorticoid receptors, JBI Joanna Briggs Institute.
CAMARADES quality assessment was performed on 12 animal studies of which 58%, 42%, 0% rated very poor-to-poor, fair, and good, respectively (Tables 2 and S8).
Mechanism for developing CSCR
Mechanical obstruction
Mechanical obstruction of the dominant vortex vein, which drains the macula, may contribute to the pathogenesis of CSCR (Table 1) [23, 30]. Two studies demonstrated that patients with CSCR had asymmetrical dilatation of the vortex vein [23, 30]. Vortex veins transverse the sclera at an oblique angle and the narrowing of the scleral channel from thickened or rigid sclera is thought to result in venous congestion thereby increasing the permeability of choriocapillaris. A recent study showed that patients with CSCR had significantly thicker sclera compared to normal eyes [31]. Applying the JBI criteria across these studies (1 case series, 2 cross-sectional) revealed fair-to-good quality research but were limited by small sample sizes, did not identify or control for confounding factors, and had inherent inability of cross-sectional studies to assert a temporal relationship between exposure and outcome. In support of this hypothesis, a single interventional case report (n = 1 patient; JBI 44%) showed effective management of chronic CSCR after partial-thickness scleral resection but larger studies are needed to confirm these findings [32].
Acquired focal choroidal excavation (FCE) is described by a concavity in the choroid (mostly in the macula) and may belong to the pachychoroid disease spectrum as it shares many clinical features including thickened choroid and choroid vessel hyperpermeability [33]. It has been proposed that FCE may contribute to the pathogenesis of CSCR by mechanical compression of the choriocapillaris and Sattler layer under the concavity lesion thereby exacerbate ischemia-induced RPE/Bruch membrane damage [33, 34]. However, FCE may also be considered a sequela of CSCR thus, its role in pachychoroid disease progression remains unclear and requires further elucidation [35].
Lastly, mechanical compression of choriocapillaris by the pachyvessels in the Haller’s layer is an important pathophysiological finding that guides the use of photodynamic therapy (PDT) in CSCR [36, 37]. Specifically, PDT exerts its therapeutic effect by damaging endothelial cells via radical formation which promotes large choroid vessel remodeling and shrinkage [36]. Ultimately, the shrinkage of large choroid vessels improves choriocapillaris blood flow by increasing pressures in the large vessels and/or alleviating the mechanical compression on capillaries [36].
Inflammation
Although there is no sufficient data linking CSCR to systemic inflammatory disease, two studies demonstrated elevated systemic inflammatory markers (neutrophil-to-lymphocyte ratio and monocyte-to-high-density lipoprotein ratio) in patients with acute CSCR (Table 1) [26, 38]. These two studies demonstrated poor-to-fair quality scores (JBI 45%, 75%) and were limited by small-to-moderate sample sizes (n = 76, 407 patients), insufficient identification/control for confounding factors, and not using multimodal imaging to diagnose CSCR. It was proposed that the pro-inflammatory milieu may result in the generation of reactive oxidative species causing oxidative damage to RPE and choroid endothelial cells. One case series (n = 12 patients; JBI 70%) demonstrated that the use of curcumin-phospholipid (anti-inflammatory properties) provided structural and functional benefits but overall, the results are inconclusive as there was no control group [39]. Two small-sized (n = 27, 57 patients) retrospective cohort studies reported structural gains with the use of topical or oral nonsteroidal anti-inflammatory drugs but the quality of the study was poor (JBI 35%, 45%) due to insufficient control for confounding factors, short follow-up time, inadequate data presentation, and lack of statistical analysis [40, 41].
Choroidal endothelial dysfunction and local inflammation have been hypothesized to activate platelets resulting in thrombogenesis and ischemia in the choroid vessels [26, 42]. One study (n = 198 patients) that treated patients with aspirin for anti-thrombotic effects demonstrated quicker resolution of CSCR but no functional benefits [43]. This was a fair quality study (JBI 55%) with its conclusion limited by the use of a historical control group.
The link between systemic inflammation and CSCR is intriguing given that elevated glucocorticoid, which is an associated risk factor for CSCR, has anti-inflammatory effects. The relation between these two opposing mechanisms and their role in CSCR development needs further evaluation.
Paracrine mediators
Studies looking at the expression profile of cytokines and growth factors (e.g. interleukins 6, 8, interferon gamma-induced protein-10, vascular endothelial growth factor) within the aqueous humor of CSCR patients have produced mixed results (Table 1) [24, 25, 44, 45]. For example, while one report showed elevated interleukin 6 and 8 in chronic CSCR [24], others showed no difference with the control group [45]. One study reported decreased platelet-derived growth factor (PDGF) levels in patients with CSCR [45]. PDGF deficiency was thought to contribute to CSCR pathogenesis by promoting RPE dysfunction, pericyte loss, and a local increase in VEGF resulting in choroid hyperpermeability [45]. These studies are limited by (1) lack of independent assessment by other research groups; (2) small sample size (n = 20–57 patients); (3) poor control of confounding factors; (4) cross-sectional analysis of paracrine factors making temporal association to CSCR difficult and; (5) sampling error since the secretome within the aqueous humor may not reflect the local concentration within the choroid. As such, the role of cytokines/growth factors in the pathogenesis of CSCR remains elusive and investigations are ongoing.
Two studies demonstrated a relationship between blood serotonin levels and CSCR (Table 1) [46, 47]. One reported that patients with lower serotonin levels were more likely to develop chronic CSCR while no difference was seen between acute CSCR and the control group (case-control, JBI 60%) [46]. While the second study (case series; JBI 90%) did not find a relationship between chronic and acute CSCR, it showed that patients with lower serotonin levels were more likely to develop multiple leakage sites, show recurrence, and have worsening recovery of vision at follow up [47]. It is thought that lower serotonin levels may result in vascular dysregulation which may influence choroid permeability but its direct effect on choroid vessels and RPE is lacking.
Lipocalin 2 (LCN2) is an acute-phase protein with both anti-and pro-inflammatory effects and is secreted by various cell types including RPE and retina Müller cells [48]. One case-control study (n = 277 patients; JBI 45%; Table 1) demonstrated a significantly lower levels of serum LCN2 in CSCR patients [48]. Moreover, a stepwise decrease in LCN2 levels was correlated with worsening CSCR severity. It was proposed that decreased serum LCN2 makes RPE susceptible to oxidative damage and alters its blood–retinal barrier function. Unfortunately, the study focused only on serum LCN2 levels which may not be reflective in ocular tissue and lacked complete identification of confounding factors such as baseline blood pressure, other medical co-morbidities, or prior use of steroids. Interestingly, glucocorticoid which is associated with increased risk for CSCR is shown to upregulate LCN2 secretion in various non-ocular cells [49]. This contradictory effect highlights the complexity of CSCR pathogenesis and questions the role of glucocorticoids in CSCR pathogenesis. It is possible that glucocorticoids may have a differential effect on ocular tissue LCN2 expression, but this remains to be proven.
The role of VEGF in the pathogenesis of CSCR is unclear but local VEGF production due to choroidal ischemia may contribute to vascular hyperpermeability [45]. Despite the poor correlation between VEGF levels and the development of CSCR, some investigators believe that intravitreal injection of anti-VEGF agents (e.g. bevacizumab, ranibizumab) would benefit patients by reducing choroid hyperpermeability [50]. Unfortunately, given the lack of large-scale RCTs, the efficacy of anti-VEGF therapy remains controversial [50,51,52,–53]; the JBI score of these studies (2 RCTs, 1 case series) ranged from very poor to good quality (Table 1). A meta-analysis (enrolled 14 studies, 266 eyes) revealed that anti-VEGF treatment for acute CSCR is not superior to observation alone [50]. Similarly, no visual gains were observed in patients with chronic CSCR (symptoms ≥ 3 months) [50]. Imbalance in proangiogenic and antiangiogenic factors may also contribute to the development of CSCR. For example, serum levels of angiopoietin-1 (promotes the formation of endothelial tight junctions) were decreased in chronic CSCR and is thought to contribute to disease progression by promoting leaky choroid vessels (n = 80 patients; JBI = 55%; Table 1) [54].
Angiogenic factors also play a role in the formation of type 1 choroidal neovascularization (CNV) which complicates chronic CSCR in 16–36% of cases [55,56,–57]. CSCR-associated CNV is considered a subset of a disease spectrum called pachychoroid neovascularization (Fig. 3) [16]. The mechanism of PNV is unclear but is thought to result from a pachychoroid-driven process i.e., a combination of choroid congestion and choriocapillaris compression resulting in ischemia and RPE damage that promotes the production of angiogenic factors (Fig. 2) [16, 58]. This is supported by imaging modalities which have allowed researchers to understand choroid hemodynamics in a patient with CSCR [59,60,61,–62]. Hypoperfusion causing ischemia can be seen with indocyanine green angiography (ICGA) as areas of delayed choroid arterial filling [59]. Saito et al. used laser speckle flowgraphy to characterize choroid hemodynamics and proposed that regions corresponding to delayed ICGA likely reflect areas of increased vascular resistance (decreasing blood flow to choriocapillaris) with compensatory passive blood overflow into surrounding choroid veins [61]. Detailed anatomical and functional assessment of vascular network using OCTA also revealed flow impairment in choriocapillaris in regions corresponding hypoperfusion on ICGA along with decreased deep retinal capillary perfusion and density in areas of subretinal fluid [60].
Choroid ischemia promoting VEGF secretion may play an important role in the development of CNV. This is supported by studies showing clinical benefits of using anti-VEGF treatment in patients with CSCR-associated CNV [63, 64]. Anti-VEGF also demonstrated similar efficacy to full-fluence PDT for treating CNV in chronic CSCR patients [64]. Intriguingly, a few studies characterizing the aqueous humor cytokine profile demonstrated lower concentrations of VEGF in patients with PNV compared to neovascular-age-related macular degeneration (nAMD) suggesting that these two diseases entities have a different mechanism for initiating neovascularization [65, 66]. The difference in VEFG levels may explain why PNV required fewer anti-VEGF injections compared to nAMD [67]. For nAMD, drusen play a crucial role in driving chronic inflammation which promotes the production of VEGF [66]. In contrast, CSCR associated CNV (or in general PNV) is likely the result of pachychoroid-driven changes resulting in chronic choroid/RPE ischemia that stimulates the production of angiogenic factors. Lower levels of VEGF in PNV compared to nAMD may be a result of: (1) Angiogenic factors other than VEGF playing a larger role in PNV; (2) PNV may have a lower threshold response to VEGF due to differences in genetics, mechanical forces (dilated and congested choroid vessels) or biochemical/cellular milieu (inflammatory vs. ischemia); (3) The concentration of VEGF in aqueous humor may not be reflective of the disease process as VEGF in CSCR maybe more confined within the choroid compared to nAMD [65]. Interestingly, another study found that the levels of plasma VEGF were significantly lower in patients with acute/chronic CSCR [54]. They hypothesized that vessel hyperpermeability and CNV formation may be secondary to a VEGF-independent process called arteriogenesis (transformation of existing vessels causing increased lumen size and thickness) rather than angiogenesis (formation of new vessels in a VEGF-dependent process) but this remains to be confirmed. Overall, further work is required to characterize the role of various angiogenic factors in the formation of CNV in pachychoroid diseases like CSCR.
Oxidative stress
A few small-sized cross-sectional studies (n = 36–116 patients) have shown a link between systemic oxidative stress and CSCR (Table 1) [68,69,70,–71]. Patients with CSCR demonstrated greater blood disulfide-to-thiol ratio (a measure of elevated oxidative stress) and decreased antioxidant activity compared to healthy controls [69, 70]. This imbalance of oxidative and anti-oxidative homeostasis may lead to reactive oxygen species-mediated damage of choroid vessels and the RPE. Risk factors that are associated with CSCR like elevated cortisol, obstructive sleep apnea, stress, and H. pylori infection are also linked to increased oxidative stress suggesting a potential common pathway for disease pathogenesis [72,73,–74]. While these cross-sectional studies demonstrated poor-to-good quality assessment, confounding factors were poorly controlled and they are inherently limited to make temporal associations. An attempt to treat CSCR with high-dose antioxidants in a single RCT (n = 58 patients, JBI 62%, Tables 1 and S7) did not show any functional or structural benefits [75].
Autonomic dysfunction
Given that choroid vessels are regulated by the autonomic nervous system [76], it is unsurprising to consider that CSCR was associated with autonomic nervous activity imbalances [77, 78]. Increased sympathetic tone and decreased parasympathetic activity may cause autonomic dysfunction induced vasospasm leading to choroid ischemia and subsequent choroid hyperpermeability [77, 78]. Likewise, the use of sympathomimetic drugs has been implicated in the formation of CSCR [77, 79]. Unfortunately, the role of sympathetic dysregulation in CSCR remains unclear as many of these studies were small-sized case series (n = 3–4 patients) or cross-sectional studies with poor-to-good quality assessment scores (Table 1). One RCT showed support for beta-blocker in resolving CSCR (JBI 15%) [80], while two demonstrated no benefits (JBI 54%, 42%, Tables 1 and S7) [81, 82]. These RCTs lacked methodology to sufficiently control for bias, i.e. not reporting randomization of protocol, lacking allocation concealment, methods not accounting for loss-to-follow up, lacking patient demographics. or suffered from imprecision.
Another mechanism by which sympathomimetic drugs and adrenergic hormones may cause CSCR is by inducing cell apoptosis (Table 2) [83]. Corticosteroids may further augment this effect by increasing the expression of adrenergic receptors on cells [84]. As such, patients with conditions with increased catecholamines and corticosteroids (i.e. obstructive sleep apnea, elevated stress) are associated risk factors for developing CSCR [85,86,87,–88]. Likewise, Type-A personality has been associated to CSCR, but this link remains controversial [89].
Mineralocorticoid receptor activation
A meta-analysis by Liu et al. identified exogenous glucocorticoid use to have a strong association with developing CSCR (n = 7 studies, odds ratio = 4.29; 95% confidence interval 2.01–9.15) but showed large data heterogeneity (I2 = 84%) [86]. Another reported a relationship between endogenous cortisol levels and CSCR (n = 5 case-control studies; standard mean deviation = 0.77; 95% confidence interval 0.55–0.99) [90]. Animal studies suggested that corticosteroids interact with mineralocorticoid receptors to upregulate choroid endothelial calcium-activated potassium channel (KCa2.3) causing smooth muscle relaxation resulting in increased choroidal permeability (Fig. 2, Table 2); limitations of animal studies is discussed below [21]. Furthermore, corticosteroids may disrupt the RPE barrier by interrupting ion transport and dysregulating choroid hemodynamics by upregulating adrenergic receptors [84, 91]. Studies in rats demonstrated that intravitreal injection of aldosterone induced a mineralocorticoid/glucocorticoid receptor-dependent upregulation of epithelial sodium channel-α (ENaC-α), potassium (Kir4.1), and aquaporin-4 (AQP4) channels in retinal Müller cells resulting in retinal swelling (Fig. 2, Table 2) [92]. Others have proposed that hypercortisolism can also lead to increased levels of plasminogen activator inhibitor-1 resulting in a hypercoagulable state secondary to platelet dysfunction causing transient obstruction of choroid circulation [43, 93, 94].
Cortisol has also been shown to decrease the expression of cadherin 5 (CDH5) on choroidal vessels, a major cell–cell adhesion protein, thereby increasing vessel permeability (Table 2) [95]. Genomic studies suggest that certain CDH5 gene variants may predispose the male patient to develop CSCR when exposed to both exogenous and endogenous corticosteroids [95]. As such, mineralocorticosteroid receptor (MR) antagonists have been proposed to treat CSCR by abrogating the interaction between corticosteroid and its receptor.
The role of corticosteroid in human CSCR pathogenesis remains uncertain as clinical studies using MR antagonist (e.g. eplerenone and spironolactone) have failed to consistently provide functional and anatomical gains. Tables 1 and S4, 5 summarizes 7 observational/descriptive studies (3 cohort, 4 case series; JBI poor-to-good) which were limited by small size (n = 12–114 patients), little control for confounding factors, and inherent bias from no blinding to treatment/analysis. Without a control in interventional case series, it is a challenge to discern if the resolution of symptoms was the result of the treatment or spontaneous disease resolution. The quality of 6 RCTs (Tables 1 and S7) ranged from poor-to-good and were limited by small sample sizes (n = 15–144), some inadequately powered, poor control for confounding factors, missing placebo control, for example using observational control, and cross-over studies did not have adequate washout periods. Also, few studies reported statistically significant improvements only compared to the baseline but not to the placebo group. The largest randomized controlled trial to date (VICI trial; n = 144) was good quality (JBI 89%) and demonstrated no benefit of using oral eplerenone in treating chronic CSCR [96]. Why MR antagonist failed to treat CSCR despite the evidence showing its potential role in disease pathogenesis is perplexing but, perhaps not all patients respond to MR antagonist equally. Different haplotypes of MR gene (NR3C2) have been associated with increased risk for chronic CSCR and these genetic variants may result in different treatment efficacy by MR antagonist [97]. This may explain why patients with phenotypic differences in choroid thickness responded differently with treatment in some studies (better treatment response with thicker baseline choroid >515 µm) [98]. In addition, patients that do not respond to eplerenone may benefit from spironolactone which has a greater binding affinity to mineralocorticosteroid receptor [99]. Further studies are required to assess whether MR haplotyping will allow clinicians to predict which patients will benefit most from MR antagonist therapy.
Medications associated with CSCR
Various medications including sympathomimetic drugs [77, 79], phosphodiesterase inhibitors (e.g. tadalafil [100] and sildenafil [101]), quinoline (i.e. antimalarial drug like mefloquine [102]), and neuroleptics [103] have shown possible associations with developing CSCR (Table 1).
Neuroleptics like quetiapine are thought to cause dysregulation of choroid perfusion by altering neuroendocrine factors like dopamine and serotonin [103]. Indeed, low serum serotonin has been reported in patients with chronic CSCR [46].
Phosphodiesterase inhibitors are thought to cause choroid hyperpermeability as a result of nitric oxide-mediated venous dilation [104]. Although this notion has been challenged by a case series study showing functional and anatomical benefits in chronic CSCR [105].
Quinolines may exert their effects by disrupting connexin 43 within the gap junctions of choroid vessels resulting in hyperpermeability [102].
These studies were mainly limited to case reports/series and quality assessment predominantly ranged from very poor-to-fair due to bias arising from the lack of control groups, randomization, and blinding to treatment/analysis. Larger adequately controlled studies are required to confirm the association between these medications and CSCR.
Genomic analysis
Certain gene variants (complement factor H [CFH], age-related maculopathy susceptibility 2 [ARMS2], nuclear receptor subfamily 3 group C member 2 [NR3C2; mineralocorticoid receptor], and CDH5) have also been associated with increased risk for CSCR [95, 97, 106, 107].
CFH is a major regulatory protein that inactivates the complement system [108]. Genomic and proteomic analysis hint that CFH is upregulated in CSCR patients suggesting that dysregulation of complement system may play a role in pathogenesis [106, 109, 110]. Additionally, CFH can bind and stabilize adrenomedullin, a member of the calcitonin peptides family which has vasodilatory effects on choroid vessels [108, 111]. Other genes involved in the complement pathway are also linked to the development of CSCR including a cluster of differentiation 46 (CD46) and complement component 4B (C4B) [106, 112].
Lastly, allele variants of transcription factor GATA-5 and tumor necrosis factor receptor superfamily member-10A are associated with CSCR where the former may play a role in choroidal endothelial cell dysfunction and the latter is thought to create an imbalance in hormone secretion by the adrenal glands [113]. Regardless of the associations predicted by the genome-wide analysis, direct evidence such as proteomic/miRNA expression profile, co-localization and functional analysis of proteins at specific tissue, and morphological features using immunohistochemistry are needed to confirm their role in the development of CSCR.
Preclinical studies
Progress in understanding CSCR pathogenesis has not accelerated and some preclinical studies have attempted to shed light on this topic. Preclinical studies date back to 1981 with a significant gap until the last decade (9 studies since 2010). Although many were exploratory studies, the CAMARADES tool demonstrated diminished quality due to high risk for bias (no randomization to treatment and lacked blinding to treatment/data analysis), small sample size, some lacking exploration of cellular mechanisms, and few limited to abstracts or preliminary reports (Tables 2 and S8).
The earliest animal model was developed about four decades ago in which administration of adrenaline in monkeys produced CSCR phenotype seen on fluorescein angiography [114]. While this was an essential step forward in understanding CSCR, the overall quality of the study was poor with small sample size, high risk for bias, no detailed exploration of molecular mechanism, and the model has not been explored since its conception. As such, the role of autonomic dysregulation in CSCR relied primarily on indirect evidence from in vitro studies [83] and a small number of human case series showing association between disease and the use of sympathetic medications as discussed above.
Only two preclinical studies (by the same research group) have produced CSCR phenotype in rats with intraocular injection of corticosteroids [21, 92]. They showed that MR activation could produce CSCR phenotype and was ameliorated with MR antagonist but, they were small sized (n = 3–8 per group), subject to performance and detection bias (not blinded to treatment/analysis), and have not been validated by other independent research groups. In support of this mechanism, mice over-expressing MR produced a phenotype similar to CSCR but was only reported as a preliminary result without further investigation [5]. While exciting, larger and more stringently controlled pre-clinical studies are need to dissect the role of MR in CSCR [95]. It should also be noted that findings in pre-clinical studies, especially small animal models, may not necessarily translate to humans due to anatomical and physiological differences. For example, mice lack macula and foveal pit (an important area of CSCR development), have thinner Bruch’s membrane and show many differences in RPE gene expression controlling processes like oxidative stress, immunoregulation, structural proteins, and cell-signaling pathways [115]. As such, larger animal models more closely modeling human eyes, such as non-human primates, are likely more appropriate for studying CSCR disease [116].
To support the hypothesis of mechanical obstruction, one study produced CSCR phenotype in mice after ligating the vortex vein [117]. However, this study was subject to detection bias, small-sized (n = 6 per group), did not report replicates for various experiments (e.g. choroid thickness measured by optical coherence tomography or gene expression analysis by microarray), and lacked appropriate immunohistochemistry staining controls. Nonetheless, this model has the potential to allow researchers to explore the role of venous congestion on choroidal circulation, RPE function, and structural alterations in the blood-retinal barrier.
Other animal models that mechanically induce retinal detachment (i.e., injecting solutions into the sub-retinal space) are likely not appropriate models for CSCR as they do not address underlying etiology [118, 119]. However, they may have application in exploring laser treatment modalities to aid with earlier fluid reabsorption.
Lastly, the new mutant mouse strain (nm3342, or also known as retinal pigment epithelium atrophy 1 [rpea1]), develops early central exudative retinal detachment followed by late-onset of RPE atrophy mimicking chronic CSCR [120, 121]. The rpea1 mice contain a mutation in the in the Prkcq gene producing an aberrant protein kinase Cθ (PKCθ; expressed mostly in RPE/choroid tissue). Aberrant PKCθ disrupts RPE cytoskeletal structure and metabolism and may also disturb choroid endothelial cell barrier function [120]. Unfortunately, the role of protein kinase C isoforms in the development of CSCR in humans remains unknown but, it provides an intriguing avenue for future research.
Conclusion
Evidence is building that CSCR is a complex and multifactorial disease with varying phenotypic presentations influenced by genetic, environmental, exogenous, and endogenous hormonal and paracrine factors. Our analysis of the literature showed that many studies are inadequately powered, missing appropriate controls, lack long-term follow up, and have methodology prone to various biases thus, the etiology of CSCR remains elusive. However, preclinical and clinical studies have provided useful insight to begin understanding the disease mechanism. Activation of mineralocorticoid receptors is postulated to play a central role in formation of CSCR but targeting this pathway for treatment has produced inconsistent results. Investigations in the future may explore other mechanisms including inflammation, oxidative stress, and venous congestion. A newer focus on vortex veins compression caused by thickened/ridged sclera is promising for understanding disease and developing future therapies. Still, a unified model is lacking to explain the formation of choroid hyperpermeability and RPE dysfunction seen in CSCR. While we explored over two decades of work investigating the etiology of CSCR, intuitively the pathogenic mechanisms causing choroid thickness, choriocapillaris ischemia, and RPE dysfunction likely overlap with the new and exciting concept of pachychoroid disease spectrum. The development of well-controlled and rigorously designed studies is needed to dissect the mechanisms leading to pachychoroid disease while opening avenues for new interventions.
References
van Rijssen TJ, van Dijk EHC, Yzer S, Ohno-Matsui K, Keunen JEE, Schlingemann RO, et al. Central serous chorioretinopathy: towards an evidence-based treatment guideline. Prog Retin Eye Res. 2019;73:100770.
Dansingani KK, Balaratnasingam C, Naysan J, Freund KB. En face imaging of pachychoroid spectrum disorders with swept-source optical coherence tomography. Retina. 2016;36:499–516.
Liew G, Quin G, Gillies M, Fraser-Bell S. Central serous chorioretinopathy: a review of epidemiology and pathophysiology. Clin Exp Ophthalmol. 2013;41:201–14.
Daruich A, Matet A, Marchionno L, De Azevedo JD, Ambresin A, Mantel I, et al. Acute central serous chorioretinopathy: factors influencing episode duration. Retina. 2017;37:1905–15.
Daruich A, Matet A, Dirani A, Bousquet E, Zhao M, Farman N, et al. Central serous chorioretinopathy: recent findings and new physiopathology hypothesis. Prog Retin Eye Res. 2015;48:82–118.
Imamura Y, Fujiwara T, Spaide RF. Fundus autofluorescence and visual acuity in central serous chorioretinopathy. Ophthalmology. 2011;118:700–5.
Gackle HC, Lang GE, Freissler KA, Lang GK. [central serous chorioretinopathy. Clinical, fluorescein angiography and demographic aspects]. Ophthalmologe. 1998;95:529–33.
Sahoo NK, Singh SR, Kammari P, Jonnadula GB, Das AV, Chhablani J. Prevalence and profile of central serous chorioretinopathy in an indian cohort. Nepal J Ophthalmol. 2019;11:5–10.
Mrejen S, Balaratnasingam C, Kaden TR, Bottini A, Dansingani K, Bhavsar KV, et al. Long-term visual outcomes and causes of vision loss in chronic central serous chorioretinopathy. Ophthalmology. 2019;126:576–88.
Sahoo NK, Singh SR, Rajendran A, Shukla D, Chhablani J. Masqueraders of central serous chorioretinopathy. Surv Ophthalmol. 2019;64:30–44.
Teussink MM, Breukink MB, van Grinsven MJ, Hoyng CB, Klevering BJ, Boon CJ, et al. Oct angiography compared to fluorescein and indocyanine green angiography in chronic central serous chorioretinopathy. Investig Ophthalmol Vis Sci. 2015;56:5229–37.
Chan SY, Wang Q, Wei WB, Jonas JB. Optical coherence tomographic angiography in central serous chorioretinopathy. Retina. 2016;36:2051–8.
Min JY, Lv Y, Yu S, Gong YY. Findings of oct-angiography compared to fluorescein and indocyanine green angiography in central serous chorioretinopathy. Lasers Surg Med. 2018;50:987–93.
Manayath GJ, Shah VS, Saravanan VR, Narendran V. Polypoidal choroidal vasculopathy associated with central serous chorioretinopathy: pachychoroid spectrum of diseases. Retina. 2018;38:1195–204.
Yang L, Jonas JB, Wei W. Optical coherence tomography-assisted enhanced depth imaging of central serous chorioretinopathy. Investig Ophthalmol Vis Sci. 2013;54:4659–65.
Yanagi Y. Pachychoroid disease: a new perspective on exudative maculopathy. Jpn J Ophthalmol. 2020;64:323–37.
Warrow DJ, Hoang QV, Freund KB. Pachychoroid pigment epitheliopathy. Retina. 2013;33:1659–72.
Castro-Correia J, Coutinho MF, Rosas V, Maia J. Long-term follow-up of central serous retinopathy in 150 patients. Doc Ophthalmol. 1992;81:379–86.
Mohabati D, van Rijssen TJ, van Dijk EH, Luyten GP, Missotten TO, Hoyng CB, et al. Clinical characteristics and long-term visual outcome of severe phenotypes of chronic central serous chorioretinopathy. Clin Ophthalmol. 2018;12:1061–70.
Takahashi A, Ooto S, Yamashiro K, Tamura H, Oishi A, Miyata M, et al. Pachychoroid geographic atrophy: clinical and genetic characteristics. Ophthalmol Retin. 2018;2:295–305.
Zhao M, Célérier I, Bousquet E, Jeanny JC, Jonet L, Savoldelli M, et al. Mineralocorticoid receptor is involved in rat and human ocular chorioretinopathy. J Clin Investig. 2012;122:2672–9.
Pang CE, Shah VP, Sarraf D, Freund KB. Ultra-widefield imaging with autofluorescence and indocyanine green angiography in central serous chorioretinopathy. Am J Ophthalmol. 2014;158:362–71e2.
Kishi S, Matsumoto H, Sonoda S, Hiroe T, Sakamoto T, Akiyama H. Geographic filling delay of the choriocapillaris in the region of dilated asymmetric vortex veins in central serous chorioretinopathy. PLoS ONE. 2018;13:e0206646.
Terao N, Koizumi H, Kojima K, Yamagishi T, Nagata K, Kitazawa K, et al. Association of upregulated angiogenic cytokines with choroidal abnormalities in chronic central serous chorioretinopathy. Investig Ophthalmol Vis Sci. 2018;59:5924–31.
Jung SH, Kim KA, Sohn SW, Yang SJ. Cytokine levels of the aqueous humour in central serous chorioretinopathy. Clin Exp Optom. 2014;97:264–9.
Erol MK, Balkarli A, Yucel O, Akar Y, Dogan B, Suren E. Neutrophil/lymphocyte ratio and mean platelet volume in central serous chorioretinopathy. Ther Clin Risk Manag. 2017;13:945–50.
Ma LL, Wang YY, Yang ZH, Huang D, Weng H, Zeng XT. Methodological quality (risk of bias) assessment tools for primary and secondary medical studies: What are they and which is better? Mil Med Res. 2020;7:1–11.
Institute JB. Critical appraisal tools. Joanna Briggs Institute, The University of Adelaide. 2020. https://jbi.global/critical-appraisal-tools.
Sena E, van der Worp HB, Howells D, Macleod M. How can we improve the pre-clinical development of drugs for stroke? Trends Neurosci. 2007;30:433–9.
Hiroe T, Kishi S. Dilatation of asymmetric vortex vein in central serous chorioretinopathy. Ophthalmol Retin. 2018;2:152–61.
Imanaga N, Terao N, Nakamine S, Tamashiro T, Wakugawa S, Sawaguchi K, et al. Scleral thickness in central serous chorioretinopathy. Ophthalmol Retina. 2021;5:285–91.
Venkatesh P, Chawla R, Tripathy K, Singh HI, Bypareddy R. Scleral resection in chronic central serous chorioretinopathy complicated by exudative retinal detachment. Eye Vis. 2016;3:23.
Verma S, Kumar V, Azad S, Bhayana AA, Surve A, Kumar S, et al. Focal choroidal excavation: review of literature. Br J Ophthalmol. 2021;105:1043–8.
Chung CY, Li SH, Li KKW. Focal choroidal excavation—morphological features and clinical correlation. Eye. 2017;31:1373–9.
Obata R, Takahashi H, Ueta T, Yuda K, Kure K, Yanagi Y. Tomographic and angiographic characteristics of eyes with macular focal choroidal excavation. Retina. 2013;33:1201–10.
Yang HS, Kang TG, Park H, Heo JS, Park J, Lee KS, et al. Quantitative evaluation of choriocapillaris using optical coherence tomography and optical coherence tomography angiography in patients with central serous chorioretinopathy after half-dose photodynamic therapy. PLoS ONE. 2020;15:e0227718.
Chan WM, Lam DS, Lai TY, Tam BS, Liu DT, Chan CK. Choroidal vascular remodelling in central serous chorioretinopathy after indocyanine green guided photodynamic therapy with verteporfin: a novel treatment at the primary disease level. Br J Ophthalmol. 2003;87:1453–8.
Sirakaya E, Duru Z, Kucuk B, Duru N. Monocyte to high-density lipoprotein and neutrophil-to-lymphocyte ratios in patients with acute central serous chorioretinopathy. Indian J Ophthalmol. 2020;68:854–8.
Mazzolani F, Togni S. Oral administration of a curcumin-phospholipid delivery system for the treatment of central serous chorioretinopathy: a 12-month follow-up study. Clin Ophthalmol. 2013;7:939–45.
Khan NA, Khan AA, Khan A, Khan A, Memon JI, Shaikh M. Treatment of central serous chorioretinopathy (csc) using diclofenac through different routes of administration, a comparative study. Adv Ophthalmol Vis Syst. 2017;6:101–5.
Bahadorani S, Maclean K, Wannamaker K, Chu ER, Gresores N, Sohn JH, et al. Treatment of central serous chorioretinopathy with topical nsaids. Clin Ophthalmol. 2019;13:1543–8.
Dursun A, Toker MI, Ozec AV, Bozali E, Kirboga K, Dursun FG, et al. Relationship between mean platelet volume and central serous chorioretinopathy. Int Ophthalmol. 2017;37:119–24.
Caccavale A, Romanazzi F, Imparato M, Negri A, Morano A, Ferentini F. Low-dose aspirin as treatment for central serous chorioretinopathy. Clin Ophthalmol. 2010;4:899–903.
Lim JW, Kim MU, Shin MC. Aqueous humor and plasma levels of vascular endothelial growth factor and interleukin-8 in patients with central serous chorioretinopathy. Retina. 2010;30:1465–71.
Shin MC, Lim JW. Concentration of cytokines in the aqueous humor of patients with central serous chorioretinopathy. Retina. 2011;31:1937–43.
Sakai T, Tsuneoka H. Reduced blood serotonin levels in chronic central serous chorioretinopathy. Ophthalmol Retina. 2017;1:145–8.
Kimura T, Araki T, Komuku Y, Iwami H, Gomi F. Central serous chorioretinopathy and blood serotonin concentrations. J Clin Med. 2021;10:558.
Matet A, Jaworski T, Bousquet E, Canonica J, Gobeaux C, Daruich A, et al. Lipocalin 2 as a potential systemic biomarker for central serous chorioretinopathy. Sci Rep. 2020;10:20175.
Conde J, Lazzaro V, Scotece M, Abella V, Villar R, López V, et al. Corticoids synergize with il-1 in the induction of lcn2. Osteoarthr Cartil. 2017;25:1172–8.
Ji S, Wei Y, Chen J, Tang S. Clinical efficacy of anti-vegf medications for central serous chorioretinopathy: a meta-analysis. Int J Clin Pharm. 2017;39:514–21.
Bae SH, Heo J, Kim C, Kim TW, Shin JY, Lee JY, et al. Low-fluence photodynamic therapy versus ranibizumab for chronic central serous chorioretinopathy: One-year results of a randomized trial. Ophthalmology. 2014;121:558–65.
Chung YR, Kim JW, Song JH, Park A, Kim MH. Twelve-month efficacy of intravitreal bevacizumab injection for chronic, atypical, or recurrent central serous chorioretinopathy. Retina. 2019;39:134–42.
Lim JW, Ryu SJ, Shin MC. The effect of intravitreal bevacizumab in patients with acute central serous chorioretinopathy. Korean J Ophthalmol. 2010;24:155–8.
Karska-Basta I, Pociej-Marciak W, Chrzaszcz M, Kubicka-Trzaska A, Debicka-Kumela M, Gawecki M, et al. Imbalance in the levels of angiogenic factors in patients with acute and chronic central serous chorioretinopathy. J Clin Med 2021;10:1087.
Shiragami C, Takasago Y, Osaka R, Kobayashi M, Ono A, Yamashita A, et al. Clinical features of central serous chorioretinopathy with type 1 choroidal neovascularization. Am J Ophthalmol. 2018;193:80–6.
Loo RH, Scott IU, Flynn HW Jr, Gass JD, Murray TG, Lewis ML, et al. Factors associated with reduced visual acuity during long-term follow-up of patients with idiopathic central serous chorioretinopathy. Retina. 2002;22:19–24.
Bousquet E, Bonnin S, Mrejen S, Krivosic V, Tadayoni R, Gaudric A. Optical coherence tomography angiography of flat irregular pigment epithelium detachment in chronic central serous chorioretinopathy. Retina. 2018;38:629–38.
Cheung CMG, Lee WK, Koizumi H, Dansingani K, Lai TYY, Freund KB. Pachychoroid disease. Eye. 2019;33:14–33.
Kitaya N, Nagaoka T, Hikichi T, Sugawara R, Fukui K, Ishiko S, et al. Features of abnormal choroidal circulation in central serous chorioretinopathy. Br J Ophthalmol. 2003;87:709–12.
Podkowinski D, Foessl B, de Sisternes L, Beka S, Mursch-Edlmayr AS, Strauss RW, et al. Early alterations in retinal microvasculature on swept-source optical coherence tomography angiography in acute central serous chorioretinopathy. Sci Rep. 2021;11:3129.
Saito M, Saito W, Hirooka K, Hashimoto Y, Mori S, Noda K, et al. Pulse waveform changes in macular choroidal hemodynamics with regression of acute central serous chorioretinopathy. Invest Ophthalmol Vis Sci. 2015;56:6515–22.
Baek J, Kook L, Lee WK. Choriocapillaris flow impairments in association with pachyvessel in early stages of pachychoroid. Sci Rep. 2019;9:5565.
Schworm B, Luft N, Keidel LF, Hagenau F, Kern C, Herold T, et al. Response of neovascular central serous chorioretinopathy to an extended upload of anti-vegf agents. Graefes Arch Clin Exp Ophthalmol. 2020;258:1013–21.
Peiretti E, Caminiti G, Serra R, Querques L, Pertile R, Querques G. Anti-vascular endothelial growth factor therapy versus photodynamic therapy in the treatment of choroidal neovascularization secondary to central serous chorioretinopathy. Retina. 2018;38:1526–32.
Hata M, Yamashiro K, Ooto S, Oishi A, Tamura H, Miyata M, et al. Intraocular vascular endothelial growth factor levels in pachychoroid neovasculopathy and neovascular age-related macular degeneration. Investig Ophthalmol Vis Sci. 2017;58:292–8.
Terao N, Koizumi H, Kojima K, Yamagishi T, Yamamoto Y, Yoshii K, et al. Distinct aqueous humour cytokine profiles of patients with pachychoroid neovasculopathy and neovascular age-related macular degeneration. Sci Rep. 2018;8:10520.
Matsumoto H, Hiroe T, Morimoto M, Mimura K, Ito A, Akiyama H. Efficacy of treat-and-extend regimen with aflibercept for pachychoroid neovasculopathy and type 1 neovascular age-related macular degeneration. Jpn J Ophthalmol. 2018;62:144–50.
Kunikata H, Sato R, Nishiguchi KM, Nakazawa T. Systemic oxidative stress level in patients with central serous chorioretinopathy. Graefes Arch Clin Exp Ophthalmol. 2020;258:1575–7.
Türkcü FM, Yüksel H, Yüksel H, Sahin A, Cinar Y, Cingü AK, et al. Serum dehydroepiandrosterone sulphate, total antioxidant capacity, and total oxidant status in central serous chorioretinopathy. Graefes Arch Clin Exp Ophthalmol. 2014;252:17–21.
Turkoglu EB, Dikci S, Çelik E, Erel Ö, Neselioglu S, Alışık M, et al. Thiol/disulfide homeostasis in patients with central serous chorioretinopathy. Curr Eye Res. 2016;41:1489–91.
Altinkaynak H, Kurkcuoglu PZ, Caglayan M, Yorgun MA, Yuksel N, Kosekahya P, et al. A novel marker in acute central serous chorioretinopathy: Thiol/disulfide homeostasis. Int Ophthalmol. 2018;38:175–81.
Yavaş GF, Küsbeci T, Kaşikci M, Günay E, Doğan M, Unlü M, et al. Obstructive sleep apnea in patients with central serous chorioretinopathy. Curr Eye Res. 2014;39:88–92.
Spiers JG, Chen HJ, Sernia C, Lavidis NA. Activation of the hypothalamic-pituitary-adrenal stress axis induces cellular oxidative stress. Front Neurosci. 2014;8:456.
Vijayvergiya R, Vadivelu R. Role of helicobacter pylori infection in pathogenesis of atherosclerosis. World J Cardiol. 2015;7:134–43.
Ratanasukon M, Bhurayanontachai P, Jirarattanasopa P. High-dose antioxidants for central serous chorioretinopathy; the randomized placebo-controlled study. BMC Ophthalmol. 2012;12:20.
McDougal DH, Gamlin PD. Autonomic control of the eye. Compr Physiol. 2015;5:439–73.
Michael JC, Pak J, Pulido J, de Venecia G. Central serous chorioretinopathy associated with administration of sympathomimetic agents. Am J Ophthalmol. 2003;136:182–5.
Tewari HK, Gadia R, Kumar D, Venkatesh P, Garg SP. Sympathetic-parasympathetic activity and reactivity in central serous chorioretinopathy: A case-control study. Invest Ophthalmol Vis Sci. 2006;47:3474–8.
Pierce KK, Lane RG. Central serous chorioretinopathy associated with the use of ephedra. Retin Cases Brief Rep. 2009;3:376–8.
Chen LC, Ma JW, Shieh PC, Horng CT. Oral treatment of central serous chorioretinopathy patients using propranolol tablets. Pharmaceuticals. 2020;13:336.
Chrapek O, Jirkova B, Kandrnal V, Rehak J, Sin M. Treatment of central serous chorioretinopathy with beta-blocker metipranolol. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2015;159:120–3.
Kianersi F, Fesharaki F. Effects of propranolol in patients with central serous chorioretinopathy. J Res Med Sci. 2008;13:103–7.
Sibayan SA, Kobuch K, Spiegel D, Eckert E, Leser R, Monzer J, et al. Epinephrine, but not dexamethasone, induces apoptosis in retinal pigment epithelium cells in vitro: possible implications on the pathogenesis of central serous chorioretinopathy. Graefes Arch Clin Exp Ophthalmol. 2000;238:515–9.
Ullian ME. The role of corticosteriods in the regulation of vascular tone. Cardiovasc Res. 1999;41:55–64.
Scarinci F, Ghiciuc CM, Patacchioli FR, Palmery M, Parravano M. Investigating the hypothesis of stress system dysregulation as a risk factor for central serous chorioretinopathy: a literature mini-review. Curr Eye Res. 2019;44:583–9.
Liu B, Deng T, Zhang J. Risk factors for central serous chorioretinopathy: a systematic review and meta-analysis. Retina. 2016;36:9–19.
Jain AK, Kaines A, Schwartz S. Bilateral central serous chorioretinopathy resolving rapidly with treatment for obstructive sleep apnea. Graefes Arch Clin Exp Ophthalmol. 2010;248:1037–9.
Schellevis RL, Altay L, Kalisingh A, Mulders TWF, Sitnilska V, Hoyng CB, et al. Elevated steroid hormone levels in active chronic central serous chorioretinopathy. Investig Ophthalmol Vis Sci. 2019;60:3407–13.
van Haalen FM, van Dijk EHC, Andela CD, Dijkman G, Biermasz NR, Pereira AM, et al. Maladaptive personality traits, psychological morbidity and coping strategies in chronic central serous chorioretinopathy. Acta Ophthalmol. 2019;97:e572–e9.
Liang ZQ, Huang LZ, Qu JF, Zhao MW. Association between endogenous cortisol level and the risk of central serous chorioretinopathy: a meta-analysis. Int J Ophthalmol. 2018;11:296–300.
Arndt C, Sari A, Ferre M, Parrat E, Courtas D, De Seze J, et al. Electrophysiological effects of corticosteroids on the retinal pigment epithelium. Investig Ophthalmol Vis Sci. 2001;42:472–5.
Zhao M, Valamanesh F, Celerier I, Savoldelli M, Jonet L, Jeanny JC, et al. The neuroretina is a novel mineralocorticoid target: Aldosterone up-regulates ion and water channels in muller glial cells. FASEB J. 2010;24:3405–15.
Caccavale A, Romanazzi F, Imparato M, Negri A, Morano A, Ferentini F. Central serous chorioretinopathy: a pathogenetic model. Clin Ophthalmol. 2011;5:239–43.
Sogutlu Sari E, Yazici A, Eser B, Erol MK, Kilic A, Ermis SS, et al. The prevalence of 4g/5g polymorphism of plasminogen activator inhibitor-1 (pai-1) gene in central serous chorioretinopathy and its association with plasma pai-1 levels. Cutan Ocul Toxicol. 2014;33:270–4.
Schubert C, Pryds A, Zeng S, Xie Y, Freund KB, Spaide RF, et al. Cadherin 5 is regulated by corticosteroids and associated with central serous chorioretinopathy. Hum Mutat. 2014;35:859–67.
Lotery A, Sivaprasad S, O'connell A, Harris RA, Culliford L, Ellis L, et al. Eplerenone for chronic central serous chorioretinopathy in patients with active, previously untreated disease for more than 4 months (vici): a randomised, double-blind, placebo-controlled trial. Lancet. 2020;395:294–303.
van Dijk EHC, Schellevis RL, van Bergen M, Breukink MB, Altay L, Scholz P, et al. Association of a haplotype in the nr3c2 gene, encoding the mineralocorticoid receptor, with chronic central serous chorioretinopathy. JAMA Ophthalmol. 2017;135:446–51.
Bousquet E, Dhundass M, Lejoyeux R, Shinojima A, Krivosic V, Mrejen S, et al. Predictive factors of response to mineralocorticoid receptor antagonists in nonresolving central serous chorioretinopathy. Am J Ophthalmol. 2019;198:80–7.
Pichi F, Carrai P, Ciardella A, Behar-Cohen F, Nucci P. Central Serous Chorioretinopathy Study G. Comparison of two mineralcorticosteroids receptor antagonists for the treatment of central serous chorioretinopathy. Int Ophthalmol. 2017;37:1115–25.
Gordon-Bennett P, Rimmer T. Central serous chorioretinopathy following oral tadalafil. Eye. 2012;26:168–9.
Fraunfelder FW, Fraunfelder FT. Central serous chorioretinopathy associated with sildenafil. Retina. 2008;28:606–9.
Jain M, Nevin RL, Ahmed I. Mefloquine-associated dizziness, diplopia, and central serous chorioretinopathy: a case report. J Med Case Rep. 2016;10:305.
Jain M. Quetiapine associated central serous chorioretinopathy: implicit role of serotonin and dopamine pathways. Indian J Ophthalmol. 2019;67:292–4.
Yuan Z, Hein TW, Rosa RH Jr., Kuo L. Sildenafil (viagra) evokes retinal arteriolar dilation: dual pathways via nos activation and phosphodiesterase inhibition. Investig Ophthalmol Vis Sci. 2008;49:720–5.
Breazzano MP, Coleman DJ, Chen RWS, Chang S, Daly S, Tsang SH. Prospective impact of sildenafil on chronic central serous chorioretinopathy: Pisces trial. Ophthalmol Retin. 2020;4:1119–23.
Schellevis RL, van Dijk EHC, Breukink MB, Altay L, Bakker B, Koeleman BPC, et al. Role of the complement system in chronic central serous chorioretinopathy: a genome-wide association study. JAMA Ophthalmol. 2018;136:1128–36.
de Jong EK, Breukink MB, Schellevis RL, Bakker B, Mohr JK, Fauser S, et al. Chronic central serous chorioretinopathy is associated with genetic variants implicated in age-related macular degeneration. Ophthalmology. 2015;122:562–70.
Ferreira VP, Pangburn MK, Cortes C. Complement control protein factor h: the good, the bad, and the inadequate. Mol Immunol. 2010;47:2187–97.
Kowalczuk L, Matet A, Dor M, Bararpour N, Daruich A, Dirani A, et al. Proteome and metabolome of subretinal fluid in central serous chorioretinopathy and rhegmatogenous retinal detachment: a pilot case study. Transl Vis Sci Technol. 2018;7:3.
Miki A, Kondo N, Yanagisawa S, Bessho H, Honda S, Negi A. Common variants in the complement factor h gene confer genetic susceptibility to central serous chorioretinopathy. Ophthalmology. 2014;121:1067–72.
Dorner GT, Garhöfer G, Huemer KH, Golestani E, Zawinka C, Schmetterer L, et al. Effects of adrenomedullin on ocular hemodynamic parameters in the choroid and the ophthalmic artery. Investig Ophthalmol Vis Sci. 2003;44:3947–51.
Breukink MB, Schellevis RL, Boon CJ, Fauser S, Hoyng CB, den Hollander AI, et al. Genomic copy number variations of the complement component c4b gene are associated with chronic central serous chorioretinopathy. Investig Ophthalmol Vis Sci. 2015;56:5608–13.
Hosoda Y, Miyake M, Schellevis RL, Boon CJF, Hoyng CB, Miki A, et al. Genome-wide association analyses identify two susceptibility loci for pachychoroid disease central serous chorioretinopathy. Commun Biol. 2019;2:468.
Yoshioka H, Katsume Y, Akune H. Experimental central serous chorioretinopathy in monkey eyes: fluorescein angiographic findings. Ophthalmologica. 1982;185:168–78.
Bennis A, Gorgels TGMF, Ten Brink JB, van der Spek PJ, Bossers K, Heine VM, et al. Comparison of mouse and human retinal pigment epithelium gene expression profiles: potential implications for age-related macular degeneration. PLoS ONE. 2015;10:e0141597.
Park HK, Jo W, Choi HJ, Kim B, Lee G, Seo J, et al. Usefulness of optical coherence tomography to detect central serous chorioretinopathy in monkeys. J Appl Toxicol. 2015;35:199–204.
Matsumoto H, Mukai R, Hoshino J, Oda M, Matsuzaki T, Ishizaki Y, et al. Choroidal congestion mouse model: could it serve as a pachychoroid model? PLoS ONE. 2021;16:e0246115.
Negi A, Marmor MF. Experimental serous retinal detachment and focal pigment epithelial damage. Arch Ophthalmol. 1984;102:445–9.
Matsumoto H, Miller JW, Vavvas DG. Retinal detachment model in rodents by subretinal injection of sodium hyaluronate. J Vis Exp. 2013:50660.
Ji X, Liu Y, Hurd R, Wang J, Fitzmaurice B, Nishina PM, et al. Retinal pigment epithelium atrophy 1 (rpea1): a new mouse model with retinal detachment caused by a disruption of protein kinase c, theta. Investig Ophthalmol Vis Sci. 2016;57:877–88.
Luna G, Lewis GP, Linberg KA, Chang B, Hu Q, Munson PJ, et al. Anatomical and gene expression changes in the retinal pigmented epithelium atrophy 1 (rpea1) mouse: a potential model of serous retinal detachment. Investig Ophthalmol Vis Sci. 2016;57:4641–54.
Author information
Authors and Affiliations
Contributions
PK, AG and MSB were responsible for designing the study protocol. PK and AG were responsible for screening studies, data acquisition, analysis, organizing tables/figures and drafting the manuscript. MSB, CG, RK, and SGC were involved in editing the manuscript, helping revise the protocol and providing feedback. The final version was of the manuscript was approved by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Kanda, P., Gupta, A., Gottlieb, C. et al. Pathophysiology of central serous chorioretinopathy: a literature review with quality assessment. Eye 36, 941–962 (2022). https://doi.org/10.1038/s41433-021-01808-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-021-01808-3
This article is cited by
-
Central serous chorioretinopathy with a golden sheen
Eye (2024)
-
Presentation and outcome of central serous chorioretinopathy with and without pachychoroid
Eye (2024)
-
Pathomechanisms in central serous chorioretinopathy: A recent update
International Journal of Retina and Vitreous (2023)
-
Central serous chorioretinopathy: updates in the pathogenesis, diagnosis and therapeutic strategies
Eye and Vision (2023)
-
Comparisons of choroidal blood flow velocity between initial-onset acute uveitis associated with Vogt–Koyanagi–Harada disease and acute central serous chorioretinopathy
Eye (2023)
